

Abstract
Sensory hair cells in the cochlea, like most neuronal populations that are postmitotic, terminally differentiated, and non-regenerating, depend on robust mechanisms of self-renewal for lifelong survival. We report that hair cell homeostasis requires a specific sub-branch of the DNA damage nucleotide excision repair pathway, termed transcription-coupled repair (TCR). Cockayne syndrome (CS), caused by defects in TCR, is a rare DNA repair disorder with a broad clinical spectrum that includes sensorineural hearing loss. We tested hearing and analyzed the cellular integrity of the organ of Corti in two mouse models of this disease with mutations in the Csb gene (CSBm/m mice) and Csa gene (Csa−/− mice), respectively. Csbm/m and Csa−/− mice manifested progressive hearing loss, as measured by an increase in auditory brainstem response thresholds. In contrast to wild-type mice, mutant mice showed reduced or absent otoacoustic emissions, suggesting cochlear outer hair cell impairment. Hearing loss in Csbm/m and Csa−/− mice correlated with progressive hair cell loss in the base of the organ of Corti, starting between 6 and 13 weeks of age, which increased by 16 weeks of age in a basal-to-apical gradient, with outer hair cells more severely affected than inner hair cells. Our data indicate that the hearing loss observed in CS patients is reproduced in mouse models of this disease. We hypothesize that accumulating DNA damage, secondary to the loss of TCR, contributes to susceptibility to hearing loss.
Keywords: Cockayne syndrome, csb/csa, DNA damage, DNA repair, hair cell, hearing loss
Introduction
Cockayne syndrome (CS) is a DNA repair disorder caused by mutations in CS group B (CSB) and CS group A (CSA) genes. This rare autosomal-recessive disorder is characterized by numerous developmental and physiological deficits, including hearing loss (Nance and Berry, 1992). Since patients with CS also exhibit traits reminiscent of normal aging, CS has been classified as a segmental progeroid syndrome (Martin, 2005). About 80% of CS cases are caused by a defect in the CSB (also called ERCC6) gene, with the remaining cases caused by a defect in the CSA (also called ERCC8) gene and other less common genetic defects (Mallery et al., 1998). Mouse mutants in the Csb or Csa gene (Csbm/m or Csa−/−, respectively) have a clear deficit in transcription-coupled repair (TCR), which removes DNA lesions from transcribed (active) genes, and are predisposed to UVB radiation-induced skin cancer, among other defects (van der Horst et al., 1997, 2002).
Environmental stresses, such as from ototoxic drugs, noise, and aging, are believed to be the major factors leading to the death of sensory hair cells in the inner ear, and a primary cause of hearing loss (Schacht et al., 2012). However, very little is known about the underlying genetic susceptibilities that lead to the significant variability in the predisposition to hearing loss (Yamasoba et al., 2013). DNA damage signaling and DNA repair pathways play a major role in maintaining the viability of cells, both during development and in the lifelong homeostatic maintenance of highly differentiated, non-regenerating cells such as neurons (McMurray, 2005) in the mature animal. Although the DNA damage-signaling pathway has been implicated in ototoxin-induced hair cell death (Zhang et al., 2003) and in regulating age-dependent proliferation capacity of nonsensory supporting cells (Laos et al., 2014), the DNA repair systems active in hair and supporting cells remain largely uncharacterized.
Nucleotide excision repair (NER) is a universal pathway involved in the repair of a wide spectrum of DNA-distorting lesions. NER operates through the following two subpathways: global genome NER (GG-NER), which repairs DNA damage throughout the genome; and transcription-coupled NER (TC-NER), which removes transcription-blocking lesions from active genes (Fousteri and Mullenders, 2008). TC-NER is triggered by lesion-stalled RNA polymerase II, resulting in the recruitment of CSB and CSA factors. Additionally, CSB and CSA proteins participate in the repair of other, non-NER types of DNA damage, such as removing transcription-blocking oxidative base lesions, termed TCR (to contrast with TC-NER; Marteijn et al., 2014).
We previously reported that Ercc1δ/− mice, defective in the downstream NER function, display a progressive and accelerated loss of hearing and vision, suggesting that unrepaired DNA damage can induce age-related decline of the auditory and visual systems (Spoor et al., 2012). Hearing loss is one of the recognized clinical features of CS (Nance and Berry, 1992). Here we report that mice with mutations in the CS genes Csb and Csa manifest progressive high-frequency hearing loss and sensory hair cell degeneration starting after 6 weeks of age, providing a working model of Cockayne syndrome-associated hearing loss and implicating the TCR/TC-NER pathway in inner-ear homeostasis.
Materials and Methods
Experimental animals.
The generation, characterization, and PCR genotyping of Csa−/− mice (knock-out mice) and Csbm/m mice (in which the CS1AN patient mutation is mimicked, resulting in a null mouse) have been previously described (van der Horst et al., 1997, 2002; Berg et al., 2000). The absence of wild-type (WT) mRNA and protein in Csbm/m and Csa−/− mice was previously documented (van der Horst et al., 1997, 2002), and the absence of wild-type Csa protein in Csa−/− cochlear cross sections was confirmed by immunostaining with an antibody to Csa (data not shown). Unless stated otherwise, all mice were bred to congeneity in C57BL/6J background (backcrossed >10 generations). Studies were conducted at laboratories in Rotterdam, The Netherlands, and Los Angeles, CA. In Rotterdam, hearing tests were performed on Csbm/m mice. As C57BL/6J mice are known to develop high-frequency hearing loss due to a hypomorphic cadherin 23 allele leading to disorganized hair bundles (Noben-Trauth et al., 2003), Csbm/m mice (n = 2 males and 4 females) were compared with age-matched WT controls (n = 2 males and 5 females) at ages 6, 8, and 12 weeks followed in time; at 36 weeks, different Csbm/m (n = 3 females) and WT (n = 4 males and 1 female) mice were used. Fixed tissue from 2-, 6-, 13- and 16-week-old homozygous Csbm/m mice and WT littermates were sent to the Los Angeles site for histological analysis. All animal studies in Rotterdam were conducted in accordance with Dutch law and were approved by an independent animal ethical committee. In Los Angeles, Csa−/− in C57BL/6J mice and Csbm/m in CBA/CaJ mice were characterized. Csa−/− mice (n = 4 males and 5 females) were compared with age-matched WT controls (n = 5 males and 6 females) for the hearing tests. To test hearing in a strain background lacking age-related hearing loss (AHL) susceptibility, Csbm/m mice were bred onto a CBA/CaJ background [N3 generation; 87.5% CBA/CaJ; n = 3 males and 5 females (Csbm/m); 4 males and 1 female (WT)]. Since AHL susceptibility requires C57BL/6J-derived modifiers that are absent from the CBA/CaJ background in addition to the Cdh23ahl allele (Kane et al., 2012), the chance of the needed modifiers being present in Csb in CBA/CaJ mice was extremely low (p = 0.073, assuming the probability for occurrence for linked loci described by Silver (1995). All animal experiments conducted in Los Angeles were approved by the House Research Institute Institutional Animal Care and Use Committee. No significant differences between male and female groups were seen in any of these experiments, which is consistent with the absence of sex bias in Cockayne syndrome (Nance and Berry, 1992).
Auditory brainstem response and distortion product otoacoustic emission recordings.
Open-field and closed-field auditory brainstem response (ABR) acoustics were used in Rotterdam and Los Angeles, respectively, leading to small systemic differences in ABR stimulus parameters between the two sites. However, all comparisons were extracted from data on cohorts of animals that were tested at the same site. ABR and distortion product otoacoustic emission (DPOAE) recordings were performed as described previously (Spoor et al., 2012), with the following modifications: mice were placed in a sound-attenuated box with the ears at a distance of 4 cm from a frontally placed Radio Shack Super Tweeter 40–1310B loudspeaker (Rotterdam) or through inserted earphones (Los Angeles). The sound pressure level (SPL) of the stimuli ranged between −10 and 110 dB (Rotterdam) or 20 and 105 dB (Los Angeles). For determining ABR thresholds, 500 responses (Rotterdam) or 300 responses (Los Angeles) with artifacts of <30 μV were averaged. Hearing level thresholds were measured at 4, 8, 16, and 32 kHz (Rotterdam), or 4, 8, 12, 16, 24, and 32 kHz (Los Angeles). When an ABR threshold was above the maximum range, it was classified as 115 dB (Rotterdam) or 105 dB (Los Angeles). The presentation of stimuli and the averaging of responses were controlled by custom-made Erasmus University Physiological Response Averager software (Rotterdam) or BioSig software (Los Angeles). Post hoc, groups were compared with two-way ANOVAs and Bonferroni tests. At 36 weeks, differences in means were tested for significance using a one-way ANOVA with Tukey's post hoc test. DPOAE recordings were performed at second stimulus frequency (f2) = 4, 6, 8, 10, 12, and 16 kHz (Rotterdam) or f2 = 8, 12, 16, and 24 kHz (Los Angeles). DPOAEs of WT animals were not above baseline between 4 and 8 kHz, and therefore only results obtained at ≥10 kHz are presented. Differences between groups were analyzed for significance with Student's t test.
Histology.
Animals were asphyxiated by CO2 inhalation, followed by cervical dislocation and 4% paraformaldehyde transcardiac perfusion. Dissected temporal bones were fixed overnight in 4% paraformaldehyde and decalcified in 300 mm EDTA, pH 8.0/PBS at 4°C. After decalcification, cochleae were dissected in PBS, and whole-mount preparations of the organ of Corti were permeabilized in 0.2% Triton X-100/PBS for 30 min, treated with 4% donkey serum/PBS for 1 h, then incubated with anti-MyosinVI (1:500, rabbit; Proteus) or anti-parvalbumin clone PARV-19 (1:500, mouse; Sigma) at 4°C overnight. Rhodamine-conjugated secondary antibodies (1:200; Jackson ImmunoResearch) were used to visualize the labeled hair cells. Basal, middle, and apical regions were imaged using a Leica TCS SP5 confocal microscope, and all MyosinVI-stained cells were counted. Inner and outer hair cell densities were calculated in each specimen by averaging counts of at least three 100 μm segments from each of the basal, middle, and apical regions using ImageJ software (National Institutes of Health). The SEM was calculated from three animals per experimental group. Decalcified cochleae were embedded in paraffin and cut into 8-μm-thick sections that were stained with hematoxylin and eosin, and photographed using a Zeiss Axio Observer.Z1 inverted microscope with AxioVision software.
Results
Progressive hearing loss in mature Csbm/m mice
To investigate the requirement of Csb in maintenance of hearing function, we compared ABRs of Csbm/m mice with those of age-matched wild-type controls on a C57BL/6J background (Fig. 1A). At 6 weeks of age, the ABR thresholds of Csbm/m mice at 16 and 32 kHz were modestly elevated compared with controls (both p < 0.01). Between 6 and 12 weeks of age, the thresholds showed a further increase, with the shifts most pronounced at the ultrasonic frequencies, which are represented in the base of the cochlea (Müller and Smolders, 2005). By 12 weeks of age, Csbm/m mice had undergone a ∼57 dB SPL threshold shift compared with age-matched controls at 16 and 32 kHz, which correspond to the middle and basal regions of the cochlea. Thresholds for wild-type mice were in the normal range for the C57BL/6J background, where a loss of ∼15 dB occurred in the 32 kHz range, as previously reported (Willott and Erway, 1998). At 36 weeks of age, the thresholds of Csbm/m mice showed a further increase at all frequencies compared with age-matched controls (p < 0.01). Additionally, Csbm/m mice backcrossed onto a CBA/CaJ background, which lacks the AHL susceptibility locus and the needed modifiers found in C57BL/6J (Kane et al., 2012), presented a similar time course for hearing threshold increases compared with littermates as Csbm/m in C57BL/6J mice (data not shown), suggesting that the AHL susceptibility is likely decoupled from the Csbm/m-induced hearing loss deficit. These measures indicate that hearing is substantially compromised in Csbm/m mice starting after 6 weeks of age during normal development.
Figure 1.

Csbm/m mice show progressively higher ABR thresholds and reduced DPOAEs in early life. A, ABR thresholds of mice carrying a homozygous mutation in the Csb gene (Csbm/m) and WT controls were measured between 6 and 36 weeks of age. In control mice, measurements at 6 and 8 weeks of age were not significantly different from 12 weeks of age (p = NS for all differences), and thus only 12 week values are shown. The 36-week-old control mice display elevated high-frequency ABR thresholds, as expected in a C57BL/6J strain background (Willott and Erway, 1998); however, thresholds in Csbm/m mice remain significantly higher than those in WT mice at this age (p < 0.01). B, 8- and 12-week-old Csbm/m mice have reduced or absent DPOAEs compared with WT controls. ABR and DPOAE data are shown as the mean ± SEM.
To assess whether the progressive hearing loss observed in Csbm/m mice originated in the cochlea, we recorded DPOAEs, an objective indicator of functioning cochlear outer hair cells. At frequencies between 10 and 16 kHz, DPOAEs in the Csbm/m mice (C57BL/6J) were reduced in amplitude or were absent, both at 8 and at 12 weeks of age, compared with age-matched wild-type controls (p < 0.05 for all differences; Fig. 1B), indicating outer hair cell impairment secondary to Csb deficiency. The combined ABR and DPOAE results provided evidence of cochlear damage in mature Csbm/m mice.
Cochlear hair and supporting cell loss in mature Csbm/m mice
The increased auditory thresholds and absence of DPOAEs in mature Csbm/m mice (Fig. 1A) suggest that the defect was primarily of cochlear origin. We therefore charted cochlear hair cell integrity in a different cohort of mature Csbm/m mice and wild-type littermates on a C57BL/6J background at various ages by performing hair cell counts on MyosinVI-stained cochlear surface preparations. We found normal numbers, and normal arrangements of outer and inner hair cells within the sensory epithelium of postnatal day 14 (data not shown) and 6-week-old Csbm/m mice (Fig. 2A,B). We observed a complete loss of outer hair cells in the basal turn of the organ of Corti in all 13-week-old mutants analyzed, with inner hair cells intact at this age. The middle and apical regions showed a normal complement of sensory hair cells at 13 weeks. As the 4 and 8 kHz ABR frequencies correspond to locations within ∼20% of the apex (Müller and Smolders, 2005), the lack of a clear effect on hair cell densities at the apex is thus in line with the normal ABR thresholds within the 4–8 kHz region at the 12 week time point (Fig. 1A). The deterioration at 32 kHz (∼20% from the base) matches the loss of outer hair cells within the basal region of 13-week-old mutants. By 16 weeks of age, the middle turns presented a variable outer hair cell loss ranging from mild to severe, depending on the exact relative positions from the round window (Fig. 2A), while the apical turns displayed normal numbers and arrangements of sensory hair cells. These observations suggest a pattern of progressive damage to outer hair cells as a result of Csb loss that follows a basal-to-apical gradient. Inner hair cells were less severely affected, but nonetheless showed significant reduction in the number in the base by 16 weeks of age (Fig. 2A). In contrast, wild-type littermates showed no abnormalities in the organ of Corti at all ages analyzed.
Figure 2.
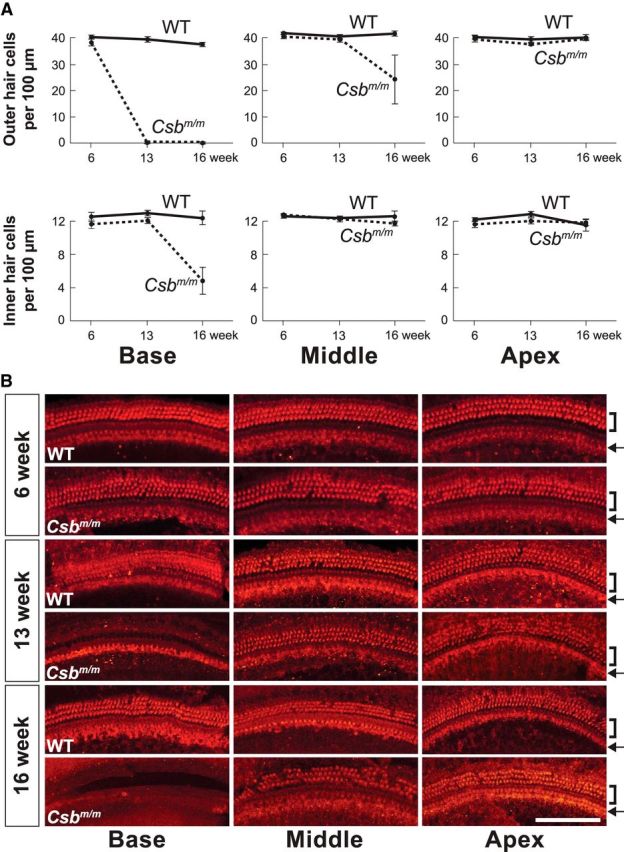
Csbm/m mice manifest progressive hair cell loss in a basal-to-apical gradient, with outer hair cells more severely affected than inner hair cells. A, Whole-mount preparations of the organ of Corti from 6-, 13-, and 16-week-old mice carrying a homozygous mutation in the Csb gene (Csbm/m) and WT littermates were stained with an antibody against the hair cell marker MyosinVI. Outer hair cells and inner hair cells were quantified independently along the length of the cochlear duct (base, middle, and apex) for each genotype and age group. Solid line, WT mice; broken line, Csbm/m mice. Error bars represent the SEM. B, Representative pictures from basal, middle, and apical regions are shown. In addition to an apparent basal-to-apical gradient of cell loss, outer hair cells (brackets) are significantly more affected than inner hair cells (arrows) in Csbm/m mice with respect to age. Scale bar, 100 μm.
Consistent with surface preparations, representative hematoxylin and eosin-stained paraffin sections through the organ of Corti from 16-week-old Csbm/m mice show a complete loss of outer and inner hair cells within the basal region (Fig. 3). Additionally, we observed severe degeneration of supporting cells (Deiters' cells and pillar cells) in the base, with the basilar membrane fully intact. The middle sections showed some signs of outer hair cell and supporting cell degeneration in the 16-week-old mutant, with an apparent collapse of Nuel's space (Fig. 3, compare Csbm/m with WT mice). The sections of 6-week-old Csbm/m and wild-type littermates displayed a normal complement of hair cells and supporting cells (Fig. 3). Together, the data indicate that Csb deficiency predisposes the mice to progressive loss of hair cells and supporting cells.
Figure 3.
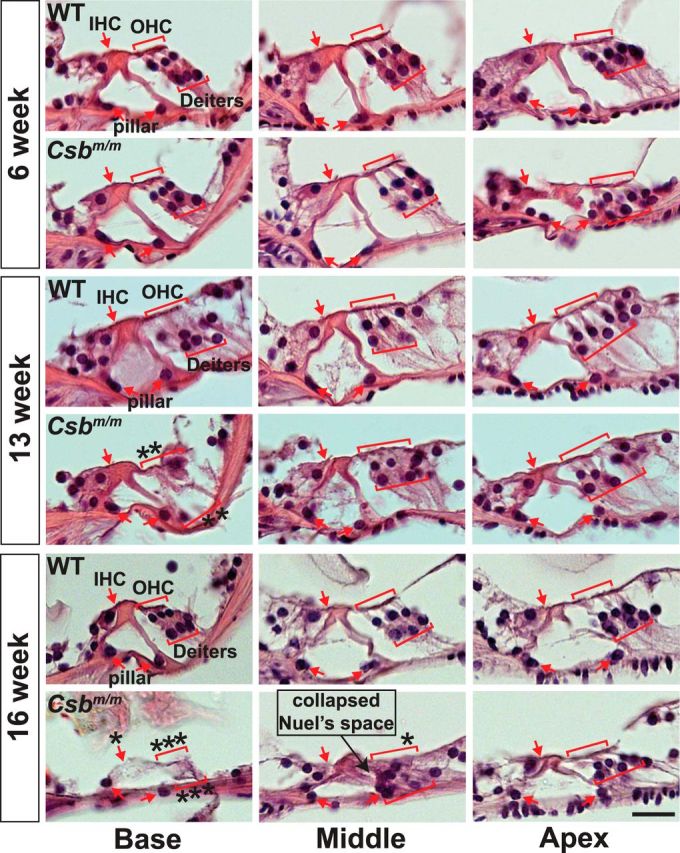
Hematoxylin and eosin-stained paraffin sections through the organ of Corti of Csbm/m mice indicate progressive hair cell and supporting cell loss. Representative sections from each cochlear position obtained at 6, 13, and 16 weeks are pictured. Outer hair cells (OHC), inner hair cells (IHC), Deiters' cells, and pillar cells are indicated in red. OHCs and IHCs are separated by two rows of pillar cells forming the tunnel of Corti. An asterisk indicates a missing cell. Notice the clear deterioration of outer hair cells and Deiters' cells at 16 weeks, leading to the collapse of Nuel's space (formed by outer pillar cells on one side and OHCs/Deiters' cells on the other). Scale bar, 20 μm.
Progressive hearing loss and cochlear hair cell death in mature Csa−/− mice
Mice with a homozygous deletion in the CS group A gene (Csa−/−) show many of the same phenotypes as Csbm/m mice (van der Horst et al., 2002). To test whether Csa−/− mice also exhibit progressive hearing loss of cochlear origin, as observed with Csbm/m mice, we measured ABRs and DPOAEs in mutant mice and age-matched wild-type controls on a C57BL/6J background (Fig. 4A,B). Auditory thresholds were not elevated at 6 and 9 weeks of age across the frequency spectrum tested (p = NS; Fig. 4A). At 13 weeks of age, Csa−/− mice presented a significant increase in hearing thresholds at ≥8 kHz (p < 0.01). By 16 weeks of age, thresholds remained significantly elevated; however, the auditory shifts were more pronounced at 24 and 32 kHz (both p < 0.001) than at 16 and 8 kHz (p < 0.05) or 12 and 4 kHz (p < 0.01). While 6-week-old Csa−/− mice had DPOAEs similar to those of wild-type controls (p = NS for all differences), by 16 weeks of age DPOAEs were reduced in amplitude (12 and 16 kHz) or were absent (24 kHz) in Csa−/− mice compared with age-matched controls (all p < 0.05; Fig. 4B), indicating outer hair cell impairment. These findings show that Csa−/− mice manifest a progressive increase in hearing thresholds of cochlear origin, especially at higher frequencies during normal development, much like their Csbm/m counterparts.
Figure 4.
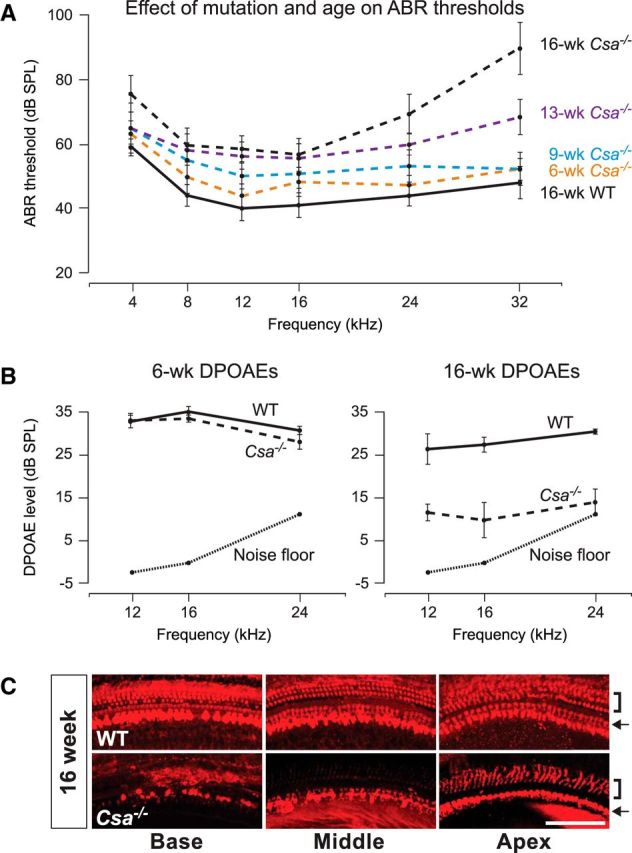
Csa−/− mice show progressive elevated ABR thresholds, reduced DPOAEs, and hair cell loss in a basal-to-apical gradient, with outer hair cells more severely affected than inner hair cells, in early life. A, ABR thresholds of mice carrying a homozygous deletion in the Csa gene (Csa−/−) and WT controls were measured between 6 and 16 weeks of age. No significant difference between control mice at any of the ages measured was detected (p = NS for all differences), and thus only 16 week values are shown. Solid line, WT mice; broken line, Csa−/− mice. B, The 6-week-old Csa−/− mice have DPOAEs similar to those of age-matched WT controls, whereas 16-week-old Csa−/− mice have reduced or absent DPOAEs compared with WT controls. C, Representative pictures of 16-week-old Csa−/− and WT cochlear whole-mount preparations stained with an antibody against the hair cell marker parvalbumin. In addition to an apparent basal-to-apical gradient of cell loss, outer hair cells (brackets) are more affected than inner hair cells (arrows) in Csa−/− mice. Scale bar, 100 μm. ABR and DPOAE data are shown as the mean ± SEM.
Whole-mount preparations of the organ of Corti from 16-week-old Csa−/− and wild-type mice were stained with an antibody to the hair cell marker parvalbumin for counting. We observed a complete loss of outer hair cells in the basal and middle turns of Csa−/− cochleae, and a gradient of inner hair cell loss from base to apex (Fig. 4C), indicating a similar pattern of damage as seen in 16-week-old Csbm/m mice (Figs. 2, 3). In addition, outer hair cells were more severely affected than inner hair cells in Csa−/− mice, as was the case in Csbm/m mice. In contrast, age-matched controls showed a normal complement of outer and inner hair cells throughout the entire cochlear turn (Fig. 4C). Together, the data indicate that Csa deficiency predisposes mature mice to hearing loss and hair cell degeneration, as observed with Csb deficiency.
Discussion
In this study, we observed progressive hearing loss in Csbm/m and Csa−/− mice, which was more profound at the higher frequencies. Csbm/m and Csa−/− mice exhibit some of the same symptoms as humans with Cockayne syndrome, except that they do not manifest a reduction in lifespan (eliminating the possibility of systemic failure in mutant mice in conjunction with a hair cell-specific effect) or reproductive problems (van der Horst et al., 1997, 2002). These mouse models for human CS recapitulate the hearing loss observed in CS patients with respect to affected frequencies, time course, and place of origin. In humans, most audiometric studies have demonstrated a progressive, sensorineural, high-frequency hearing loss (Iwasaki and Kaga, 1994; Rapin et al., 2006). Inner and outer hair cell loss, as well as loss of spiral ganglion neurons have been observed in CS patients (Shemen et al., 1984; Rapin et al., 2006). These changes took place at the level of the basal turn, which is responsible for high-frequency hearing. This is in accordance with our data, since the reduction or absence of otoacoustic emissions, and the histological results, indicate a cochlear involvement in the hearing loss of Csbm/m and Csa−/− mice.
Hearing loss is a very common consequence of aging. Accumulation of DNA damage due to the action of reactive oxygen species (ROS) is generally thought to contribute to loss of hair cells (McFadden et al., 2001; Jiang et al., 2007). The role of accumulated endogenous DNA damage in the pathogenesis of CS is well established (Schumacher et al., 2009; Coppedè and Migliore, 2010). Based on this study, we postulate that CSB and CSA inactivation in the inner ear predisposes the sensory hair cells to cell death due to accumulation of unrepaired DNA damage, leading to the early demise of the hair cells. Additionally, stalled RNA Pol II and the loss of CSB or CSA may lead to a sustained p53 response and subsequent apoptosis. The differential vulnerabilities in outer hair cells and inner hair cells of mutant mice during normal development further suggest an additional inherent difference between outer hair cells and inner hair cells in terms of DNA repair and/or apoptotic responses.
The hair cell loss that we have observed in Csbm/m and Csa−/− mice may not be spontaneous. Instead, it may be secondary to increased susceptibility to low-level environmental stress as well as endogenous stress (e.g., ROS and other reactive side products from metabolism) as a result of the mutation. Csbm/m and Csa−/− mice challenged with low levels of cisplatin presented elevated ABR threshold shifts in the high frequencies and outer hair cell degeneration in a basal-to-apical gradient at a much earlier age (Segil laboratory, unpublished observations). Thus, one interpretation could be that the loss of CSB or CSA is not itself responsible for the progressive damage, but that it predisposes the mice, and potentially Cockayne syndrome patients, to hair cell damage. This also raises the possibility that a variation in DNA repair efficiency may underlie susceptibility to hearing loss in the human population.
Hair cells of the inner ear are hypersensitive to environmental stress of a variety of types, including mechanical (loud noise) and ototoxic (e.g., certain antibiotics and chemotherapy agents; Schacht et al., 2012). In each of these cases, it is postulated that DNA damage, caused either directly (in the case of cisplatin) or indirectly through the generation of free radicals, may contribute to cell death. Given that hair cells are terminally differentiated nondividing cells, and thus not subject to replication stress, it seems likely that transcriptional stress resulting from unrepaired DNA damage is responsible for signaling apoptosis. Based on the observation that GG-NER is downregulated in some highly differentiated cells (Nouspikel and Hanawalt, 2003), we postulate that hair cells may be particularly dependent on the TC-NER/TCR pathway for their long-term survival. Csbm/m and Csa−/− mice represent a model for testing the contribution of DNA damage to hearing loss, and the mechanisms by which Cockayne syndrome factors contribute to hair cell homeostasis warrant further study.
Footnotes
This work was supported by National Institutes of Health (NIH)-Ruth L. Kirschstein National Research Service Award F32DC010125 (to R.N.R.), the Sidgmore Family Foundation, NIH Grant R01-DC-007173 (to N.S.), the Dutch Fund for Economic Structure Reinforcement (Grant 0908 for the “NeuroBasic PharmaPhenomics project,” to J.G.G.B.), and the Heinsius-Houbolt Fund (A.P.N.). We thank Juan Llamas, Welly Makmura, and Francesca Della Ripa for excellent technical support; and Caroline Abdala for assistance with the ABR and DPOAE analyses.
The authors declare no competing financial interests.
References
- Berg RJ, Rebel H, van der Horst GT, van Kranen HJ, Mullenders LH, van Vloten WA, de Gruijl FR. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res. 2000;60:2858–2863. [PubMed] [Google Scholar]
- Coppedè F, Migliore L. DNA repair in premature aging disorders and neurodegeneration. Curr Aging Sci. 2010;3:3–19. doi: 10.2174/1874609811003010003. [DOI] [PubMed] [Google Scholar]
- Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Kaga K. Chronological changes of auditory brainstem responses in Cockayne's syndrome. Int J Pediatr Otorhinolaryngol. 1994;30:211–221. doi: 10.1016/0165-5876(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol Aging. 2007;28:1605–1612. doi: 10.1016/j.neurobiolaging.2006.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane KL, Longo-Guess CM, Gagnon LH, Ding D, Salvi RJ, Johnson KR. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear Res. 2012;283:80–88. doi: 10.1016/j.heares.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laos M, Anttonen T, Kirjavainen A, af Hallstrom T, Laiho M, Pirvola U. DNA damage signaling regulates age-dependent proliferative capacity of quiescent inner ear supporting cells. Aging (Albany NY) 2014;6:496–510. doi: 10.18632/aging.100668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, Stefanini M, Lehmann AR. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62:77–85. doi: 10.1086/301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- Martin GM. Genetic modulation of senescent phenotypes in Homo sapiens. Cell. 2005;120:523–532. doi: 10.1016/j.cell.2005.01.031. [DOI] [PubMed] [Google Scholar]
- McFadden SL, Ding D, Salvi R. Anatomical, metabolic and genetic aspects of age-related hearing loss in mice. Audiology. 2001;40:313–321. doi: 10.3109/00206090109073128. [DOI] [PubMed] [Google Scholar]
- McMurray CT. To die or not to die: DNA repair in neurons. Mutat Res. 2005;577:260–274. doi: 10.1016/j.mrfmmm.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Müller M, Smolders JW. Shift in the cochlear place-frequency map after noise damage in the mouse. Neuroreport. 2005;16:1183–1187. doi: 10.1097/00001756-200508010-00010. [DOI] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer's disease. Bioessays. 2003;25:168–173. doi: 10.1002/bies.10227. [DOI] [PubMed] [Google Scholar]
- Rapin I, Weidenheim K, Lindenbaum Y, Rosenbaum P, Merchant SN, Krishna S, Dickson DW. Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol. 2006;21:991–1006. doi: 10.1177/08830738060210110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacht J, Talaska AE, Rybak LP. Cisplatin and aminoglycoside antibiotics: hearing loss and its prevention. Anat Rec (Hoboken) 2012;295:1837–1850. doi: 10.1002/ar.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Hoeijmakers JH, Garinis GA. Sealing the gap between nuclear DNA damage and longevity. Mol Cell Endocrinol. 2009;299:112–117. doi: 10.1016/j.mce.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Shemen LJ, Mitchell DP, Farkashidy J. Cockayne syndrome—an audiologic and temporal bone analysis. Am J Otol. 1984;5:300–307. [PubMed] [Google Scholar]
- Silver LM. Mouse genetics: concepts and applications. Oxford, UK: Oxford UP; 1995. [Google Scholar]
- Spoor M, Nagtegaal AP, Ridwan Y, Borgesius NZ, van Alphen B, van der Pluijm I, Hoeijmakers JH, Frens MA, Borst JG. Accelerated loss of hearing and vision in the DNA-repair deficient Ercc1(delta/−) mouse. Mech Ageing Dev. 2012;133:59–67. doi: 10.1016/j.mad.2011.12.003. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, Bootsma D, Hoeijmakers JH. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–435. doi: 10.1016/S0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, Meira L, Gorgels TG, de Wit J, Velasco-Miguel S, Richardson JA, Kamp Y, Vreeswijk MP, Smit B, Bootsma D, Hoeijmakers JH, Friedberg EC. UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 2002;1:143–157. doi: 10.1016/S1568-7864(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Willott JF, Erway LC. Genetics of age-related hearing loss in mice. IV. Cochlear pathology and hearing loss in 25 BXD recombinant inbred mouse strains. Hear Res. 1998;119:27–36. doi: 10.1016/S0378-5955(98)00029-X. [DOI] [PubMed] [Google Scholar]
- Yamasoba T, Lin FR, Someya S, Kashio A, Sakamoto T, Kondo K. Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear Res. 2013;303:30–38. doi: 10.1016/j.heares.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Liu W, Ding D, Salvi R. Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience. 2003;120:191–205. doi: 10.1016/S0306-4522(03)00286-0. [DOI] [PubMed] [Google Scholar]