

Abstract
Approximately 350,000 individuals in the United States rely on maintenance hemodialysis treatment because of end-stage renal disease. Despite improvements in dialysis technology, the mortality rate for patients treated with maintenance dialysis is still exceptionally high, with a 5-year survival rate of only 35%. Many patients succumb to conditions resulting at least in part from the chronic induction of inflammation. Among the triggers of inflammation, the complement system is of particular importance, being a well-appreciated mediator of inflammatory processes that is involved in many pathologic states. Here we used a refined pre-clinical model of hemodialysis in cynomolgus monkeys to confirm that even modern, polymer-based hemodialysis filters activate complement and to evaluate the potential of Cp40, a peptidic C3 inhibitor, to attenuate hemodialysis-induced complement activation. Our data show marked induction of complement activation even after only a single session of hemodialysis. Importantly, complete inhibition of complement activation was achieved in response to two distinct Cp40 treatment regimens. Further, we show that application of Cp40 during hemodialysis resulted in increased levels of the anti-inflammatory cytokine IL-10, indicating that Cp40 may be a potent and cost-effective treatment option for attenuating chronic inflammatory conditions in dialysis-dependent patients.
Keywords: Complement, C3, hemodialysis, compstatin, Cp40
Introduction
Approximately 500,000 individuals in the United States require kidney replacement therapy for end-stage renal disease (ESRD). Replacement therapy is the most desirable treatment option for ESRD patients, but compatibility issues and limited availability of kidneys have forced hundreds of thousands of patients annually to rely on hemodialysis (HD) (NKUDIC, 2012). Despite improvements in HD techniques and materials, the mortality rate for patients undergoing HD remains ~16% (NKUDIC, 2012). ESRD patients suffer from various morbidity-associated complications, including anemia, malnutrition, oxidative stress, endothelial and immune cell dysfunction, leukopenia, anaphylactoid reactions, and increased risk for artherosclerosis and myocardial infarction (Jofre et al., 2006; Nilsson et al., 2007; Cavalli et al., 2010). It is generally accepted that chronic inflammation contributes to these adverse conditions (Galli, 2007). Indeed, one of the major causes of mortality in ESRD patients is cardiovascular disease (CVD), yet the survival rate of HD patients has not improved after decades of treating underlying risk factors such as hypercholesterolemia and hypertension (Zaritsky & Kalantar-Zadeh, 2012). Additionally, chronic inflammation is considered a contributor to resistance to erythropoietin treatment (Macdougall & Cooper, 2002). This involvement of inflammatory responses in several aspects of HD-related complications suggests that anti-inflammatory therapies would greatly benefit the clinical outcome and improve the quality-of-life of patients.
While the underlying disease is an important contributor to chronic inflammation in ESRD, the HD procedure itself induces inflammation (Stenvinkel & Alvestrand, 2002). Indeed, higher levels of acute-phase proteins and pro-inflammatory cytokines are strongly correlated with mortality in HD patients (Kimmel et al., 1998; Yeun et al., 2000; Rao et al., 2005). Given that patients requiring maintenance HD usually undergo three or more treatments per week for long periods of time, it is likely that these “chronic acute” inflammatory triggers contribute to their morbidity and mortality.
The complement system is intricately involved in promoting inflammation, modulating protein and cellular networks in response to activation by foreign and damaged cells or by artificial surfaces (Ricklin et al., 2010; Ricklin & Lambris, 2013a). Therefore, biomaterials commonly used in the clinic trigger substantial levels of complement activation, with cellulose-based HD filters being particularly problematic (Ekdahl et al., 2011; DeAngelis et al., 2012). In an extracorporeal circuit HD model with human blood, we have shown that even modern, polysulfone-based HD filters induce complement C5a generation and increased expression of tissue factor, the primary initiator of coagulation; this mechanism may contribute to the elevated risk of thrombosis in HD patients (Kourtzelis et al., 2010). Importantly, complement inhibition using the peptidic C3 inhibitor compstatin (Qu et al., 2011) significantly reduces tissue factor expression, cell activation, and release of inflammatory markers, suggesting that therapeutic complement inhibition may have wide-ranging beneficial effects in HD (Kourtzelis et al., 2010).
Here we used a pre-clinical model of HD in cynomolgus monkeys to confirm that even modern, polymer-based HD filters activate complement and to evaluate the potential of Cp40, a peptidic C3 inhibitor, to attenuate HD-induced complement activation.
Material and Methods
Study Animals
All procedures were conducted according to a written protocol approved by an institutional animal care and use committee and with strict compliance to accepted animal welfare standards, including the Public Health Service Policy on Humane Use of Laboratory Animals, the ILAR Guide for the Care and Use of Laboratory Animals, and the Animal Welfare Act.
Purpose-bred, experimentally naïve cynomolgus monkeys (Macaca fascicularis) with an activated partial thromboplastin time (APTT) in the normal range (17.8 – 20.4 s) at the Simian Conservation Breeding and Research Center (SICONBREC, Makati City, Philippines), a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), were used in this study. Animals were approximately 4 to 5 years of age and weighed 4.7 to 5.6 kg. The study animals underwent a comprehensive health screening prior to use, including a complete physical examination by a staff veterinarian and evaluation of a standard panel of serum chemistry and hematological and coagulation parameters.
Hemodialysis procedure
HD was performed using an AK 96 Dialysis machine and pediatric Polyflux® 2H filter (Gambro). Monkeys were fasted overnight with water provided ad libitum. Animals were sedated with a mixture of 10.0 mg/kg ketamine plus 1.0 mg/kg xylazine and isoflurane (3% for induction, 1% for maintenance). Sedated monkeys were wrapped in a water-heating blanket for body temperature maintenance during the procedure, then intubated. A venous catheter was placed into each femoral vein for blood access, and a sample of blood was collected from each catheter to obtain baseline values prior to the start of the experiment. Before the system was connected, the blood lines were flushed with 200-500 ml of saline, and 300 ml of 2 IU/ml low molecular weight heparin in saline was introduced into the system. Because of the low blood volume of the monkeys as compared to the size of the filter, ~7-8% of the total monkey blood volume was supplemented with either blood from a donor monkey (data shown in Fig. 1) or saline (data shown in Figs. 2-4). For animals treated with Cp40, a bolus dose (2 mg/kg) of the compound was injected into one of the femoral catheters. A mixture of 50 IU heparin in 25 ml of saline was injected into the outflow blood line, not yet connected to the animal or the dialysis filter. The outflow blood line was connected to the animal, and 24 ml of blood was withdrawn into the blood line. The outflow blood line was then removed from the catheter and attached to the end of the inflow blood line to set up an extracorporeal circuit. The blood/saline/heparin mixture was circulated for 1 min to prime the system. The ends of the blood lines were unattached and connected to the femoral catheters to set up the HD circuit (including the filter), as shown in Fig. 1A. Immediately prior to the initiation of extracorporeal circulation, a bolus dose of 30 IU/kg heparin was injected into the access port located at the inflow blood line, and a 4-h constant rate infusion of 50 ml heparin at 20 IU/kg/h was initiated. Simultaneously, infusion of Cp40 (4 μg/kg/min at 0.21 ml/min) was performed in some of the treatment animals. The extracorporeal blood circulation (i.e., dialysis) was initiated at a rate of 300 ml/min using bicarbonate-buffered dialysate (Gambro BiCart – sodium bicarbonate) at a rate of 50 ml/min. Blood samples were collected as soon as dialysis was started (0 h), then at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, and 4 h after the start of the dialysis. Blood was collected from two access ports (outflow and inflow sampler) present in the bloodline immediately after passing through the dialyzer (inflow, “in-circuit” conditions), or immediately after exit from the body (outflow, “systemic” conditions). After the 4 h blood collection, the extracorporeal circulation and heparin and Cp40 (in treated animals) infusion were stopped, the blood lines and catheters removed, and the monkeys returned to their cage and allowed to recover. Further single blood collections were taken from the femoral vein at 5, 6, 16-20, and 28 h after the start of HD. A schematic diagram of the HD system, including the blood lines and catheters, is shown in Fig. 1A.
Figure 1.

Modern hemodialysis filters induce complement activation. (A) Schematic diagram of hemodialysis circuit. Access lines were set up on the femoral veins of a cynomolgus monkey (Macaca fascicularis) for blood outflow and inflow. Blood was sampled from two access ports (outflow & inflow sampler) located on each blood line to obtain samples before and after filtering. Cp40 and heparin were introduced into the system immediately prior to blood circulation via a third port. Sterile bicarbonate buffer was used as dialysate. (B) Hemodialysis was performed in cynomolgus monkeys for a period of four hours and blood samples were collected prior to, during, and after blood circulation as indicated. (C) Complement C3 activation was measured in plasma samples by ELISA and represented as optical density (O.D.) values. Three sessions of hemodialysis were performed with a 2-day interval between each session. The figures depict data from blood samples collected from the inflow sampler (Fig. 1A). Similar results were obtained with samples collected from the outflow sampler.
Figure 2.

Cp40 prevents hemodialysis-induced complement activation. (A) Hemodialysis was performed in cynomolgus monkeys for a period of four hours and blood samples were collected prior to, during, and after blood circulation as indicated. Cp40 was injected as an i.v. bolus of 2 mg/kg followed by an i.v. infusion of 4 μg/kg/min. (B) Blood samples were collected from a monkey that was untreated (black line) or treated with Cp40 (gray line) and the activation of complement C3 was measured in plasma samples by ELISA, represented as optical density (O.D.) values. The figure depicts data from blood samples collected from the inflow sampler (Fig. 1A). Similar results were obtained with samples collected from the outflow sampler. (C) Levels of Cp40 were measured by mass spectrometry in the plasma of a monkey treated with Cp40. Samples were collected from both samplers as indicated in Fig. 1A.
Figure 4.
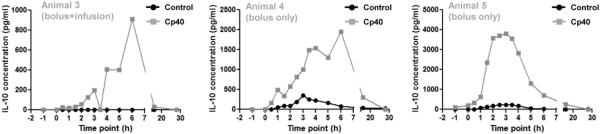
The use of Cp40 during hemodialysis induces increased plasma levels of the anti-inflammatory cytokine IL-10. Hemodialysis was performed in cynomolgus monkeys for a period of four hours and blood samples were collected prior to, during, and after blood circulation. Cp40 was injected as an i.v. bolus of 2 mg/kg (Animals 4 and 5) or as an i.v. bolus of 2 mg/kg followed by i.v. infusion of 4 μg/kg/min (Animal 3). Blood samples were collected from untreated (black line) or Cp40-treated (gray line) monkeys and plasma levels of IL-10 were measured by multiplex ELISA.
Blood samples
Blood samples (0.5 ml) were collected in 2-ml EDTA Vacutainer blood-collecting tubes, kept on ice, and centrifuged at 800 × g for 10 min to obtain plasma. Plasma samples were immediately frozen and shipped to the University of Pennsylvania for further analysis.
Measurement of activated C3 in the plasma
Concentrations of activated fragments of monkey C3 were measured in plasma by a sandwich ELISA. Microtiter plates were coated with 50 ml of a 1 μg/ml solution of capture antibody C3-28 in PBS. This antibody specifically recognizes neo-epitopes exposed on molecules of activated C3 (C3b, iC3b, C3c) (Soulika et al., 2000). The wells were then blocked with 1% BSA in PBS for 1 h, followed by addition of diluted plasma samples and serial dilutions (curve) of plasma activated with cobra venom factor (100% of activated C3), which were incubated for 1 h. The plates were washed thrice with 0.05% PBS–Tween 20, and peroxidase-conjugated polyclonal rabbit anti-human C3b IgG (MP Biomedicals) was added. After 1 h of incubation, wells were washed, and bound C3 fragments were detected by adding hydrogen peroxide plus tetramethylbenzidine according to the manufacturer’s instructions (R&D Systems). The reaction was stopped with 2 N sulfuric acid, and the plates were read at 450 nm. All steps were performed at room temperature.
The conversion of optical density (O.D.) values to percentage of total C3 activation was performed using the following formula: x (%) = [O.D. curve/(O.D. sample * 100)] : (dilution curve/dilution sample).
Measurement of plasma IL-10
IL-10 concentrations were measured in plasma using the Milliplex MAP Non-Human Primate Cytokine Magnetic Bead Panel - Immunology Multiplex Assay (EMD Millipore) according to the manufacturer’s instructions. Measurements were taken using a Bio-Plex 200 instrument (Bio-Rad).
Analysis of plasma levels of Cp40 by UPLC-MS
Cp40 present in the plasma samples was extracted by solid-phase extraction (SPE) in a 96-well plate format (HLB Oasis 30 μm, 10 mg; Waters, Milford, MA). The SPE material was thoroughly conditioned using acetonitrile and water. Plasma samples were diluted 1:1 with 4% phosphoric acid, and the compstatin analog Cp20 was spiked into all samples at 5 pM concentration as an internal standard (Qu et al., 2011). The samples were loaded on the SPE plate and washed with 10% acetonitrile in 0.1% formic acid. Extracted peptides were eluted with 200 μl of 65% acetonitrile in 0.1% formic acid and collected in a LoBind tube (Eppendorf) to avoid peptide adsorption. Finally, 5 μl of each eluent was diluted with 45 μl of 0.1% formic acid and injected into the UPLC-MS system, consisting of an online ACQUITY UPLC coupled to a SYNAPT G2-S instrument equipped with an ESI source and controlled by MassLynx 4.1 software (Waters). Each sample was injected in quadruplicate. Reversed-phase (RP) liquid chromatography was used for peptide separation with a 1.7-μm UPLC BEH130 C18 column (2.1 μm × 150 mm; Waters) at a column temperature of 40 °C. Peptides were separated at a flow rate of 0.15 ml/min with a linear gradient of 10-60% acetonitrile in water containing 0.1% formic acid over 8 min. Eluted peptides were directly analyzed by MS; the ESI source capillary voltage was set to 3.2 kV, the cone voltage to 30 V, and the source temperature to 120 °C. [Glu1]-fibrinopeptide B (Sigma) was used for lock-mass correction with a sampling rate of 30 s. Mass spectra were acquired in positive mode over an m/z range of 50-1950 Da at a scan rate of 1 s.
Peptide synthesis
The 13-residue cyclic peptide Cp40 ((D)Tyr-Ile-[Cys-Val-Trp(Me)-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle-NH2, with Sar = sarcosine/N-methyl glycine, mIle = N-methyl isoleucine) was synthesized by Fmoc solid-phase methodology using diisopropylcarbodiimide and Oxyma as coupling reagents, as described previously (Qu et al., 2011). The crude peptide was purified by RP-HPLC, and the resulting trifluoroacetate salt form of each peptide was converted to the acetate salt form using anion exchange resin in sodium acetate buffer (Roux et al., 2008). The purified peptides were >95% pure as determined by analytical RP-HPLC (Luna C18, 100 Å column, 250 × 4.60 mm; Phenomenex, Torrance, CA). The mass of each peptide was confirmed using a MALDI micro MX instrument (Waters, Milford, MA) or a SYNAPT G2-S mass spectrometry instrument (Waters).
Results & Discussion
To better mimic the clinical situation, we have established a pre-clinical model of HD in cynomolgus monkeys for confirming the presence of complement activation during HD in vivo. We set up an HD circuit in the monkeys (Fig. 1A) and collected blood samples before, during, and after HD (Fig. 1B). Each monkey underwent three sessions of HD, with a 2-day interval between sessions. We used a pediatric HD filter with high biocompatibility, and all monkeys received standard heparin treatment. Despite these measures, we detected marked complement activation in the monkeys’ plasma during HD (Fig. 1C). Once blood circulation was initiated (0 h), the levels of activated C3 increased and remained elevated during the 4-h HD session (0.5 to 4 h), then slowly decreased after the HD procedure was halted and finally returned to near-basal levels (5 to 28 h) (Fig. 1C). Since C3 activation levels were similar in all three HD sessions (Fig. 1C), only one session was performed per individual for each treatment regimen in the following experiments.
We then asked whether such HD-induced complement activation could be attenuated by the C3 inhibitor compstatin (Ricklin & Lambris, 2008). The analog Cp40 was selected as the benchmark drug based on previous pharmacokinetic studies showing a 12-h plasma half-life and an activity profile (binding affinity of 0.5 nM) well suited for this proposed study (Qu et al., 2013). In order to achieve full complement inhibition during the HD procedure, the molar concentration of Cp40 had to be maintained at levels equivalent to or above those of the C3 target protein (1-2 mg/ml or 6-12 μM in monkeys) (Qu et al., 2013). Therefore, the monkeys were injected with an initial i.v. bolus dose of 2 mg/kg, followed by an i.v. 4 μg/kg/min infusion of Cp40 during the 4-h HD procedure (Fig. 2A). As a control, the same monkeys underwent HD without Cp40 treatment, with sufficient rest between experiments. In these controls, the levels of activated C3 followed the same pattern observed in the multiple-session experiments described above (Fig. 2B, black line). Cp40 treatment completely abolished HD-induced complement activation, which remained at basal levels throughout the procedure (Fig. 2B, gray line). The absence of C3 activation was confirmed in three other monkeys treated with the same Cp40 regimen (Supplementary Fig. 1).
Maintenance of target-saturating levels of Cp40 throughout the procedure was demonstrated by mass spectrometry measurements of plasma drug concentrations. The Cp40 levels remained constant at 5.2 μM (similar to the expected C3 concentration) during the first 6 h and decreased afterwards at a rate consistent with the established half-life of Cp40 (Fig. 2C); no difference was observed between the inflow and outflow samples. This observation is particularly important because it verifies that despite its low molecular weight (~1.5 kDa), Cp40 is not substantially removed by the HD filter when bound to C3.
Since the HD process did not accelerate the elimination of bound Cp40, and because the half-life of Cp40 is much longer than the HD session, we hypothesized that a single i.v. bolus injection of 2 mg/kg Cp40 at the beginning of HD would be enough to block HD-induced complement activation throughout the procedure (Fig. 3A); this treatment regimen was indeed sufficient to abrogate C3 activation during the entire session in both monkeys injected with Cp40 (Fig. 3B). Here, to determine the physiological extent of complement activation during HD in control experiments, levels of activated C3 were represented as percentages of activation (see Methods). As before, levels increased upon initiation of HD, reaching as high as 5% of activated C3 molecules (Fig. 3B). Although this level is markedly lower than that previously observed with cellulose-based filters (~20-25%) (Oppermann et al., 1988), activation of 5% of total C3 represents a considerable stimulus. Considering the plasma C3 concentration (~1 mg/ml), 5% would generate up to 2.5 μg/ml and 200 ng/ml of the pro-inflammatory C3a and C5a, respectively. Even in immune complex-mediated diseases or during cardiopulmonary bypass surgery, complement activation levels typically do not exceed 10% (Soulika et al., 2000). To further verify that a single dose of Cp40 was enough to saturate C3 and completely inhibit its activation, we used mass spectrometry to measure the plasma concentrations of Cp40 in monkeys receiving the bolus regimen. As seen in the bolus-plus-infusion experiments, constant levels of the inhibitor (~6 μM and ~8 μM) similar to the expected levels of C3 were observed during the first 6 h after HD initiation, followed by a slow decrease thereafter (Fig. 3C). These studies confirmed that a single injection of Cp40 before the HD session is sufficient to prevent HD-induced complement activation.
Figure 3.

A single dose of Cp40 at the beginning of hemodialysis is sufficient to prevent complement activation. (A) Hemodialysis was performed in cynomolgus monkeys for a period of four hours and blood samples were collected prior to, during, and after blood circulation as indicated. Cp40 was injected as an i.v. bolus of 2 mg/kg. (B) Blood samples were collected from untreated (black line) and Cp40-treated (gray line) monkeys and the activation of complement C3 was measured in plasma samples by ELISA, represented as a percentage of total C3 activation (%). The figures depict data from blood samples collected from the inflow sampler (Figure 1A). Similar results were obtained with samples collected from the outflow sampler. (C) Levels of Cp40 were measured by mass spectrometry in the plasma of monkeys treated with Cp40. Samples were collected from both samplers as indicated in Fig. 1A.
Increased levels of acute phase proteins and pro-inflammatory cytokines have been detected in ESRD patients and are strongly correlated with mortality in HD patients (Bologa et al., 1998; Yeun et al., 2000; Rao et al., 2005). Given the immunomodulatory functions of complement (Ricklin et al., 2010), we asked whether the observed C3 activation and consequent C3a/C5a elevation during HD would induce cytokine production and whether complement inhibition would have modulatory effects. Although we detected the presence of pro-inflammatory cytokines during HD, their release profile was not consistent among different monkeys. It is likely that a single session of HD is not sufficient to induce substantial levels of pro-inflammatory cytokines in healthy animals. In contrast, the HD procedure resulted in increased levels of the anti-inflammatory cytokine IL-10 in two of three tested monkeys (Fig. 4, black line). Surprisingly, the levels were higher in all three animals when the monkeys were treated with Cp40 according to either treatment regimen (bolus only or bolus plus infusion; Fig. 4, gray line). While IL-10 was absent from the plasma of all three monkeys at the beginning of HD, the levels peaked at 3 h after HD initiation in the control experiments, reaching concentrations of 346 and 235 pg/ml. During Cp40 treatment, IL-10 levels peaked at 3-6 h, with concentrations reaching 1953, 910, and 3810 pg/ml.
Our results emphasize the capacity of modern “biocompatible” HD membranes to activate complement and show that a single dose of the C3 inhibitor Cp40 is able to abrogate such activation. Further, our data indicate that inhibition of complement activation by Cp40 may reduce the inflammatory stimuli triggered by the HD procedure. Given the immune-regulatory properties of IL-10 (Stenvinkel et al., 2005), it is likely that Cp40 inhibits the complement-mediated production of inflammatory cytokines during HD, tipping the balance toward an anti-inflammatory environment. Interestingly, a potential correlation between complement inhibition and IL-10 profiles has been suggested in a colitis model (Jain et al., 2013). Further investigation is needed to better characterize the role of Cp40 in modulating inflammatory factors during HD. The non-human primate model of HD introduced here represents an important tool for evaluating inflammation and treatment regimens in a close-to-clinical context.
Compstatin analogs have proven to be attractive options for treating complement-related disorders such as sepsis, periodontitis, transplantation, age-related macular degeneration, paroxysmal nocturnal hemoglobinuria, and biomaterial-induced inflammation (Silasi-Mansat et al., 2010; Maekawa et al., 2014; Risitano et al., 2014; Kourtzelis et al., 2013; Chi et al., 2010; Fiane et al., 1999). Considering the prevalence of ESRD and the high frequency and cost of its treatment, any added therapeutic must be cost-effective. Complement inhibitors currently in the clinic are protein-based and target individual initiation or effector pathways (Ricklin & Lambris, 2013b). In contrast, compstatin analogs block complement comprehensively by preventing activation of C3 by all initiation pathways (classical, lectin and alternative pathways) and impairing generation of pro-inflammatory complement effectors (Ricklin and Lambris, 2008). This is of particular importance in the case of HD, as different filter materials may trigger distinct initiation pathways (DeAngelis et al., 2012), and since initial deposition of C3 activation fragments can fuel an amplification loop that exacerbates activation. As a small peptide that can be synthesized at comparatively low cost, Cp40 has significant potential, especially since a single dose of the drug prior to the HD session can offer sufficient protection. Moreover, any potential reducing effect of complement-targeted treatment on the overall inflammatory status of ESRD patients may indirectly affect treatment cost by reducing medication required to controlling adverse effects such as cardiovascular disease or anemia.
In summary, we established a refined NHP model of hemodialysis at close-to-clinical conditions that allows to investigate pathophysiological aspects of the procedure, such as filter-induced inflammatory complications, and evaluate suitable treatment options. The broad inhibitory profile, high efficacy in controlling complement activation during HD sessions, and expected low production cost let compstatin Cp40 appear as an interesting candidate for add-on therapy for patients undergoing HD. Of note, the study presented here was performed in healthy animals without underlying kidney disease, and both the inflammatory baseline condition and its therapeutic modulation are expected to be more complex in ESRD. It will therefore be important to evaluate the benefit of attenuating complement- and inflammation-induced adverse events during HD on the morbidity and quality-of-life in ESRD patients.
Supplementary Material
Acknowledgements
We thank Dr. Deborah McClellan for editorial assistance, Dr. Yijun Huang for his help in the synthesis of complement inhibitors and controls, and Dr. Benjamin Laskin for critical discussion. This work was supported by National Institutes of Health grants AI068730, AI030040, AI097805, and the European Community's Seventh Framework Programme under grant agreement number 602699 (DIREKT).
Abbreviations
- APTT
activated partial thromboplastin time
- CVD
cardiovascular disease
- ESRD
end-stage renal disease
- HD
hemodialysis
- O.D.
optical density
- SPE
solid-phase extraction
- RP
reversed-phase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
J.D.L. and D.R. are the inventors of patents and/or patent applications that describe the use of complement inhibitors for therapeutic purposes. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors for clinical applications. The remaining authors declare no competing financial interests.
References
- Bologa RM, Levine DM, Parker TS, Cheigh JS, Serur D, Stenzel KH, Rubin AL. Interleukin-6 predicts hypoalbuminemia, hypocholesterolemia, and mortality in hemodialysis patients. Am. J Kidney Dis. 1998;32:107–114. doi: 10.1053/ajkd.1998.v32.pm9669431. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Del Vecchio L, Manzoni C, Locatelli F. Hemodialysis: yesterday, today and tomorrow. Minerva. Urol. Nefrol. 2010;62:1–12. [PubMed] [Google Scholar]
- Chi ZL, Yoshida T, Lambris JD, Iwata T. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv. Exp. Med. Biol. 2010;703:127–135. doi: 10.1007/978-1-4419-5635-4_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis RA, Reis ES, Ricklin D, Lambris JD. Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology. 2012;217:1097–1105. doi: 10.1016/j.imbio.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl KN, Lambris JD, Elwing H, Ricklin D, Nilsson PH, Teramura Y, Nicholls IA, Nilsson B. Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv. Drug Deliv. Rev. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiane AE, Mollnes TE, Videm V, Hovig T, Hogasen K, Mellbye OJ, Spruce L, Moore WT, Sahu A, Lambris JD. Compstatin, a peptide inhibitor of C3, prolongs survival of ex vivo perfused pig xenografts. Xenotransplantation. 1999;6:52–65. doi: 10.1034/j.1399-3089.1999.00007.x. [DOI] [PubMed] [Google Scholar]
- Galli F. Protein damage and inflammation in uraemia and dialysis patients. Nephrol. Dial. Transplant. 2007;22(Suppl 5):v20–v36. doi: 10.1093/ndt/gfm294. [DOI] [PubMed] [Google Scholar]
- Jain U, Woodruff TM, Stadnyk AW. The C5a receptor antagonist PMX205 ameliorates experimentally induced colitis associated with increased IL-4 and IL-10. Br. J. Pharmacol. 2013;168:488–501. doi: 10.1111/j.1476-5381.2012.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jofre R, Rodriguez-Benitez P, Lopez-Gomez JM, Perez-Garcia R. Inflammatory syndrome in patients on hemodialysis. J. Am. Soc. Nephrol. 2006;17:S274–S280. doi: 10.1681/ASN.2006080926. [DOI] [PubMed] [Google Scholar]
- Kimmel PL, Phillips TM, Simmens SJ, Peterson RA, Weihs KL, Alleyne S, Cruz I, Yanovski JA, Veis JH. Immunologic function and survival in hemodialysis patients. Kidney Int. 1998;54:236–244. doi: 10.1046/j.1523-1755.1998.00981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtzelis I, Markiewski MM, Doumas M, Rafail S, Kambas K, Mitroulis I, Panagoutsos S, Passadakis P, Vargemezis V, Magotti P, Qu H, Mollnes TE, Ritis K, Lambris JD. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood. 2010;116:631–639. doi: 10.1182/blood-2010-01-264051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtzelis I, Rafail S, DeAngelis RA, Foukas PG, Ricklin D, Lambris JD. Inhibition of biomaterial-induced complement activation attenuates the inflammatory host response to implantation. FASEB J. 2013;27:2768–2776. doi: 10.1096/fj.12-225888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Cooper AC. Erythropoietin resistance: the role of inflammation and pro-inflammatory cytokines. Nephrol. Dial. Transplant. 2002;17(Suppl 11):39–43. doi: 10.1093/ndt/17.suppl_11.39. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, Lambris JD, Hajishengallis G. Genetic and intervention studies implicating complement c3 as a major target for the treatment of periodontitis. J. Immunol. 2014;192:6020–6027. doi: 10.4049/jimmunol.1400569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol. Immunol. 2007;44:82–94. doi: 10.1016/j.molimm.2006.06.020. [DOI] [PubMed] [Google Scholar]
- NKUDIC Kidney and urologic diseases statistics for the United States. 2012. National Kidney & Urologic Diseases Information Clearinghouse (NKUDIC) - A service of the National Institute of Diabetes and Digestive Kidney Diseases (NIDDK) - National Institutes of Health (NIH). 2-15-2012. 5-30-2012.
- Oppermann M, Haubitz M, Quentin E, Gotze O. Complement activation in patients with renal failure as detected through the quantitation of fragments of the complement proteins C3, C5, and factor B. Klin. Wochenschr. 1988;66:857–864. doi: 10.1007/BF01728947. [DOI] [PubMed] [Google Scholar]
- Qu H, Magotti P, Ricklin D, Wu EL, Kourtzelis I, Wu YQ, Kaznessis YN, Lambris JD. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol. Immunol. 2011;48:481–489. doi: 10.1016/j.molimm.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, Tzekou A, DeAngelis RA, Resuello RR, Lupu F, Barlow PN, Lambris JD. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Guo D, Perianayagam MC, Tighiouart H, Jaber BL, Pereira BJ, Balakrishnan VS. Plasma interleukin-6 predicts cardiovascular mortality in hemodialysis patients. Am. J Kidney Dis. 2005;45:324–333. doi: 10.1053/j.ajkd.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Compstatin: a complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 2008;632:273–292. doi: 10.1007/978-0-387-78952-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J. Immunol. 2013a;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J. Immunol. 2013b;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, Lin Z, Pascariello C, Raia M, Sica M, Del VL, Pane F, Lupu F, Notaro R, Resuello RR, DeAngelis RA, Lambris JD. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Zekri E, Rousseau B, Paternostre M, Cintrat JC, Fay N. Elimination and exchange of trifluoroacetate counter-ion from cationic peptides: a critical evaluation of different approaches. J. Pept. Sci. 2008;14:354–359. doi: 10.1002/psc.951. [DOI] [PubMed] [Google Scholar]
- Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, Ivanciu L, Lupu C, Mollnes TE, Taylor FB, Kinasewitz G, Lambris JD, Lupu F. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulika AM, Khan MM, Hattori T, Bowen FW, Richardson BA, Hack CE, Sahu A, Edmunds LH, Jr., Lambris JD. Inhibition of heparin/protamine complex-induced complement activation by Compstatin in baboons. Clin. Immunol. 2000;96:212–221. doi: 10.1006/clim.2000.4903. [DOI] [PubMed] [Google Scholar]
- Stenvinkel P, Alvestrand A. Inflammation in end-stage renal disease: sources, consequences, and therapy. Semin. Dial. 2002;15:329–337. doi: 10.1046/j.1525-139x.2002.00083.x. [DOI] [PubMed] [Google Scholar]
- Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, Heimburger O, Cederholm T, Girndt M. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005;67:1216–1233. doi: 10.1111/j.1523-1755.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- Yeun JY, Levine RA, Mantadilok V, Kaysen GA. C-Reactive protein predicts all-cause and cardiovascular mortality in hemodialysis patients. Am. J Kidney Dis. 2000;35:469–476. doi: 10.1016/s0272-6386(00)70200-9. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Kalantar-Zadeh K. The crossroad of RAAS modulation, inflammation, and oxidative stress in dialysis patients: light at the end of the tunnel? J. Am. Soc. Nephrol. 2012;23:189–191. doi: 10.1681/ASN.2011121208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.