

Abstract
Brain-derived neurotrophic factor (BDNF) and endocannabinoids (eCBs) have been individually implicated in behavioral effects of cocaine. The present study examined how BDNF-eCB interaction regulates cocaine-induced synaptic plasticity in the ventral tegmental area and behavioral effects. We report that BDNF and selective tyrosine kinase receptor B (TrkB) agonist 7,8-dihydroxyflavone (DHF) activated the TrkB receptor to facilitate two forms of eCB-mediated synaptic depression, depolarization-induced suppression of inhibition (DSI), and long-term depression (I-LTD) of IPSCs in ventral tegmental area dopamine neurons in mouse midbrain slices. The facilitation appears to be mediated by an increase in eCB production via phospholipase Cγ pathway, but not by an increase in CB1 receptor responsiveness or a decrease in eCB hydrolysis. Using Cre-loxP technology to specifically delete BDNF in dopamine neurons, we showed that eCB-mediated I-LTD, cocaine-induced reduction of GABAergic inhibition, and potentiation of glutamatergic excitation remained intact in wild-type control mice, but were impaired in BDNF conditional knock-out mice. We also showed that cocaine-induced conditioned place preference was attenuated in BDNF conditional knock-out mice, in vivo pretreatments with DHF before place conditioning restored cocaine conditioned place preference in these mice, and the behavioral effect of DHF was blocked by a CB1 receptor antagonist. Together, these results suggest that BDNF in dopamine neurons regulates eCB responses, cocaine-induced synaptic plasticity, and associative learning.
Keywords: BDNF, cocaine, conditioned place preference, DSI, endocannabinoid, I-LTD
Introduction
Drugs of abuse, such as cocaine, induce long-term neuroadaptations in the reward circuit that lead to addictive behavior (Hyman et al., 2006). Brain-derived neurotrophic factor (BDNF) and endocannabinoids (eCBs) are important for activity-dependent synaptic plasticity and drug-induced neuroadaptations (Gerdeman et al., 2003; Lupica and Riegel, 2005; Heifets and Castillo, 2009; McGinty et al., 2010; Park and Poo, 2013). BDNF and eCBs are released in response to neuronal activity and are key regulators of long-term synaptic plasticity (Heifets and Castillo, 2009; Park and Poo, 2013). BDNF in the mesolimblic dopamine system is elevated following cocaine withdrawal (Grimm et al., 2003); the increased BDNF following cocaine withdrawal facilitates LTP in the ventral tegmental area (VTA) (Pu et al., 2006) and prefrontal cortex (Lu et al., 2010). Pharmacological and genetic manipulations of BDNF and its receptor tyrosine kinase receptor B (TrkB) altered cocaine self-administration, conditioned place preference (CPP), and behavioral sensitization (Berglind et al., 2007; Graham et al., 2007; Bahi et al., 2008; Lobo et al., 2010; Lu et al., 2010; Huang et al., 2011). The eCB system has been implicated in habit learning and cocaine seeking (for review, see Gerdeman et al., 2003; Lupica and Riegel, 2005). eCBs mediate short-term and long-term depression (LTD) at excitatory and inhibitory synapses (I-LTD) (Gerdeman et al., 2002; Marsicano et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003). eCB-mediated synaptic plasticity represents a widespread and fundamental mechanism by which synaptic strength and behavior can be regulated (Gerdeman et al., 2003; Heifets and Castillo, 2009). Recent studies suggest a strong interaction between BDNF and the eCB system (Luongo et al., 2014). BDNF increases CB1 receptor expression in cultured cerebellar granule neurons (Maison et al., 2009). BDNF depresses IPSCs in neocortical layer 2/3 pyramidal neurons, and this depression is mediated by CB1 receptors (Lemtiri-Chlieh and Levine, 2010; Zhao and Levine, 2014). In contrast, BDNF inhibited CB1 agonist-induced depression of IPSCs in the striatum through a mechanism mediated by altered cholesterol metabolism and membrane lipid raft function (De Chiara et al., 2010). However, it remains poorly understood whether and how BDNF interacts with eCBs to regulate cocaine-induced synaptic plasticity.
We have shown that repeated cocaine exposure in vivo reduces GABAergic inhibition onto VTA dopamine neurons, and eCB-mediated I-LTD might constitute a mechanism for cocaine-induced reduction of GABAergic inhibition (Liu et al., 2005; Pan et al., 2008b). The present study investigated the role and mechanism by which BDNF-eCB interaction regulates cocaine-induced synaptic plasticity in VTA dopamine neurons. Using Cre-loxP technology to specifically delete BDNF in dopamine neurons, we showed that eCB-mediated I-LTD, cocaine-induced reduction of GABAergic inhibition, and potentiation of glutamatergic excitation were impaired following conditional knock-out (cKO) of BDNF in dopamine neurons. These results suggest that BDNF-eCB interaction is required for cocaine-induced inhibitory synaptic plasticity in VTA dopamine neurons. In addition, BDNF cKO mice exhibited impaired CPP to cocaine, suggesting that cue-drug associated learning was impaired in these mice.
Materials and Methods
Animals.
Animal maintenance and use were in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. C57BL/6J mice (Jax stock #000664), homozygous BDNF-floxed mice (BdnfloxP/loxP, Jax stock #004339), heterozygous DAT-Cre+/− mice (Jax stock #006660), and ROSA26-flox-STOP-flox-lacZ (R26R) reporter mice (Jax stock #003474) were obtained from The Jackson Laboratory. R26R mice were crossed with DAT-Cre+/− mice to examine the Cre expression and effectiveness of recombination. Expression of Cre recombinase was identified by immunofluorescence staining of β-galactosidase (β-gal). To generate dopamine neuron-specific BDNF cKO mice, we crossed BdnfloxP/loxP mice with DAT-Cre+/− mice to produce a compound, DAT-Cre+/−/BdnfloxP/wt mouse line. BDNF cKO (DAT-Cre+/−/BdnfloxP/loxP) mice were generated by crossing this compound, DAT-Cre+/−/BdnfloxP/wt mice with BdnfloxP/loxP mice. DAT-Cre+/−/BDNFloxP/wt or DAT-Cre−/−/BdnfloxP/loxP mice were used as wild-type controls. Genotyping analysis was performed by using standard PCR technique on tail biopsies. BDNF mRNA levels in the VTA were further determined by real-time PCR.
Brain slice preparation.
C57BL/6J mice, BDNF cKO mice, and their wild-type littermates (P18-P30) of either sex were used for slice electrophysiology. Mice were anesthetized by isoflurane inhalation and decapitated. Horizontal midbrain slices were prepared at 250 μm thickness using a Leica vibrating slicer as described previously (Pan et al., 2008b). Slices were prepared at 4°C-6°C in a sucrose-based solution containing the following (in mm): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 10 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. The slices were immediately transferred to oxygenated (95% O2/5% CO2) ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The slices were allowed to recover at 35°C for 30 min and then incubated at room temperature for at least 30 min before patch-clamp recordings.
Electrophysiology.
Whole-cell patch-clamp recordings were made using patch-clamp amplifiers (Multiclamp 700B) under infrared-differential interference contrast microscopy. Data acquisition and analysis were performed using DigiData 1440A digitizer and analysis software pClamp 10 (Molecular Devices). Signals were filtered at 2 kHz and sampled at 10 kHz. Dopamine neurons in the VTA were identified by long duration (>1.5 ms) of spontaneous action potentials in cell-attached configuration (Chieng et al., 2011) and the presence of large Ih currents, rhythmic firing at low frequency, and prominent afterhyperpolarization in whole-cell mode (Johnson and North, 1992; Jones and Kauer, 1999; Liu et al., 2005). Neurons were voltage-clamped at −70 mV unless stated otherwise. For recording of evoked IPSCs, electrical stimulation was delivered by a bipolar tungsten stimulation electrode (WPI) that was placed ∼150 μm rostral to the recorded dopamine neuron. Glutamate receptor antagonists CNQX (20 μm) and d-AP-5 (50 μm) were present in the ACSF throughout the experiments. For the experiments of depolarization-induced suppression of inhibition (DSI), glass pipettes (3–5 mΩ) were filled with an internal solution containing the following (in mm): 80 Cs-methanesulfonate, 60 CsCl, 2 QX-314, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine (pH 7.2 with CsOH). To induce DSI, neurons were depolarized from −70 to 0 mV for 5 s, and IPSCs were evoked at 4 s intervals. For recording of maximal IPSCs (see Fig. 8A), neurons were voltage-clamped at −20 mV, and glass pipettes were filled with an internal solution containing the following (in mm): 135 Cs-methanesulfonate, 5 CsCl, 10 HEPES, 1 EGTA, 2 MgCl2, 4 Mg-ATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine (pH 7.2 with CsOH). Maximal IPSCs were elicited by gradually increasing the stimulation intensity to recruit saturating IPSCs (Huang et al., 1999; Liu et al., 2005; Pan et al., 2008b). For recording of (S)-3,5-dihydroxyphenylglycine (DHPG)-induced inward currents, the recording pipette was filled with same internal solution as that of maximal IPSCs. A glass pipette (3–5 mΩ) was filled with ACSF containing 10 μm DHPG, the tip of the pipette was positioned ∼100 μm to the recorded neurons. DHPG was pressure injected via a Spritzer (Toohey). GABAA receptor blocker picrotoxin (50 μm) and AMPA receptor antagonist CNQX (10 μm) were present in the ACSF. For I-LTD and mIPSC experiments, glass pipettes were filled with an internal solution containing the following (in mm): 100 K-gluconate, 50 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 Mg-ATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine (pH 7.2 with KOH). mIPSCs were recorded in the presence of Na+ channel blocker tetrodotoxin (TTX, 0.5 μm).
Figure 8.

Endogenous BDNF is required for cocaine-induced reduction of GABAergic inhibition. Wild-type control and BDNF cKO mice were given intraperitoneal injections of saline or cocaine (15 mg/kg) for 5 d. Midbrain slices were prepared from these four groups of mice the following day. A, Top, Sample IPSCs in response to stimuli of incremental intensities (20–140 μA). The neurons were voltage-clamped at −20 mV. Bottom, Summarized data showed that the mean amplitude of maximal IPSCs was significantly decreased in cocaine-treated wild-type control mice compared with saline-treated wild-type control mice (**p < 0.01). BDNF cKO had no significant effects on the mean amplitude of maximal IPSCs in saline-injected groups (p > 0.05) but blocked the decreases in the mean amplitude of maximal IPSCs in cocaine-injected groups (p > 0.05). n = 10 or 11 for each group. B, Same as in A, except that mIPSCs were recorded at a holding potential −70 mV. n = 11 or 12 for each group. ***p < 0.001.
AMPA/NMDA receptor (AMPAR/NMDAR) ratio was measured based on published studies (Saal et al., 2003; Liu et al., 2005). Evoked EPSCs were recorded from dopamine neurons in the presence of GABAA receptor blocker picrotoxin (50 μm). Glass pipettes were filled with an internal solution containing the following (in mm): 140 Cs-methanesulfonate, 10 HEPES, 1 EGTA, 5 tetraethylammonium chloride (TEA-Cl), 2.5 MgATP, 0.3 Na2GTP, and 2 MgCl2 (pH 7.2 with CsOH). Neurons were voltage-clamped at 40 mV to record dual-component EPSCs containing both AMPAR- and NMDAR-EPSCs that were isolated pharmacologically. After a stable baseline recording of total EPSCs, NMDAR antagonist d-AP5 (50 μm) was then applied in the bath for 6–10 min to isolate fast AMPAR EPSCs. NMDAR-EPSCs were calculated as the subtraction of AMPAR-EPSCs from the total EPSCs from the same neuron. An average of 10–20 consecutive EPSCs was collected for the each type of EPSCs. The AMPAR/NMDAR ratio was calculated by dividing the peak of the AMPAR-EPSC by the peak of the NMDAR-EPSC.
Series resistance (15–30 mΩ) was monitored throughout all recordings, and data were discarded if the resistance changed by >20%. All recordings were performed at 32 ± 1°C using an automatic temperature controller (Warner Instruments).
Immunofluorescence staining.
The experimental procedure was conducted as we have previously described (Zhong et al., 2014). Mice were anesthetized by isoflurane, and the brain was fixed by transcardial perfusion with 0.1 m sodium PBS followed by 4% PFA in 4% sucrose-PBS, pH 7.4. The brain was then removed, postfixed in the same fixative for 4 h at 4°C, dehydrated in gradual concentrations of sucrose (20% and 30%) in 0.1 m PBS at 4°C, and frozen on dry ice. Coronal VTA sections were made at 20 μm thickness with a cryostat. The sections were incubated with primary antibodies against β-gal (chicken, 1:1000, Abcam) and TH (rabbit, 1:300, Santa Cruz Biotechnology) at 4°C for 48 h. After rinsing three times at 10 min each in PBS, the sections were incubated in the different secondary antibodies: anti-chicken IgG AlexaFluor-568 conjugate (1:1000, Invitrogen) and anti-rabbit IgG AlexaFluor-488 conjugate (1:500, Cell Signaling Technology) for 4 h at room temperature in the dark. The sections were analyzed using a Nikon Eclipse TE-2000U confocal microscope.
RNA isolation, cDNA synthesis, and real-time PCR.
To examine BDNF mRNA levels in the VTA, BDNF cKO and wild-type control mice were anesthetized by isoflurane inhalation and decapitated. VTA punches were dissected out in RNase-free environment and were homogenized for total RNA isolation using an RNA mini kit (Invitrogen) according to the manufacturer's instructions. Extracted total RNA was treated by DNase (Bio-Rad) for 15 min to eliminate possible DNA contamination. RNA was reverse transcribed into cDNA with an iScript cDNA synthesis kit (Bio-Rad). Levels of BDNF and β-actin mRNAs were quantified by real-time quantitative PCR using the following primers: BDNF, 5′-CTCAGGCAGAATGAGCAATG-3′ and 5′-AGCCGTCTGTGCTCTTCACT-3′; β-actin: 5′-GTGACGTTGACATCCGTAAAGA-3′ and 5′-GCCGGACTCATCGTACTCC-3′. Real-time quantitative PCR was performed on a Bio-Rad CFX96 RT system using a SYBR Green supermix kit (Bio-Rad). The ΔΔCT method was used to quantify BDNF expression levels based on normalization to β-actin.
CPP.
Cocaine CPP experiments were performed with a three-chamber apparatus (Med Associates). The CPP protocol consisted of the following sessions: (1) Pretest (day 1): BDNF cKO and wild-type control mice (2–3 months old) were allowed to explore three chambers freely for 20 min, and time spent in every chamber was recorded. Mice showing unconditioned side preference (≥180 s disparity) were excluded. (2) Conditioning (day 2–7): Cocaine conditioning. On days 2, 4, and 6, mice received cocaine injection (15 mg/kg, i.p.) and were immediately confined to one chamber for 30 min. On days 3, 5, and 7, mice received saline injection (0.9% NaCl, 1 ml/kg, i.p.) and were immediately confined to the opposite chamber for 30 min. Saline conditioning. Mice received daily saline injection (0.9% NaCl, 1 ml/kg, i.p.) and were immediately confined to one chamber for 30 min on days 3, 5, and 7 and were confined to the opposite chamber for 30 min on days 2, 4, and 6. (3) CPP test (day 8): All of the animals were allowed to explore freely for 20 min between the two sides, and time spent on each side was recorded (see Figs. 10 and 11). In separate experiments (see Figs. 10C,D and 11), control and BDNF cKO mice received intraperitoneal injections of vehicle, 7,8-dihydroxyflavone (DHF; 5 mg/kg), AM251 (3 mg/kg), and DHF + AM251 30 min before each saline or cocaine conditioning, and CPP was performed as described above.
Figure 10.

BDNF cKO attenuated CPP to cocaine. A, Wild-type control and BDNF cKO mice exhibited no significant unconditioned preference in each chamber during pretest (n = 9–11 for each group; p > 0.05). B, Mice received saline (0.9% NaCl, i.p.) and cocaine (15 mg/kg) place conditioning once daily for 6 d. CPP was tested 24 h after the last conditioning. BDNF cKO significantly decreased the CPP score in cocaine-conditioned mice (**p < 0.01) but did not affect the CPP score in saline-conditioned mice (p > 0.05). C, D, Same as in A, B, wild-type control and BDNF cKO mice underwent cocaine conditioning. There was no unconditioned place preference during pretest (n = 9 or 10 for each group; p > 0.05; C). The mice were pretreated with vehicle or DHF (5 mg/kg, i.p.) before each saline or cocaine conditioning. CPP was significantly attenuated in vehicle-pretreated BDNF cKO mice compared with vehicle-pretreated wild-type mice (***p < 0.001), whereas DHF pretreatment significantly increased cocaine CPP in BDNF cKO mice (***p < 0.001), but not in control mice (p > 0.05).
Figure 11.
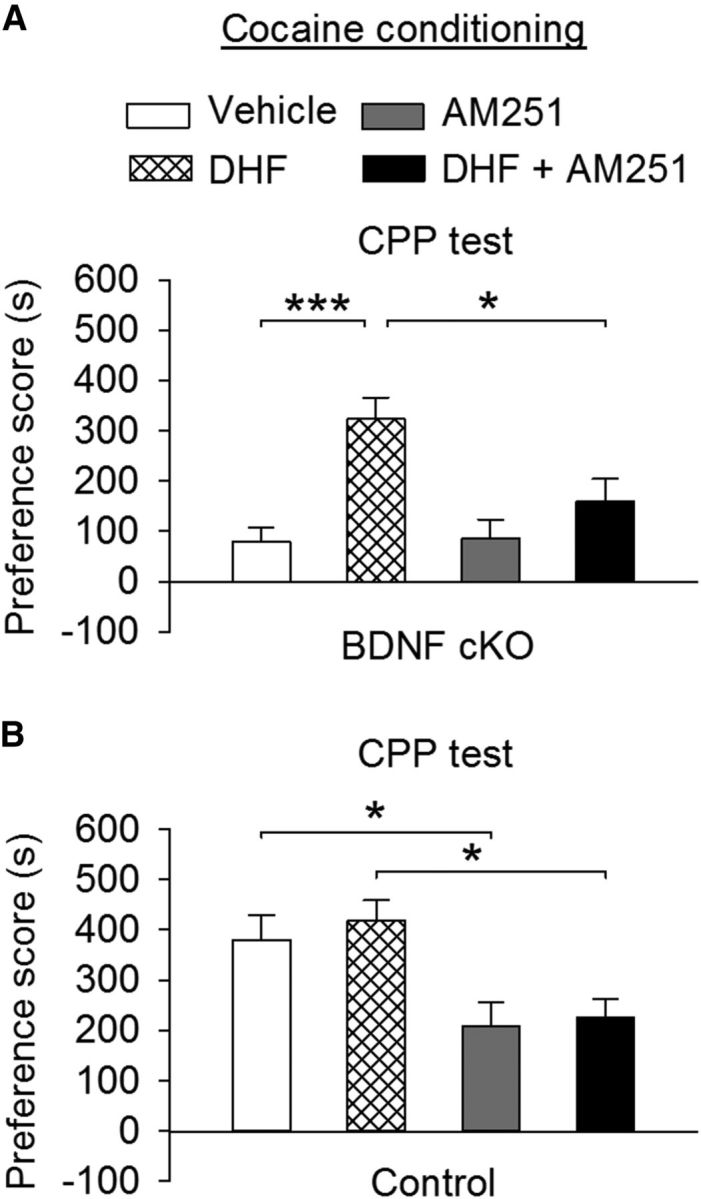
CB1 receptor antagonism blocked DHF-induced restoration of cocaine CPP in BDNF cKO mice and attenuated cocaine CPP in wild-type mice. A, Before each place conditioning, BDNF cKO mice received intraperitoneal injections of AM251 (3 mg/kg) or AM251 + DHF (5 mg/kg). The preference scores were plotted together with vehicle and DHF groups in BDNF cKO mice from Figure 10D. DHF restored cocaine CPP in BDNF cKO mice (***p < 0.001). AM251 blocked DHF-induced restoration of cocaine CPP in BDNF cKO mice (*p < 0.05) but had no significant effect on the preference score compared with that in vehicle-treated BDNF cKO mice (n = 10–12 for each group; p > 0.05). B, Before each place conditioning, wild-type control mice received intraperitoneal injections of AM251 or AM251 + DHF. The preference scores were plotted together with vehicle and DHF groups in wild-type control mice from Figure 10D. AM251 significantly decreased the preference score in wild-type control mice compared with vehicle treatment group (*p < 0.05). The AM251 + DHF group exhibited significant decrease in the preference score compared with the DHF group (n = 9 or 10 for each group; *p < 0.05).
Statistics.
Data are presented as mean ± SEM. The magnitude and decay constant (τ) of DSI and I-LTD were calculated as we have described (Pan et al., 2008b, 2009). mIPSCs were analyzed using Mini-analysis (Synaptosft) (Pan et al., 2008a). CPP scores were calculated as the time spent in cocaine-conditioned chamber minus that in saline-conditioned chamber (Zhong et al., 2012; Yu et al., 2013). Datasets were compared with either Student's t test, one-way or two-way ANOVA followed by Tukey's post hoc analysis. Results were considered to be significant at p < 0.05.
Chemicals.
Cocaine hydrochloride, picrotoxin, CNQX, DHF, and all other common chemicals were obtained from Sigma-Aldrich. d-AP5, BDNF, DHPG, cyclotraxin B (CTX-B), 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP hydrochloride), and TTX were obtained from Tocris Bioscience.
Results
TrkB receptor agonists facilitated eCB-mediated DSI and I-LTD
Depolarization of hippocampal and cerebellar neurons induces transient suppression of inhibition (DSI) and excitation (DSE), which are mediated by CB1 receptors (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). We determined whether DSI could be induced in mouse VTA dopamine neurons and whether BDNF altered DSI induction. Midbrain slices were prepared from C57BL/6J mice, and whole-cell voltage-clamp recordings were made from VTA dopamine neurons. IPSCs were evoked by electrical stimulation of synaptic afferents in the presence of glutamate receptor antagonists CNQX (20 μm) and d-AP-5 (50 μm). We found that depolarization of dopamine neurons from −70 to 0 mV for 5 s did not induce significant depression of IPSCs. Following 10 min perfusion of BDNF (20 ng/ml), robust DSI was induced (vehicle, 3.9 ± 6.0%, n = 9; BDNF, 29.7 ± 6.3%, n = 8; Student's t test, t(15) = 2.97, p = 0.01; Fig. 1A,B). Similarly, TrkB receptor agonist DHF (10 μm) (Jang et al., 2010) enabled DSI in VTA dopamine neurons (vehicle, 2.7 ± 5.1%, n = 10; DHF, 32.3 ± 6.2%, n = 11, t(19) = 3.64, p = 0.002; Fig. 1C,D). DSI induced in DHF was blocked by the CB1 receptor antagonist AM251 (2 μm; 2.2 ± 2.8%, n = 9; t(18) = 4.12, p < 0.001 vs DHF), the TrkB receptor inhibitor K252a (200 nm; 3.8 ± 4.2%, n = 9; t(18) = 3.64, p = 0.002 vs DHF; Fig. 1D), or the TrkB receptor antagonist CTX-B (100 nm; 3.0 ± 6.7%, n = 12; t(21) = 3.18, p = 0.004 vs DHF). Thus, BDNF and DHF enabled DSI in VTA dopamine neurons via activation of the TrkB receptor.
Figure 1.

TrkB receptor agonists enabled DSI in VTA dopamine neurons. A, A brief depolarization (5 s from −70 to 0 mV) did not induce significant depression of IPSCs in VTA dopamine neurons, whereas bath application of BDNF (20 ng/ml) enabled DSI. Shown are sample traces of evoked IPSCs (top) and averaged DSI (bottom). Solid lines indicate single exponential fitting curves of the decay of DSI. B, Summary of the magnitude of DSI in the presence of vehicle or BDNF (20 ng/ml) (n = 8 or 9). *p < 0.05. C, D, Bath application of TrkB receptor agonist DHF (10 μm) enabled DSI (n = 10 or 11; **p < 0.01), which was blocked by the CB1 receptor antagonist AM251 (2 μm; n = 9; ***p < 0.001), the TrkB receptor inhibitor K252a (200 nm; n = 9; **p < 0.01), or the TrkB receptor antagonist CTX-B (100 nm; n = 12; **p < 0.01). Error bars indicate SEM.
The CB1 receptor also mediates LTD at excitatory and inhibitory synapses (I-LTD) (Gerdeman et al., 2002; Chevaleyre and Castillo, 2003; Safo and Regehr, 2005). We next determined whether TrkB agonists affected I-LTD in VTA dopamine neurons. Repeated synaptic stimulation (10 Hz stimulation for 5 min) did not induce significant depression of IPSCs (90.7 ± 6.1% of baseline, n = 9; t(16) = 1.30, p = 0.21; Fig. 2A), suggesting that this stimulation is subthreshold for I-LTD induction. However, the presence of BDNF (20 ng/ml; 64.7 ± 7.5% of baseline, n = 7; t(12) = 2.72, p = 0.002; Fig. 2B) or TrkB agonist DHF (10 μm; 63.8 ± 8.5% of baseline, n = 9; t(16) = 3.42, p = 0.004; Fig. 2C) enabled the 10 Hz stimulation to induce I-LTD. Bath application of BDNF (20 ng/ml; 91.8 ± 6.9% of baseline, n = 6; t(10) = 0.96, p = 0.36) or DHF (10 μm; 88.6 ± 9.1% of baseline, n = 7; t(12) = 1.15, p = 0.27) in the absence of the 10 Hz stimulation had no significant effects on basal IPSCs (Fig. 2D). Thus, BDNF and DHF enabled previously subthreshold stimulation to induce I-LTD. I-LTD induced in the presence of BDNF was blocked by the CB1 receptor antagonist AM251 (2 μm; 88.8 ± 5.7% of baseline, n = 7; t(12) = 2.58, p = 0.024) or by TrkB receptor inhibitor K252a (200 nm; 92.5 ± 6.3% of baseline, n = 6; t(11) = 2.79, p = 0.018; Fig. 2E). Similarly, I-LTD induced in the presence of DHF was blocked by AM251 (2 μm; 90.9 ± 6.9% of baseline, n = 8; t(15) = 2.42, p = 0.028) or K252a (200 nm; 89.2 ± 7.2% of baseline, n = 8; t(15) = 2.25, p = 0.04; Fig. 2F). Together, these results indicate that BDNF and DHF facilitated eCB-mediated DSI and I-LTD in VTA dopamine neurons via activation of the TrkB receptor.
Figure 2.

TrkB receptor agonists enabled I-LTD induction in VTA dopamine neurons. A, The 10 Hz, 5 min stimulation (indicated by arrow) did not induce I-LTD (n = 9; p > 0.05). B, C, The presence of BDNF (20 ng/ml; n = 7; p < 0.01; B) or DHF (10 μm; n = 9; p < 0.01; C) before and during the 10 Hz stimulation led to I-LTD induction. D, Bath application of BDNF (20 ng/ml; n = 6; p > 0.05) or DHF (10 μm; n = 7; p > 0.05) in the absence of the 10 Hz stimulation had no significant effects on basal IPSCs. E, I-LTD induced in the presence of BDNF was blocked by the CB1 receptor antagonist AM251 (2 μm; n = 7; p < 0.05) or by TrkB receptor inhibitor K252a (200 nm; n = 6; p < 0.05). F, I-LTD induced in the presence of DHF was blocked by AM251 (n = 8; p < 0.05) or K252a (n = 8; p < 0.05).
Signaling mechanisms for TrkB receptor agonist-induced facilitation of eCB-mediated synaptic depression
Activation of Gq/11-coupled receptors, such as Group I metabotropic glutamate receptors (mGluRs), enhances DSI (Varma et al., 2001; Edwards et al., 2006) through an increase in the production of eCB ligand 2-AG (Maejima et al., 2001; Varma et al., 2001; Jung et al., 2005). Activation of Group I mGluRs is required for I-LTD in the hippocampus (Chevaleyre and Castillo, 2003) and VTA (Pan et al., 2008a, b). Group I mGluRs are positively coupled to phospholipase Cβ (PLCβ), which cleaves phosphatidylinositol 1,4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), and DAG is subsequently converted into 2-AG by DAG lipase (Di Marzo et al., 1998; Piomelli, 2003). The binding of BDNF to TrkB receptors stimulates PLCγ, which cleaves PIP2 into IP3 and DAG (Reichardt, 2006). BDNF and DHF might facilitate eCB-mediated synaptic depression via enhancement of 2-AG production. To test this possibility, we examined whether selective membrane-permeable broad-spectrum PLC inhibitor U73122 affected I-LTD induction. The inactive analog U73343 was served as a negative control. Slices were treated with U73122 (5 μm) or U73343 (5 μm) for ≥1 h; U73122 or U73343 was also bath applied throughout the experiment (Chevaleyre and Castillo, 2003; Edwards et al., 2006). As shown in Figure 3A, I-LTD induced in the presence of DHF (10 μm) was blocked by U73122 (91.8 ± 7.7% of baseline, n = 9; t(16) = 2.44, p = 0.027 vs I-LTD control in Fig. 2C) but was not significantly affected by the inactive analog U73343 (65.9 ± 7.2% of baseline, n = 7; t(14) = 0.18, p = 0.86 vs I-LTD control). Next, we examined whether DAG lipase inhibitors RHC-80267 and tetrahydrolipstatin (THL) affected I-LTD. RHC-80267 (30 μm) or THL (10 μm) was bath applied throughout the experiment. Figure 3B shows that both RHC-80267 and THL significantly attenuated I-LTD (RHC-80267, 87.5 ± 6.0% of baseline, n = 9; t(16) = 2.27, p = 0.037 vs I-LTD control; THL, 88.2 ± 7.0% of baseline, n = 8; t(15) = 2.18, p = 0.046 vs I-LTD control). These results suggest that DHF facilitated I-LTD by activating the PLC/DAG lipase pathway.
Figure 3.

DHF-facilitated I-LTD was blocked by PLC and DAG lipase inhibitors. A, The PLC inhibitor U73122 (5 μm) blocked I-LTD induced in the presence of DHF (n = 9; p < 0.05), whereas the inactive analog U73343 (5 μm) did not significantly alter I-LTD (n = 7; p > 0.05). B, DAG lipase inhibitors RHC-80267 (n = 9; p < 0.05) and THL (n = 8; p < 0.05) significantly attenuated I-LTD. C, Coapplication of mGluR1 antagonist CPCCOEt (80 μm) and mGluR5 antagonist MPEP (10 μm) did not significantly affect I-LTD (n = 8; p > 0.05). Arrows indicate the application of a 10 Hz, 5 min stimulation. D, Application of CPCCOEt (80 μm) and MPEP (10 μm) blocked the inward current induced by pressure injection of DHPG (10 μm) via a patch pipette (n = 6; **p < 0.01).
Currently available PLC inhibitors, such as U73122, cannot discriminate between PLCβ and PLCγ. There is a possibility that the TrkB receptor agonists enhance the release of glutamate, which then activates the mGluR/PLCβ pathway to enhance I-LTD. We examined whether Group I mGluR antagonism affected the facilitation of DHF on I-LTD. Bath application of mGluR1 antagonist CPCCOEt (80 μm) and mGluR5 antagonist MPEP (10 μm) did not significantly affect I-LTD induced in the presence of DHF (69.8 ± 6.2% of baseline, n = 8; t(15) = 0.56, p = 0.58 vs I-LTD control; Fig. 3C). Rapid application of Group I mGluR agonist DHPG induced an inward current in many neuronal cell types, including midbrain dopamine neurons; this current was mediated by the activation of transient receptor potential-like channels (Kim et al., 2003; Tozzi et al., 2003; Gong et al., 2011). As a positive control, we examined the effect of CPCCOEt and MPEP on the DHPG-induced inward current in the VTA. We found that a brief pressure injection (3 s) of DHPG (10 μm) via a patch pipette induced robust inward currents in all VTA dopamine neurons tested (357.5 ± 63.3 pA, n = 6). Bath application of CPCCOEt (80 μm) and MPEP (10 μm) rapidly abolished (within 10 min) the DHPG-induced currents (17.7 ± 2.9 pA, n = 6; t(5) = 5.60; p = 0.003; Fig. 3D). Thus, CPCCOEt and MPEP were effective in blocking Group I mGluR-mediated currents in the VTA. The finding that CPCCOEt and MPEP did not alter DHF-enabled I-LTD suggests that Group I mGluRs are not involved in DHF-induced facilitation of I-LTD.
We examined whether a higher concentration of DHF (30 μm) affected basal IPSCs in VTA dopamine neurons. Bath application of DHF (30 μm) for 20 min induced significant depression of evoked IPSCs (66.6 ± 5.8% of baseline, n = 8; t(14) = 4.88, p < 0.001), whereas this depression was significantly attenuated by the TrkB receptor antagonist CTX-B (100 nm; 91.9 ± 5.3% of baseline, n = 8; t(14) = 3.20, p = 0.006 vs control), the CB1 receptor antagonist AM251 (2 μm; 89.0 ± 6.0% of baseline, n = 8; t(14) = 2.67, p = 0.018 vs control), or PLC inhibitor U73122 (5 μm; 86.5 ± 7.1% of baseline, n = 7; t(13) = 2.18, p = 0.048 vs control; Fig. 4A). These results suggest that DHF at a higher concentration depresses IPSCs via a TrkB- and CB1 receptor-dependent mechanism.
Figure 4.

DHF at a higher concentration depressed IPSCs via a TrkB- and CB1 receptor-dependent mechanism. A, Bath application of DHF (30 μm) significantly decreased the amplitude of IPSCs (n = 8), and this depression was abolished by the continuous presence of the TrkB receptor antagonist CTX-B (100 nm; n = 8; p < 0.01), CB1 receptor antagonist AM251 (2 μm; n = 8; p < 0.05), or PLC inhibitor U73122 (5 μm; n = 7; p < 0.05). B, C, Bath application of CB1 receptor agonist WIN 55212–2 at 2 μm (B) and 0.5 μm (C) decreased the amplitude of IPSCs (n = 8 each group), whereas the continuous presence of DHF (10 μm) had no significant effect on this depression (n = 8 or 9; p > 0.05).
2-AG is hydrolyzed primarily by monoacylglycerol lipase (MAGL) (Cravatt et al., 1996; Blankman et al., 2007). Incubation of BDNF for 4–24 h increases the expression of CB1 receptor transcripts and decreases expression of MAGL transcripts in cultured cerebellar granule neurons (Maison et al., 2009). It is thus possible that BDNF and DHF facilitated DSI and I-LTD by enhancing CB1 receptor responsiveness or decreasing MAGL activity. We have shown that bath application of CB1 receptor agonist WIN55212–2 induced depression of IPSCs in VTA dopamine neurons (Pan et al., 2008b). An increase in CB1 receptor expression and function would enhance the depression of IPSCs induced by exogenous CB1 receptor agonists. However, we found that DHF (10 μm) had no significant effects on the depression of IPSCs induced by WIN 55212–2 at a submaximal concentration (2 μm) (vehicle, 65.9 ± 7.8% of baseline, n = 8; DHF, 62.1 ± 8.1% of baseline, n = 9; t(15) = 0.34, p = 0.74; Fig. 4B) and at a lower concentration (0.5 μm) (vehicle, 83.9 ± 5.4% of baseline, n = 8; DHF, 79.4 ± 5.5% of baseline, n = 8; t(14) = 0.58, p = 0.57; Fig. 4C), suggesting that, under the present experimental conditions, DHF does not significantly alter CB1 receptor responsiveness.
To test the possibility that BDNF and DHF facilitated DSI and I-LTD by decreasing MAGL activity, we compared the time course of DSI induced in the presence of Group I mGluR agonist DHPG, MAGL inhibitor JZL184, and TrkB agonists BDNF and DHF. We have shown that MAGL inhibitor JZL184 significantly prolongs the decay time constant of DSI in the hippocampal neurons (Pan et al., 2009). DHPG is known to enhance DSI by increasing 2-AG synthesis (Jung et al., 2005; Edwards et al., 2006). We obtained the decay time constant (τ) and magnitude of DSI induced in the presence of either BDNF or DHF from Figure 1 (τ: BDNF, 7.2 ± 1.5 s, n = 8; DHF, 7.5 ± 1.8 s, n = 11) and found that τ of DSI from either group was not significantly different from that of DHPG (1 μm; DHPG, 7.1 ± 1.7 s, n = 14; t(20) = 0.059, p = 0.95 vs BDNF group; t(23) = 0.17, p = 0.87 vs DHF group; Fig. 5A,C) but was significantly less than that of JZL184 (100 nm; JZL184, 14.7 ± 2.3 s, n = 11; t(17) = 2.48, p = 0.024 vs BDNF; t(20) = 2.41, p = 0.026 vs DHF; Fig. 5B,C). There was no significant difference of the magnitude of DSI induced in the presence of BDNF, DHF, DHPG, and JZL184 (DHPG, 28.5 ± 4.8%, n = 11; JZL184, 32.9 ± 6.3%, n = 11; Fig. 5D). DSI induced in the presence of DHPG and JZL184 was blocked by AM251 (2 μm; DHPG + AM251, 4.0 ± 3.2%, n = 9; t(18) = 4.06, p < 0.001 vs DHPG; JZL184 + AM251, 3.5 ± 5.5%, n = 8; t(17) = 3.36, p = 0.004 vs JZL184). BDNF is coupled to PLCγ pathway, which cleaves PIP2 into IP3 and DAG (Reichardt, 2006). DAG is a precursor of eCB Iigand 2-AG (Di Marzo et al., 1998; Piomelli, 2003). BDNF is therefore linked to 2-AG-synthesizing machinery. Comparing the time course of DSI, it is apparent that BDNF and DHF behave like DHPG, which increases 2-AG synthesis (Jung et al., 2005), but not like JZL184, which inhibits 2-AG hydrolysis (Long et al., 2009). It is thus likely that BDNF and DHF enhance DSI via an increase in 2-AG synthesis rather than a reduction of 2-AG hydrolysis.
Figure 5.

Time course comparison of DSI implies that BDNF and DHF enable DSI via an increase in 2-AG production but not via a decrease in 2-AG hydrolysis. A, B, DSI was induced in VTA dopamine neurons in the continuous presence of Group I mGluR agonist DHPG (1 μm; n = 11; A) and MAGL inhibitor JZL184 (100 nm; n = 11; B). Solid lines indicate single exponential fitting curves of the decay of DSI. C, Summary of the time constant (τ) of DSI induced in the presence of BDNF, DHF, DHPG, and JZL184. τ of DSI in BDNF (n = 8) and DHF (n = 11) groups were obtained from Figure 1A, C and was not significantly different from that of the DHPG group (p > 0.05) but was significantly less than that of the JZL184 group (*p < 0.05). D, There was no significant difference in the magnitude of DSI induced in the presence of BDNF, DHF, DHPG, and JZL184 (p > 0.05).
Effects of conditional knock-out of BDNF in dopamine neurons on I-LTD
To test the role of endogenous BDNF in I-LTD, we used Cre-loxP technology to selectively delete BDNF from dopamine neurons and examined its impact on I-LTD. We used DAT-Cre+/− mice that express Cre recombinase controlled by dopamine transporter (DAT) promoter (Bäckman et al., 2006). To examine the effectiveness and specificity of Cre recombinase in DAT-Cre+/− mice, we crossed this mouse line with ROSA26 LacZ reporter mice. Cre recombinase was selectively expressed in the midbrain dopamine neurons as shown by colocalization of β-gal with dopamine synthetic enzyme and dopamine cell marker tyrosine hydroxylase (TH) (Fig. 6A,B). These results confirm the effectiveness and specificity of the expression of Cre recombinase in DAT-Cre+/− mice.
Figure 6.
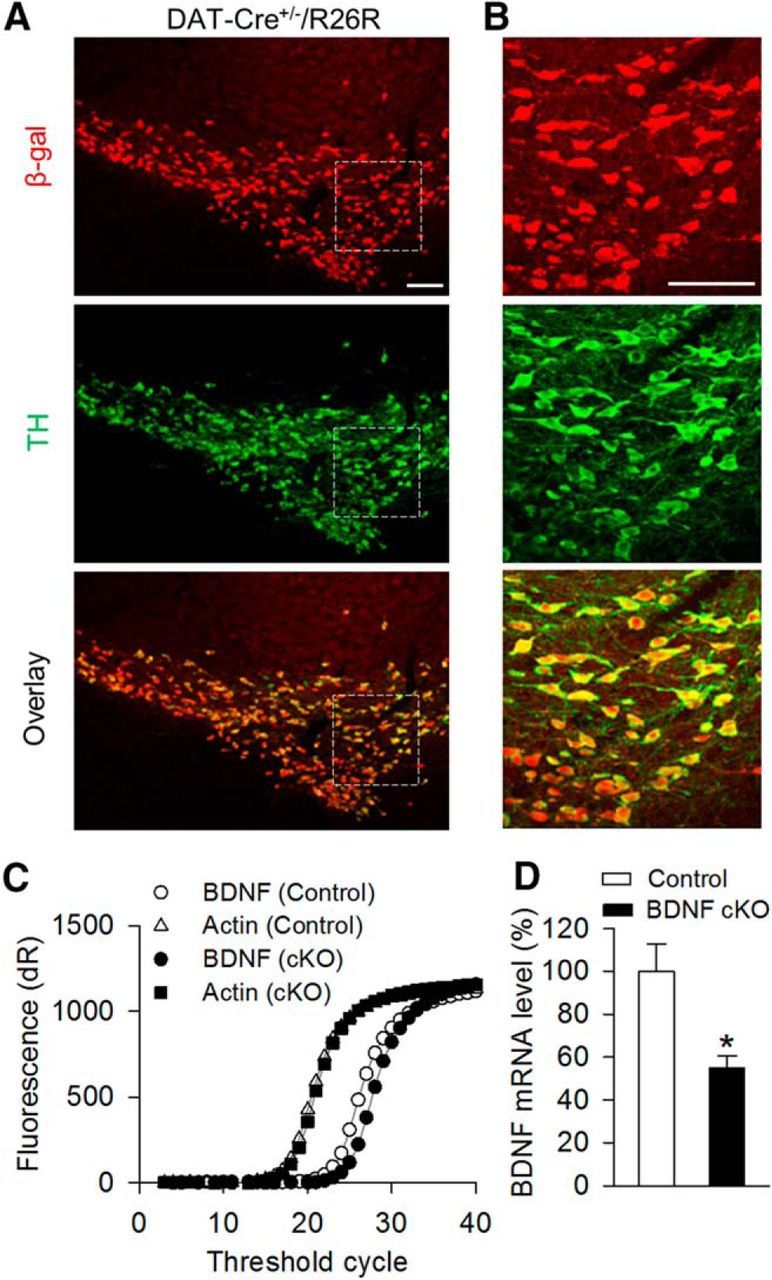
Generation of dopamine neuron-specific BDNF cKO mice. A, B, Cre recombinase was expressed in midbrain dopamine neurons in the offspring (DAT-Cre+/−/R26R) of DAT-Cre+/− mice crossed with R26R lacZ reporter mice. Immunofluorescence staining of midbrain sections showed that β-galactosidase (red) was exclusively expressed in TH+ (green) dopamine neurons in DAT-Cre+/−[r]/R26R mice (A). n = 3 mice. High-power view of the dashed boxes in A showed colocalization of β-galactosidase and TH (B). Scale bar, 100 μm. C, D, BDNF cKO mice were generated by crossing BdnfloxP/loxP mice with DAT-Cre+/− mice. Representative amplification curves for BDNF and β-actin mRNA from individual mouse VTA sample (C) and summarized data (D) of real-time quantitative PCR indicated that BDNF mRNA level in the VTA of BDNF cKO (DAT-Cre+/−/BdnfloxP/loxP) mice was significantly decreased compared with that in wild-type control (DAT-Cre−/−/BdnfloxP/loxP and DAT-Cre−/−/BDNFloxP/wt) mice (*p < 0.05).
We generated dopamine neuron-specific BDNF knock-out mice by crossing homozygous BdnfloxP/loxP mice with heterozygous DAT-Cre+/− mice. Among the offspring generated, DAT-Cre+/−/BdnfloxP/loxP mice were used as BDNF cKO group. DAT-Cre−/−/BdnfloxP/loxP and DAT-Cre−/−/BDNFloxP/wt mice were used as wild-type control groups. Real-time quantitative PCR indicates that the BDNF mRNA level in the VTA of BDNF cKO mice was significantly decreased compared with that in wild-type control mice (t(8) = 3.21, p = 0.012; Fig. 6C,D). These data confirmed the effectiveness of BDNF deletion under the present experimental conditions.
We have shown that a pathophysiologically relevant concentration of cocaine (3 μm) enabled subthreshold stimulation (10 Hz, 5 min) to induce I-LTD in rat VTA dopamine neurons, and this I-LTD was blocked by CB1 receptor antagonists (Pan et al., 2008a, b). We examined whether endogenous BDNF is required for this form of I-LTD. Repeated synaptic stimulation (10 Hz, 5 min) did not induce significant depression of IPSCs in either wild-type control mice (89.1 ± 6.6% of baseline, n = 7; t(12) = 1.40, p = 0.19) or BDNF cKO mice (91.8 ± 6.2% of baseline, n = 6; t(10) = 1.11, p = 0.29; Fig. 7A). The presence of cocaine (3 μm) during 10 Hz stimulation enabled I-LTD in wild-type control mice (63.0 ± 8.6% of baseline, n = 7; t(12) = 2.40, p = 0.033 vs control) but did not induce significant depression of IPSCs in BDNF cKO mice (92.8 ± 7.6% of baseline, n = 8; t(12) = 0.10, p = 0.92 vs control; Fig. 7B). This I-LTD was blocked by the CB1 receptor antagonist AM251 (2 μm; 89.7 ± 7.8% of baseline, n = 7; t(12) = 2.29, p = 0.041 vs I-LTD control). Next, we determined whether TrkB agonist DHF could rescue I-LTD in BDNF cKO mice. In the presence of cocaine (3 μm) plus DHF (10 μm), robust I-LTD was induced in BDNF cKO mice (68.4 ± 7.7% of baseline, n = 8; t(14) = 3.43, p = 0.004), and this I-LTD was blocked by AM251 (2 μm; 93.8 ± 7.8% of baseline, n = 7; t(13) = 2.24, p = 0.043 vs I-LTD in BDNF cKO mice; Fig. 7C). Thus, DHF rescued I-LTD in VTA dopamine neurons in BDNF cKO mice. In wild-type control mice, the presence of DHF and cocaine did not significantly increase the magnitude of I-LTD compared with that induced by cocaine alone (63.4 ± 8.0% of baseline, n = 6; t(11) = 0.033, p = 0.97 vs I-LTD control in Fig. 7B,D), suggesting that endogenous BDNF is sufficient to support I-LTD.
Figure 7.

Endogenous BDNF is required for cocaine-induced I-LTD. A, The 10 Hz, 5 min stimulation had no significant effect on evoked IPSCs in either wild-type control mice (n = 7; p > 0.05) or BDNF cKO mice (n = 6; p > 0.05). Sample traces of IPSCs before and after the 10 Hz stimulation are superimposed on the top. B, The presence of cocaine (3 μm) 3 min before and during the 10 Hz stimulation enabled I-LTD in wild-type control mice (n = 7; p < 0.05) but did not induce I-LTD in BDNF cKO mice (n = 8; p > 0.05). C, The presence of cocaine (3 μm) and DHF (10 μm) 3 min before and during the 10 Hz stimulation led to I-LTD induction in BDNF cKO mice (n = 8; p < 0.01), and this I-LTD was blocked by AM251 (2 μm; n = 7; p < 0.05). D, In wild-type control mice, the presence of cocaine and DHF did not significantly increase the magnitude of I-LTD compared with that induced by cocaine alone (n = 6; p > 0.05). Arrows indicate the application of a 10 Hz, 5 min stimulation.
BNDF is required for cocaine-induced inhibitory synaptic plasticity
We have shown that daily intraperitoneal injections of cocaine to rats for 5–7 d reduces the mean amplitude of maximal IPSCs and miniature IPSCs (mIPSCs) in VTA dopamine neurons of midbrain slices, indicating that repeated cocaine exposure in vivo reduces the strength of GABAergic inhibition to VTA dopamine neurons (Liu et al., 2005). Our previous studies suggest that repeated cocaine exposure in vivo reduces the strength of GABAergic inhibition onto dopamine neurons by inducing an I-LTD-like synaptic plasticity (Pan et al., 2008b). Having shown that BDNF is required for I-LTD induction in VTA dopamine neurons, we next determined whether cocaine-induced reduction of GABAergic inhibition was altered by dopamine neuron-specific deletion of BDNF. Wild-type control and BDNF cKO mice were given intraperitoneal injections of saline or cocaine (15 mg/kg) for 5 d. Midbrain slices were prepared from these four groups of mice the next day, maximal IPSCs and mIPSCs were recorded as described in Materials and Methods. Maximal IPSCs were elicited by gradually increasing the stimulation intensity to recruit saturating IPSCs (Huang et al., 1999; Liu et al., 2005), which yield a good estimate of total inhibitory inputs to the recorded neuron. mIPSCs recorded in the presence of TTX (0.5 μmm) represent quantal GABA release from presynaptic axonal terminals (Zhu and Lovinger, 2005). Together, maximal IPSCs and mIPSCs provide a good indication of the strength of GABAergic inhibition. We found that cocaine treatment and genotype had significant effects on the mean amplitude of maximal IPSCs (cocaine: F(1,39) = 6.27, p = 0.017; genotype: F(1,39) = 4.23, p = 0.046; cocaine × genotype interaction: F(1,39) = 6.89, p = 0.012; Fig. 8A) and mIPSCs (cocaine: F(1,43) = 9.45, p = 0.004; genotype: F(1,43) = 11.12, p = 0.002; cocaine × genotype interaction: F(1,43) = 4.55, p = 0.039; Fig. 8B). Tukey's post hoc tests indicated that the mean amplitude of maximal IPSCs (p = 0.001) and mIPSCs (p < 0.001) was significantly decreased in cocaine-treated wild-type control mice compared with saline-treated wild-type control mice. BDNF cKO had no significant effects on the mean amplitude of maximal IPSCs (p = 0.69) and mIPSCs (p = 0.41) in saline-injected groups but blocked the decreases in the mean amplitude of maximal IPSCs (p = 0.93) or mIPSCs (p = 0.51) in cocaine-injected groups. These results suggest that cocaine-induced reduction of GABAergic inhibition was blocked in BDNF conditional knock-out mice.
A consequence of cocaine-induced reduction of GABAergic inhibition to VTA dopamine neurons is to facilitate synaptic potentiation in vivo (Liu et al., 2005). Single or repeated cocaine intraperitoneal injections increase the AMPAR/NMDAR ratio, an indicator for synaptic strength, in VTA dopamine neurons (Ungless et al., 2001; Borgland et al., 2004; Liu et al., 2005; Mameli et al., 2009). We determined whether endogenous BDNF is required for the increase in the AMPAR/NMDAR ratio in VTA dopamine neurons. Wild-type control and BDNF cKO mice were given intraperitoneal injections of saline or cocaine (15 mg/kg) for 5 d. Midbrain slices were prepared from these four groups of mice the next day; AMPAR/NMDAR ratio in VTA dopamine neurons was measured as described in our previous studies (Liu et al., 2005, 2014). Two-way ANOVA showed that cocaine treatments and genotype had significant effects on the AMPAR/NMDAR ratio in VTA dopamine neurons (cocaine: F(1,45) = 8.65, p = 0.005; genotype: F(1,45) = 4.24, p = 0.045); there was a significant interaction between cocaine treatments and genotype (F(1,45) = 10.20, p = 0.003; Fig. 9). Tukey's post hoc tests indicated that cocaine treatments significantly increased the AMPAR/NMDAR ratio in wild-type control mice (p < 0.001), whereas this increase was blocked in BDNF cKO mice (p = 0.86). Together, the above results suggest that BDNF is required for cocaine-induced I-LTD, reduction of GABAergic inhibition, and increase in the AMPAR/NMDAR ratio in VTA dopamine neurons.
Figure 9.

Endogenous BDNF is required for cocaine-induced potentiation of the AMPAR/NMDAR ratio. Wild-type control and BDNF cKO mice were given intraperitoneal injections of saline or cocaine (15 mg/kg) for 5 d. Midbrain slices were prepared from these four groups of mice the next day. A, Representative AMPAR- and NMDAR-mediated evoked EPSCs recorded from VTA dopamine neurons. B, Cocaine treatments significantly increased the AMPAR/NMDAR ratio in wild-type control mice (***p < 0.001), and this increase was blocked in BDNF cKO mice (p > 0.05). n = 12 or 13 for each group.
BDNF conditional knock-out attenuated CPP to cocaine
Cocaine-induced synaptic plasticity in the VTA has been implicated in the formation of drug-associated memories (Kauer, 2004). We examined the effect of dopamine neuron-specific knock-out of BDNF on cocaine-induced CPP. During the baseline preference test (pretest), six mice showing unconditioned side preference (≥180 s disparity) were excluded. The remaining mice exhibited no significant unconditioned place preference (Fig. 10A). Then the saline (0.9% NaCl, i.p.) and cocaine (15 mg/kg) place conditioning was conducted once daily for 3 d (see Materials and Methods). CPP was tested 24 h after the last place conditioning. Two-way ANOVA revealed that cocaine conditioning (F(1,35) = 18.39, p < 0.001) and BDNF cKO (F(1,35) = 7.55, p = 0.009) had significant effects on CPP scores and there was a significant interaction between cocaine conditioning and genotype (F(1,35) = 4.19, p = 0.048; Fig. 10B). Tukey's post hoc test showed that BDNF cKO led to a significant decrease in the CPP score in cocaine-conditioned mice (p = 0.001) but did not have significant effect on the CPP score in saline-conditioned mice (p = 0.64). Thus, BDNF cKO attenuated CPP to cocaine.
We next determined whether the impairment of cocaine CPP in BDNF cKO mice could be rescued by systemic administration of TrkB agonist DHF. Wild-type control and BDNF cKO mice underwent cocaine conditioning as described above. There was no unconditioned place preference during pretest (Fig. 10C). The mice were then pretreated with vehicle or DHF (5 mg/kg, i.p.) before each saline or cocaine conditioning. Two-way ANOVA revealed that genotype (F(1,34) = 24.10, p < 0.001) and drug pretreatments (F(1,34) = 12.28, p = 0.001) had significant main effects on CPP scores and there was a significant interaction between cocaine conditioning and genotype (F(1,34) = 6.49, p = 0.016; Fig. 10D). Tukey's post hoc test showed that CPP was significantly attenuated in vehicle-pretreated BDNF cKO mice compared with vehicle-pretreated wild-type mice (p < 0.001), and DHF pretreatment significantly increased cocaine CPP in BDNF cKO mice (p < 0.001), but not in control mice (p = 0.51). These results suggest that the impairment of cocaine CPP in BDNF cKO mice could be attributed to lack of BDNF.
We examined whether the CB1 receptor is required for DHF-induced restoration of cocaine CPP in BDNF cKO mice. BDNF cKO mice underwent cocaine conditioning as described above. BDNF cKO mice were then given intraperitoneal injections of either the CB1 antagonist AM251 (3 mg/kg) or AM251 (3 mg/kg) plus DHF (5 mg/kg) before each place conditioning. For comparison, the CPP scores from these two groups were analyzed together with those from BDNF cKO mice that received intraperitoneal injections of vehicle or DHF before conditioning in Figure 10D. One-way ANOVA revealed that the preference scores were significantly different among the four treatment groups (i.e., vehicle, DHF, AM251, and AM251 + DHF; F(3,38) = 7.94, p < 0.001). Tukey's post hoc test indicated that the DHF-induced restoration of cocaine CPP in BDNF cKO mice was significantly attenuated by AM251 (p = 0.025), whereas AM251 did not further decrease the preference score in BDNF cKO mice compared with the vehicle group (p = 0.99) (Fig. 11A). These results suggest that the CB1 receptor is one of the downstream effectors responsible for the DHF-induced restoration of cocaine CPP in BDNF cKO mice.
We also repeated the above AM251 experiments in wild-type control mice. These mice were given intraperitoneal injections of AM251 or AM251 plus DHF before place conditioning. For comparison, the preference scores from these two groups were also analyzed together with those from wild-type control mice in Figure 10D. One-way ANOVA showed that the preference scores were significantly different among the four treatment groups in wild-type control mice (F(3,33) = 5.85, p = 0.003). Tukey's post hoc test indicated that AM251 significantly decreased the preference score in wild-type control mice compared with vehicle treatment group (p = 0.042); the AM251 + DHF treatment group exhibited a significant decrease in the preference score compared with the DHF treatment group (p = 0.023) (Fig. 11B).
Discussion
Here we have found that TrkB agonists BDNF and DHF activated the TrkB receptor to enable eCB-mediated DSI and I-LTD in VTA dopamine neurons. BDNF cKO from dopamine neurons attenuated cocaine-induced inhibitory and excitatory synaptic plasticity and CPP. Systemic administration of DHF rescued cocaine CPP in BDNF cKO mice; this behavioral effect of DHF was attenuated by the CB1 antagonist AM251. These results uncovered a key role of BDNF-eCB interaction in regulating cocaine-induced synaptic plasticity and associative learning.
Facilitation of DSI and I-LTD by BDNF
Depolarization of VTA dopamine neurons was not sufficient to induce DSI, and electrical stimulation at 10 Hz for 5 min was subthreshold for I-LTD induction. However, the presence of BDNF or DHF enabled DSI and I-LTD, and the effects of BDNF and DHF were blocked by TrkB inhibitor K252a or TrkB antagonist CTX-B. DSI and I-LTD are mediated by 2-AG-induced activation of CB1 receptors (Chevaleyre and Castillo, 2003; Pan et al., 2009; Gao et al., 2010; Tanimura et al., 2010). An increase in 2-AG production might explain BDNF-induced facilitation of DSI and I-LTD. PLC and DAG lipases are key enzymes for 2-AG synthesis. PLC cleaves PIP2 into DAG and IP3. DAG is subsequently converted into 2-AG by diacylglycerol lipase (DAGL) (Stella et al., 1997; Di Marzo et al., 1998; Piomelli et al., 2000). There are five types of PLC (β, γ, δ, ε, and ζ) (Rebecchi and Pentyala, 2000). Gq/11-coupled receptors, such as Group I mGluRs, activate PLCβ to enhance 2-AG production (Maejima et al., 2001; Hashimotodani et al., 2005; Di Marzo, 2011). BDNF is coupled to PLCγ (Reichardt, 2006); the activation of PLCγ-DAG pathway may also enhance 2-AG production. BDNF depresses IPSCs in neocortical layer 2/3 pyramidal neurons, and this depression is blocked by a CB1 antagonist or PLC inhibitor (Lemtiri-Chlieh and Levine, 2010; Zhao and Levine, 2014). We showed that I-LTD- and BDNF-induced depression of IPSCs was blocked by PLC and DAG lipase inhibitors. These results suggest that BDNF facilitates DSI and I-LTD by enhancing 2-AG synthesis via the PLCγ-DAG pathway. However, this conclusion should be interpreted with caution since currently available PLC and DAG lipase inhibitors have off-target effects (Walker et al., 1998; Hoover et al., 2008).
Incubation of BDNF for 4–24 h increases CB1 receptor transcripts and decreases MAGL transcripts in cultured cerebellar granule neurons (Maison et al., 2009), whereas acute bath application of BDNF inhibited CB1 agonist-induced depression of IPSCs in striatal slices through a mechanism mediated by altered cholesterol metabolism and membrane lipid raft function (De Chiara et al., 2010). We also considered the possibility that BDNF and DHF might facilitate DSI and I-LTD by enhancing CB1 receptor responsiveness or decreasing MAGL activity. However, we found that DHF did not significantly alter WIN 55212–2-induced depression of IPSCs, suggesting that acute treatment with TrkB agonists does not significantly alter CB1 receptor function in the VTA. In addition, time course comparison of DSI induced in BDNF, DHF, DHPG, and JZL184 suggests that BDNF and DHF do not significantly alter MAGL activity.
The requirement of BNDF-eCB interaction in regulating cocaine-induced synaptic plasticity
We have shown that repeated intraperitoneal cocaine injections for 5–7 d reduce the strength of GABAergic inhibition to VTA dopamine neurons (Liu et al., 2005; Pan et al., 2008b). Our previous studies suggest that eCB-dependent I-LTD constitutes a mechanism for cocaine-induced reduction of GABAergic inhibition (Pan et al., 2008a, b). Given that BDNF plays a critical role in I-LTD, we examined whether it is required for the reduction of GABAergic inhibition to VTA dopamine neurons induced by repeated cocaine exposure in vivo. We found that dopamine neuron-specific knock-out of BDNF blocked cocaine-induced decreases in the amplitude of maximal IPSCs and mIPSCs. Our results appear to support a model in which BDNF-eCB interaction is required for cocaine-induced reduction of GABAergic inhibition to VTA dopamine neurons. A direct consequence of reduction of GABAergic inhibition is to prime excitatory synapses for potentiation. Indeed, BDNF cKO prevented the increase in the AMPAR/NMDAR ratio induced by repeated cocaine exposure in vivo. These results suggest a critical role of BDNF in regulating cocaine-induced excitatory and inhibitory synaptic plasticity in the VTA. BDNF cKO did not significantly alter the amplitude of maximal IPSCs and mIPSCs and AMPAR/NMDAR ratio in saline-treated mice, suggesting that BDNF is not essential for basal inhibitory and excitatory synaptic transmission. Either acute loss of BDNF or associated developmental changes could explain the effects of BDNF cKO on cocaine-induced synaptic plasticity in the VTA. Future studies are needed to distinguish between these two possibilities.
The effects of BDNF cKO on cocaine CPP
BDNF has been implicated in behavioral effects of cocaine (for review, see Corominas et al., 2007; McGinty et al., 2010). BDNF protein levels are increased in the VTA, NAc, and amygdala after 30–90 d withdrawal from cocaine self-administration (Grimm et al., 2003). Local infusion of BDNF produces brain region-specific effects on cocaine seeking. Infusion of BDNF into subcortical structures, such as VTA and NAc, enhances cocaine seeking (Lu et al., 2004; Graham et al., 2007), whereas infusion of BDNF antibody into the NAc or localized BDNF deletion in the NAc attenuates cocaine-induced reinstatement (Graham et al., 2007) and cocaine CPP (Graham et al., 2009). In contrast, BDNF infusion into the dorsomedial PFC suppresses cocaine seeking (Berglind et al., 2007). Lentiviral expression of BDNF or TrkB in the NAc of the rat enhanced cocaine-induced behavioral sensitization and CPP, delayed CPP-extinction upon repeated measurements, and increased CPP reinstatement. Conversely, lentiviral expression of a truncated form of TrkB inhibited these behavioral changes (Bahi et al., 2008). Most relevant to the present study is the finding that genetic deletion of BDNF with adeno-associated viral (AAV) vector-carrying the Cre recombinase (AAV-Cre-GFP) in the VTA of BdnfloxP/loxP mice reduced cocaine CPP (Graham et al., 2009). However, the AAV-Cre-GFP deletes BDNF in both dopamine neurons and nondopamine neurons in the VTA. In the present study, we generated dopamine neuron-specific BNDF knock-out mice by crossing homozygous BdnfloxP/loxP mice with heterozygous DAT-Cre+/− mice. Consistent with the AAV-Cre-GFP approach (Graham et al., 2009), we found that cocaine CPP was greatly attenuated in BNDF cKO mice compared with wild-type mice. This attenuation of cocaine CPP could be rescued by systemic administration of TrkB agonist DHF. Together, the above studies suggest that the behavioral deficiency in cocaine-cue learning can be attributed to the lack of BDNF, although we cannot rule out the possibility that developmental changes after BDNF cKO may also play a role. Long-term synaptic plasticity in brain's reward circuit may represent a putative mechanism for the learned association between environmental cues and rewarding effects of cocaine (Kauer, 2004; Hyman et al., 2006). The impairment of cocaine-induced synaptic plasticity might help explain the deficiency in cocaine CPP in BDNF cKO mice.
The eCB system has been implicated in behavioral effects of cocaine (Lupica and Riegel, 2005; Wiskerke et al., 2008). Systemic or local infusions of CB1 antagonists into the nucleus accumbens attenuated cocaine seeking (Xi et al., 2006, 2008). CB1 agonists trigger relapse to cocaine seeking after prolonged cocaine withdrawal, whereas CB1 antagonists attenuate relapse induced by cocaine or cocaine-associated cues (De Vries et al., 2001). CB1 knock-out mice exhibited reduced cocaine self-administration (Soria et al., 2005) but normal CPP to cocaine (Martin et al., 2000; Houchi et al., 2005). In contrast, intraperitoneal injections of a CB1 antagonist blocked the acquisition, but not the expression, of cocaine CPP (Chaperon et al., 1998). Consistent with the latter finding, we have shown that intra-VTA injections of the CB1 antagonist AM251 before each place conditioning attenuated the acquisition of CPP to cocaine (Pan et al., 2011). Constitutive knock-out of genes is known to produce compensatory effects. It is unclear whether a compensatory mechanism could explain differential effects on cocaine CPP by the CB1 antagonist and CB1 knock-out. Nevertheless, the above studies suggest that the eCB/CB1 signaling system is critically involved in cocaine reward and seeking. We have shown that intraperitoneal injection of CB1 antagonist AM251 attenuated cocaine CPP in wild-type mice and blocked DHF-induced restoration of cocaine CPP in BDNF cKO mice. These results suggest that the CB1 receptor is an important downstream target of BDNF and that BDNF-CB1 signaling in the VTA is critically involved in cocaine-cue associative learning. Given that BDNF and CB1 receptors are important modulators of synaptic plasticity, we suspect that modulation of cocaine-induced synaptic plasticity may be responsible for the behavioral effects of DHF and AM251. However, the TrkB receptor can activate a variety of downstream targets (Reichardt, 2006); we cannot exclude the possibility that DHF may activate other targets to restore CPP to cocaine.
In conclusion, we have shown that BDNF and DHF facilitate eCB-mediated DSI and I-LTD, and BDNF conditional knock-out leads to impairments of eCB-mediated I-LTD and cocaine-induced excitatory and inhibitory synaptic plasticity in VTA dopamine neurons. In addition, BDNF-CB1 signaling is also involved in cocaine-cue associated learning as indicated by cocaine CPP experiments. Together, the present study suggests that the BDNF-eCB interaction plays a critical role in mediating cocaine-induced long-term synaptic plasticity and behavioral effects.
Footnotes
This work was supported by National Institutes of Health Grants DA035217 and MH101146 and in part by the Research and Education Initiative Fund, a component of the Advancing a Healthier Wisconsin endowment at the Medical College of Wisconsin.
The authors declare no competing financial interests.
References
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Chandrasekar V, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology. 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaperon F, Soubrié P, Puech AJ, Thiébot MH. Involvement of central cannabinoid (CB1) receptors in the establishment of place conditioning in rats. Psychopharmacology. 1998;135:324–332. doi: 10.1007/s002130050518. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/S0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corominas M, Roncero C, Ribases M, Castells X, Casas M. Brain-derived neurotrophic factor and its intracellular signaling pathways in cocaine addiction. Neuropsychobiology. 2007;55:2–13. doi: 10.1159/000103570. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- De Chiara V, Angelucci F, Rossi S, Musella A, Cavasinni F, Cantarella C, Mataluni G, Sacchetti L, Napolitano F, Castelli M, Caltagirone C, Bernardi G, Maccarrone M, Usiello A, Centonze D. Brain-derived neurotrophic factor controls cannabinoid CB1 receptor function in the striatum. J Neurosci. 2010;30:8127–8137. doi: 10.1523/JNEUROSCI.1683-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries TJ, Shaham Y, Homberg JR, Crombag H, Schuurman K, Dieben J, Vanderschuren LJ, Schoffelmeer AN. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med. 2001;7:1151–1154. doi: 10.1038/nm1001-1151. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat Neurosci. 2011;14:9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/S0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Gong R, Ding C, Hu J, Lu Y, Liu F, Mann E, Xu F, Cohen MB, Luo M. Role for the membrane receptor guanylyl cyclase-C in attention deficiency and hyperactive behavior. Science. 2011;333:1642–1646. doi: 10.1126/science.1207675. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Graham DL, Krishnan V, Larson EB, Graham A, Edwards S, Bachtell RK, Simmons D, Gent LM, Berton O, Bolanos CA, DiLeone RJ, Parada LF, Nestler EJ, Self DW. Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover HS, Blankman JL, Niessen S, Cravatt BF. Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg Med Chem Lett. 2008;18:5838–5841. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houchi H, Babovic D, Pierrefiche O, Ledent C, Daoust M, Naassila M. CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology. 2005;30:339–349. doi: 10.1038/sj.npp.1300568. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yeh CM, Wu MY, Chang AY, Chan JY, Chan SH, Hsu KS. Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. J Neurosci. 2011;31:4194–4203. doi: 10.1523/JNEUROSCI.5239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/S0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci. 1999;19:9780–9787. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005;68:1196–1202. doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/S0896-6273(01)00246-X. [DOI] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, Levine ES. BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. J Neurophysiol. 2010;104:1923–1932. doi: 10.1152/jn.00472.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Liu Y, Zhong P, Wilkinson B, Qi J, Olsen CM, Bayer KU, Liu QS. CaMKII activity in the ventral tegmental area gates cocaine-induced synaptic plasticity in the nucleus accumbens. Neuropsychopharmacology. 2014;39:989–999. doi: 10.1038/npp.2013.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo L, Maione S, Di Marzo V. Endocannabinoids and neuropathic pain: focus on neuron-glia and endocannabinoid-neurotrophin interactions. Eur J Neurosci. 2014;39:401–408. doi: 10.1111/ejn.12440. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Riegel AC. Endocannabinoid release from midbrain dopamine neurons: a potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology. 2005;48:1105–1116. doi: 10.1016/j.neuropharm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/S0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maison P, Walker DJ, Walsh FS, Williams G, Doherty P. BDNF regulates neuronal sensitivity to endocannabinoids. Neurosci Lett. 2009;467:90–94. doi: 10.1016/j.neulet.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O. Cocaine, but not morphine, induces conditioned place preference and sensitization to locomotor responses in CB1 knockout mice. Eur J Neurosci. 2000;12:4038–4046. doi: 10.1046/j.1460-9568.2000.00287.x. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/S0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008a;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008b;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D, Liu QS. Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J Neurosci. 2011;31:11244–11255. doi: 10.1523/JNEUROSCI.1040-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/S0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Safo PK, Regehr WG. Endocannabinoids control the induction of cerebellar LTD. Neuron. 2005;48:647–659. doi: 10.1016/j.neuron.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Soria G, Mendizábal V, Touriño C, Robledo P, Ledent C, Parmentier M, Maldonado R, Valverde O. Lack of CB1 cannabinoid receptor impairs cocaine self-administration. Neuropsychopharmacology. 2005;30:1670–1680. doi: 10.1038/sj.npp.1300707. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci. 2003;18:2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EM, Bispham JR, Hill SJ. Nonselective effects of the putative phospholipase C inhibitor, U73122, on adenosine A1 receptor-mediated signal transduction events in Chinese hamster ovary cells. Biochem Pharmacol. 1998;56:1455–1462. doi: 10.1016/S0006-2952(98)00256-1. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wiskerke J, Pattij T, Schoffelmeer AN, De Vries TJ. The role of CB1 receptors in psychostimulant addiction. Addict Biol. 2008;13:225–238. doi: 10.1111/j.1369-1600.2008.00109.x. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci. 2006;26:8531–8536. doi: 10.1523/JNEUROSCI.0726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Spiller K, Pak AC, Gilbert J, Dillon C, Li X, Peng XQ, Gardner EL. Cannabinoid CB1 receptor antagonists attenuate cocaine's rewarding effects: experiments with self-administration and brain-stimulation reward in rats. Neuropsychopharmacology. 2008;33:1735–1745. doi: 10.1038/sj.npp.1301552. [DOI] [PubMed] [Google Scholar]
- Yu F, Zhong P, Liu X, Sun D, Gao HQ, Liu QS. Metabotropic glutamate receptor I (mGluR1) antagonism impairs cocaine-induced conditioned place preference via inhibition of protein synthesis. Neuropsychopharmacology. 2013;38:1308–1321. doi: 10.1038/npp.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Levine ES. BDNF-endocannabinoid interactions at neocortical inhibitory synapses require phospholipase C signaling. J Neurophysiol. 2014;111:1008–1015. doi: 10.1152/jn.00554.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Wang W, Yu F, Nazari M, Liu X, Liu QS. Phosphodiesterase 4 inhibition impairs cocaine-induced inhibitory synaptic plasticity and conditioned place preference. Neuropsychopharmacology. 2012;37:2377–2387. doi: 10.1038/npp.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Liu X, Zhang Z, Hu Y, Liu SJ, Lezama-Ruiz M, Joksimovic M, Liu QS. Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J Neurosci. 2014;34:6352–6366. doi: 10.1523/JNEUROSCI.3673-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Retrograde endocannabinoid signaling in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurosci. 2005;25:6199–6207. doi: 10.1523/JNEUROSCI.1148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]