

Abstract
The link between autoimmune diseases and primary immunodeficiency syndromes has been increasingly appreciated. Immunologic evaluation of a young man with autoimmune enterocolopathy and unexplained infections revealed evidence of immunodeficiency, including IgG subclass deficiency, impaired antigen-induced lymphocyte proliferation, reduced cytokine production by CD8+ T lymphocytes, and decreased numbers of natural killer (NK) cells. Genetic evaluation identified haploinsufficiency of NFAT5, a transcription factor regulating immune cell function and cellular adaptation to hyperosmotic stress, as a possible cause of this syndrome. Inhibition or deletion of NFAT5 in normal human and murine cells recapitulated several of the immune deficits identified in the patient. These results provide evidence of a primary immunodeficiency disorder associated with organ-specific autoimmunity linked to NFAT5 deficiency.
Introduction
It has been increasingly appreciated that patients with primary immunodeficiency syndromes exhibit not only an increased susceptibility to infections, but also paradoxical manifestations of autoimmunity (1, 2). Patients with well-recognized disorders such as common variable immunodeficiency (CVID) are susceptible to bacterial infections but can also present with a wide spectrum of autoimmune manifestations including vitiligo, hemolytic anemia, rheumatoid arthritis, and gastroenteropathy (3). It has been suggested that infections that fail to be cleared in an immunodeficient individual may initiate a compensatory, dysregulated inflammatory response that eventually leads to autoimmunity (4). However, an underlying primary immunodeficiency may be overlooked when patients present with predominant autoimmune manifestations. Alternatively, many patients can present with signs and symptoms suggestive of an immune deficiency, but fail to meet the diagnostic criteria for any known disorder. These observations raise the possibility that many immunodeficiency syndromes remain to be identified.
Autoimmune enteropathy (AIE) is a rare disease that presents with intractable diarrhea, histologic changes in the intestinal mucosa, and the presence of autoantibodies against gut enterocytes or goblet cells (5-7). Although the exclusion of immunodeficiency has historically been part of the diagnostic criteria for AIE, recent studies demonstrate that AIE patients frequently present with a concomitant immunodeficiency disorder and may have an increased risk of opportunistic infections when treated with immunosuppressive medications (8). These observations suggest that further studies are needed to better understand the pathophysiology of autoimmune enteropathy and to improve the therapeutic options for this condition.
Nuclear Factor of Activated T cells 5 (NFAT5), also known as tonicity enhancer binding protein (TonEBP), is a DNA-binding protein that is activated in response to osmotic stress, translocates to the nucleus, and initiates the transcription of downstream targets, including genes required for cell cycle progression and inflammation (9-13). In T lymphocytes, NFAT5 exists as a constitutive dimer and its transcriptional regulatory activity can be induced independently by either T cell receptor stimulation or hyperosmotic stress (14, 15). NFAT5 directly binds to the TNFα promoter in vivo, suggesting a critical role for this transcription factor in mediating inflammation and regulating immune responses (14). T lymphocytes with reduced NFAT5 function exhibit impaired proliferation and survival (16, 17). Importantly, lymphoid tissues have been shown to be hyperosmolar compared to blood, suggesting that the ability of lymphocytes, via induction of NFAT5 and related pathways, to adapt to osmotic stress may be important in the initiation of immune responses (18). However, NFAT5 deficiency has not previously been reported to be associated with human disease.
Here we describe a patient with a diagnosis of AIE who presented with symptoms of autoimmunity. Immunologic evaluation demonstrated defects in innate and adaptive immunity, while genetic testing revealed de novo haploinsufficiency of NFAT5. We confirmed that the patient had significantly impaired induction of NFAT5 mRNA and protein in response to osmotic stress. Using both dominant negative and RNA interference approaches in human and murine lymphocytes, we demonstrate that reduced NFAT5 activity disrupted the ability of T cells to produce TNFα and to survive in hyperosmolar conditions. Analysis of colonic tissue from patients with active inflammatory bowel disease, another immune-mediated disease, revealed reduced NFAT5 expression at the mRNA level. Together these results suggest that NFAT5 may play an important role in immune responses and that NFAT5 deficiency may be linked to human autoimmunity.
Materials and Methods
Study participants
The study protocols with informed consent were approved by the Institutional Review Board/Human Research Protections Program at the University of California, San Diego. Written informed consent was obtained from participants.
Human PBMC isolation and cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated from whole human blood on a Ficoll-Paque PLUS (GE Healthcare) gradient. Cell numbers were quantitated using an Accuri C6 Flow Cytometer (BD Biosciences). PBMCs were cultured in AIM V media (Life Technologies) with human IL-2 (Peprotech) and activated with anti-human CD3 (clone HIT3a) and CD28 (clone CD28.2) antibodies (eBioscience). Jurkat cells were grown in RPMI-1640 media (Mediatech) with 10% FBS, 2 mM L-glutamine (Sigma), 50 μM 2-mercaptoethanol (Gibco), and penicillin and streptomycin (Sigma). To evaluate lymphocyte function, PBMCs were cultured with anti-CD3 and anti-CD28 antibodies in AIM-V media for 24 hours; 0.25 μg/ml phorbol 12-myristate 13-actated, 2.5 μg/ml ionomycin, and 5 μg/ml brefeldin A (Sigma) were added during the final 6 hours of culture prior to fixation and staining for intracellular cytokine analysis as described below.
Survival in hyperosmotic conditions
Jurkat cells or human PBMCs were cultured in conventional media (280 mOsm/kg, “isotonic”) or media adjusted to 420 mOsm/kg (“hypertonic”) with sterile 4M sodium chloride (Gibco). PBMCs were also activated with anti-CD3 and anti-CD28 antibodies as well as human IL-2, as described above. Cells were cultured in isotonic or hypertonic conditions for 5 days, then assessed for viability by flow cytometry. The percentage of live lymphocytes was determined by forward scatter and side scatter properties (Supplemental Fig. 2). Viability in hypertonic conditions was expressed as percent cell death due to hypertonicity: the difference between the percentage of live cells in isotonic media and percentage of live cells in hypertonic media, normalized to the percentage of live cells in isotonic media (% live lymphocytes in isotonic culture - % live lymphocytes in hypertonic culture / % live lymphocytes in isotonic culture). As a secondary, confirmatory method to evaluate cell survival, cells were stained with anti-Annexin V and propidium iodide (eBioscience). Percent cell death was presented as the difference in Annexin V+P.I.+ cells in hypertonic versus isotonic conditions. Each experiment was repeated at least twice with at least 3 biological replicates each in Jurkat cells and at least 10 biological replicates each in human PBMCs.
Mice
All animal work was done in accordance with Institutional Animal Care and Use Guidelines of the University of California, San Diego. Mice heterozygous and null for Nfat5 have been described previously (9, 19). Nfat5+/- mice were maintained in an isogenic 129/sv background and were crossed to obtain Nfat5-/- and control Nfat5+/+ littermates.
Generation of bone marrow chimeras
Donor mice were treated with 5-fluorouracil; after four days, bone marrow cells were harvested and cultured with IL-3, stem cell factor (SCF), and IL-6 (Peprotech) overnight, then retrovirally transduced with control or dominant negative NFAT5 constructs daily for 3 days. Lethally irradiated (1000 rads) RAG1-deficient mice were injected intravenously with transduced bone marrow cells. Blood from recipient mice was obtained for FACS analysis after 8 weeks.
SNP arrays and sequencing
The samples analyzed consisted of 4 cases affected with autoimmune enterocolopathy and 7 unaffected controls. HumanCoreExome SNP Arrays were performed. We used the Genotyping module (1.9.4) within Illumina's GenomeStudio (GS) V2011.1. Data was normalized using default parameters and LogR Ratios and B Allele Frequencies were exported for further analysis in R. Because the platform is designed to detect additional rare variants (Exome) that may interfere with our copy number analysis and measurement of heterozygosity, we used only probes measuring common SNPs corresponding to Illumina's HumanCore BeadChip (287,064/526,811 probes). An interval of 139 kb was sequenced using 16 long-range PCR amplicons (Supplemental Table II) in 11 individuals. The sequencing generated between 245 × 103 and 1,594 × 103 reads per sample, of which more than 96% mapped to the genome. The targeted sequencing identified all variants in the interval and was therefore used to more accurately measure the rate of heterozygosity. The F statistic was measured using the 451 SNPs genotyped by sequencing and using those present in dbSNP (20).
DNA methylation analysis
DNA was bisulfite-modified using the EZ DNA methylation Gold Kit (Zymo Research). Methylation of NFAT5 promoter regions was quantified by bisulfite pyrosequencing (PSQ HS 96A pyrosequencing system, Qiagen), as described previously (21). Methylation levels of CpG sites were analyzed and mean methylation values for NFAT5 promoter regions were determined. The following primers were used: NFAT5-Methylation-Forward (5′-GGTAGGGGAGAAAGTATT-3′), NFAT5-Methylation-Reverse (5′-AACTACTACTATTCCTAAACC-3′), Sequencing primer (5′-TTATTGTAAATTTTAAAGGGT-3′).
Plasmid constructs and retroviral transduction
The dominant negative effect of the IDD5 domain of NFAT5 has been previously reported (14). GFP or IDD5-GFP were first cloned into the vector pENTR11 (Life Technologies), and subsequently transferred to the KMV-DV retroviral vector, yielding control (KMV-DV-GFP) and dominant negative NFAT5 (KMV-DV-IDD5-GFP) constructs. A short hairpin RNA (shRNA) to silence NFAT5 gene expression was generated using previously described methods (22). The following oligonucleotides (Valuegene), including restriction sites, were used: shLuciferase non-targeting control (5′-TGCTGTTGACAGTGAGCG CCCGCCTGAAGTCTCTGATTAA TAGTGAAGCCACAGATGTA TTAATCAGAGACTTCAGGCGGT TGCCTACTGCCTCGGA-3′), shNFAT5 (5′ - TGCTGTTGACAGTGAGCG AACGAGTGAAACCACATGGATA TAGTGAAGCCACAGATGTA TATCCATGTGGTTTCACTCGTC TGCCTACTGCCTCGGA-3′). The constructs were digested with EcoRI and XhoI restriction enzymes (New England Biolabs) to generate sticky ends and cloned into the MSCV-LTRmiR30-PIG (LMP) vector (Thermo Scientific) to generate control shRNA and NFAT5 shRNA plasmids. Viral packaging in transfected 293T cells was performed as previously described (23). To transduce cells with retrovirus containing the constructs, PBMCs were isolated and activated as described above for 2 days. Jurkat cells or PBMCs were resuspended in retroviral supernatants and polybrene at 0.8 μg/ml (American Bioanalytical) and centrifuged at 2500 rpm at room temperature for 90 minutes. Cells were subsequently cultured for 2 days prior to assessment of cytokine production or survival in hypertonic conditions. Cells expressing constructs were detected on the basis of GFP expression.
Flow cytometry
Cells were stained with the following fluorochrome-conjugated antibodies: anti-human CD4 (clone RPA-T4), anti-human CD8a (clone SK1), anti-human CD16 (clone B73.1), anti-human CD56 (clone HCD56), anti-human TNFα (clone MAb11), anti-mouse CD4 (clone GK1.5), anti-mouse CD8a (clone 53-6.7), anti-mouse CD49b (clone DX5), anti-mouse CD122 (clone TM-b1), anti-mouse TNFα (clone MP6-XT22, all from Biolegend). For intracellular antigens, cells were fixed with 4% paraformadehyde (Electron Microscopy Services) and permeabilized prior to intracellular flow cytometry with PBS (Mediatech) containing 1% FBS (Life Technologies), 1% saponin (Sigma) and 0.1% sodium azide (Sigma). Cells were acquired on an Accuri C6 Cytometer (BD Biosciences) and data analyzed with FlowJo software (TreeStar).
RNA extraction from cells and colonic biopsies
Total RNA was extracted from PBMCs or Jurkat cells using TRIzol (Life Technologies). One μl of GlycoBlue (Life Technologies) was added before RNA precipitation with isopropanol. RNA was washed with 70% ethanol and resuspended in nuclease-free water. Equal amounts of total RNA were reverse-transcribed to generate cDNA using MultiScribe Reverse Transcriptase and random primers (Applied Biosystems). To extract RNA from colonic biopsies, each sample was immediately placed into RNA Stat-60 (Tel-Test) then placed on ice. Samples were stored at -80°C, and assays were performed within 6 months of acquisition. Colonic pinch biopsies were homogenized manually in the presence of RNA Stat-60 using chilled Kontes-Duall tissue grinders (Fisher Scientific). After the addition of chloroform and centrifugation, a crude RNA extract was obtained. The resultant aqueous phase was mixed with an equal volume of 70% ethanol; the samples were then loaded onto RNeasy Lipid Tissue Mini columns (Qiagen) according to the manufacturer's protocol. RNA was eluted in 40 μl of TE buffer. RNA was quantified using Ribo-green reagent (Life Technologies).
Measurement of mRNA levels
Quantitative real-time PCR was performed in triplicate using 30 ng of cDNA and SsoAdvanced SYBR Green Supermix on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Data analysis was done using Bio-Rad CFX Manager 2.1. The relative amount of NFAT5 mRNA was determined on the basis of the threshold cycle for each PCR product (Cq). Gene expression in PBMCs was normalized with β-actin mRNA levels. Gene expression in colonic biopsies was normalized using an average of calnexin (CANX) and ribosomal protein, large P0 (RPLP0). β-actin expression has been shown to vary in inflamed gastrointestinal mucosa; in contrast, expression of RPLP0 and CANX has been shown to be relatively stable in gastrointestinal tissue, even in the setting of inflammation (24, 25). The following primers were used to detect their respective gene transcripts: NFAT5-forward (5′-AGTGGACATTGAAGGCACTAC-3′), NFAT5-reverse (5′-TTGGAACCAGCAATTCCTATTC-3′), β-actin-forward (5′-CATGTACGTTGCTATCCAGGC-3′), β-actin-reverse (5′-CTCCTTAATGTCACGCACGAT-3′), CANX-forward (5′-GGAACTCTGTCAGGGTGGATT-3′), CANX-reverse (5′-CTTGGCCCGAGACATCAACA-3′), RPLP0-forward (5′-TGGTGCTGATGGGCAAGAAC-3′), RPLP0-reverse (5′-CAGCAGCTGGCACCTTATTG-3′).
Immunoblotting
Proteins were extracted with lysis buffer (50mM HEPES, pH 7.4, 80mM NaCl, 5mM MgCl2, 10mM EDTA pH 8.0, 5mM Sodium Pyrophosphate, 1% Triton X-100) and protease inhibitor cocktail (Sigma-Aldrich). Fifty μg of protein was resolved on a 4-20% SDS polyacrylamide gel (Invitrogen), transferred onto nitrocellulose membranes (Bio-Rad), and analyzed by immunoblotting using anti-NFAT5 (Thermo Scientific), anti-NFAT1 (Thermo Scientific), and anti-β-actin (Sigma-Aldrich) primary antibodies, followed by anti-mouse IRdye 800 and anti-rabbit DyLight 680-conjugated secondary antibodies (Rockland).
Statistical analysis
Statistical analysis of the majority of experimental data was determined by a paired Student's t test. The Mann-Whitney U test was used to compare expression of NFAT5 in colonic tissue. P values of <0.05 were considered significant.
Results
Clinical presentation and evaluation
A 19-year-old man born to non-consanguinous parents of Northern European descent was affected with numerous sinopulmonary infections during the first few years of life. At 7 years of age he began to experience frequent episodes of abdominal pain, intermittent fevers, and non-bloody diarrhea associated with an eczematous rash on his extremities. He was given a presumptive diagnosis of inflammatory bowel disease and treated over the next several years with courses of mesalamine derivatives and various immunosuppressives, including corticosteroids, azathioprine, and 6-mercaptopurine, without clear sustained benefit. Endoscopic evaluation of his upper and lower gastrointestinal tracts were remarkable only for mild nodularity in the terminal ileum (Fig. 1A) and rectum (Fig. 1B). Hematoxylin and eosin staining of biopsy specimens from the duodenum (Fig. 1C) and colon (Fig. 1D) revealed intraepithelial lymphocytosis, absence of goblet cells, and abundant apoptotic enterocytes, consistent with autoimmune enteropathy. This diagnosis was confirmed by the presence of anti-goblet cell antibodies, and additional laboratory evaluation was negative for anti-enterocyte antibodies and did not otherwise reveal evidence of a known primary immunodeficiency or systemic autoimmune disorder (Table I).
Figure 1.

Endoscopic and histologic features of gastrointestinal tissue from the proband patient diagnosed with autoimmune enterocolopathy. Upper endoscopy and colonoscopy were performed on the patient; images from the terminal ileum (A) and rectum (B) are shown. Hematoxylin and eosin staining of biopsy specimens (20× magnification) from the duodenum (C) and colon (D) are shown. Arrows indicate apoptotic enterocytes.
Table I. Immunologic Laboratory Features.
| Variable | Reference Range, Adults | Patient |
|---|---|---|
| White-cell count (Thousand/mcL) | 4.0 - 10.0 | 5.8 |
| Neutrophils (%) | 34 – 71 | 57.7 |
| Lymphocytes (%) | 19 – 53 | 30.6 |
| Monocytes (%) | 5 – 12 | 11.4 |
| Eosinophils (%) | < 5.5 | 0.0 |
| Basophils (%) | < 2.0 | 0.3 |
| B lymphocytes | ||
| % CD19+ of CD45+ | 2.8-17.4 | 12.8 |
| Absolute (Cells/mcL) | 90-539 | 112.2 |
| T lymphocytes | ||
| % CD3+ | 59 – 83 | 93 |
| Absolute (Cells/mcL) | 677 – 2383 | 815 |
| NK cells | ||
| % CD16+CD56+ | 6 – 27 | 1 |
| Absolute (cells/mcL) | 101 – 678 | 5 |
| B cell developmental subsets (% of CD19+ cells) | ||
| IgM+ B cells (IgM+) | 26.0 – 76.0 | 85.3 |
| T1 B cells (CD21-) | 0.2 – 8.6 | 22.5 |
| T2 B cells (CD21+) | 92.1 – 99.6 | 77.8 |
| Transitional B cell (CD38+IgM+) | 1.2 – 50.7 | 0.6 |
| Plasmablasts (CD38+IgM-) | 4.1 - 42.2 | 1.4 |
| Immunoglobulin G, total (mg/dL)1 | 694 – 1618 | 889 |
| Sub-Class 1 (mg/dL) | 382 – 929 | 594 |
| Sub-Class 2 (mg/dL) | 241 – 700 | 134 |
| Sub-Class 3 (mg/dL) | 22 – 178 | 150 |
| Sub-Class 4 (mg/dL) | 4 – 26 | < 1.0 |
| CD8+ T lymphocyte function (% of CD8+ cells) | ||
| IFN gamma2 | 20-48 | 11 |
| TNF alpha2 | 17-61 | 9 |
| Degranulation | 8.5 – 49.1 | 7.3 |
| (%CD107a/b) | ||
| CD4+ T lymphocyte function (% of CD4+ cells)2 | ||
| IFN gamma | 8-24 | 17 |
| TNF alpha | 39-67 | 55 |
| Lymphocyte proliferation | ||
| Maximum proliferation as % of CD45 | ||
| Mitogen (PHA) | ≥ 49.9 | 58.2 |
| Candida albicans | ≥ 5.7 | 0.2 |
| Tetanus Toxoid | ≥ 5.2 | 0.0 |
| Maximum proliferation as % of CD3 | ||
| Mitogen (PHA) | ≥ 58.5 | 63.9 |
| Candida albicans | ≥ 3.0 | 0.2 |
| Tetanus Toxoid | ≥ 3.3 | 0.0 |
denotes reference range from Quest Diagnostics;
denotes reference range from Cincinnati Children's Hospital Medical Center.
All other values reflect reference range from Mayo Medical Laboratories. Abbreviations: mcL = microliter; IgM = immunoglobulin M; mg = milligram; dL = deciliter; PHA = phytohemagglutanin.
Given the patient's history of unexplained infections, an extensive immunologic evaluation was performed to rule out a known primary immunodeficiency syndrome or systemic autoimmune disease (Supplemental Table I). Diagnostic tests excluded severe combined immunodeficiency (SCID), immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX), CVID, chronic granulomatous disease (CGD), systemic lupus erythematosis, and celiac disease, among other disorders. Normal absolute numbers and percentages of naïve and memory B and T lymphocytes, as well as regulatory T cells, were observed. Levels of serum immunoglobulins (IgG, IgA, and IgM) and antibodies to childhood vaccinations were within normal limits.
Immunologic evaluation reveals immune deficiency
More comprehensive immunologic testing, however, revealed deficits in various components of innate and adaptive immunity (Table I). Although the numbers of mature B cell subsets (naïve, memory, class-switched) in the blood were within normal limits, abnormalities in certain B cell subsets were observed. These included a decrease in the number of antigen-experienced B cell populations including CD21+ T2 B cells, transitional B cells, and CD38+IgM- plasmablasts (26, 27), along with a corresponding increase in the number of IgM+ naïve B cells and CD21- T1 B cells. Although total serum IgG was normal, the patient exhibited a selective IgG subclass deficiency, with a reduction of IgG2 and a complete absence of IgG4 that persisted over time (Supplemental Table II).
Although lymphocyte proliferation in response to mitogens was normal, antigen-specific proliferation in response to Candida albicans or tetanus toxoid was absent (Table I). Moreover, CD8+, but not CD4+ T lymphocytes, appeared to be functionally impaired, with a reduced ability to degranulate and to produce the pro-inflammatory cytokines IFNγ and TNFα. Finally, innate immunity was impaired, with a reduction in the absolute number and percentage of CD56+CD16+ NK cells. Together this constellation of immune deficits involving disparate components of both innate and adaptive immunity suggested the presence of a previously unrecognized primary immunodeficiency disorder.
At the time of the immunologic evaluation, the patient was taking azathioprine (AZA) at a low, sub-therapeutic dose that is unlikely to have biologic effects. The therapeutic efficacy of AZA is most closely associated with the 6-thioguanine nucleotide (6-TGN) metabolite levels in the blood. The patient's 6-TGN level was 169 pmol/8 × 108 red blood cells, a concentration that is well below the established therapeutic range associated with a higher likelihood of clinical response, 230 to 400 pmol/8 × 108 red blood cells. This range has been determined by clinical studies that have identified metabolite concentrations associated with therapeutic responses (28-30). Nonetheless, while the patient's AZA dose was sub-therapeutic, we cannot entirely exclude the possibility that the drug may have influenced the results of the immunologic evaluation.
Genetic evaluation reveals hemizygous loss of NFAT5
To explore a genetic cause for these immunologic abnormalities, we looked for potential copy number variation using an oligo-single nucleotide polymorphism (SNP) array. This analysis revealed a 559-kilobase deletion at 16q22.1 (69,580,702-70,139,542) in one chromosome that contained eight genes NFAT5, MIR1538, NQO1, NOB1, WWP2, MIR140, CLEC18A, and PDXDC2 (Fig. 2A). We tested both parents, neither of whom harbored a similar deletion, indicating that the deletion in the proband was de novo. None of the deleted genes have been previously associated with human disease, and with the exception of NFAT5, little is known about the function of the genes in this region.
Figure 2.

NFAT5 deficiency associated with autoimmune enterocolopathy. (A) Copy number status of NFAT5. The log ratio (upper panel) and B allele frequency (lower panel) of the Infinium HumanCore BeadChip array located ∼10Mb around the NFAT5 gene (marked by an asterisk) are displayed. Expression of NFAT5 protein (B) and mRNA (C) from PBMCs isolated from the proband and a healthy control measured after stimulation for 3 days with anti-CD3 and anti-CD28 antibodies, followed by culture in hypertonic media for 1 day.
NFAT5 is a transcription factor and a member of the family of Rel-like domain-containing factors, which includes NF-kBs and the calcineurin-dependent NFATc proteins (12, 14). NFAT5 has been demonstrated to play a critical role in regulating responses to extracellular hypertonicity in a variety of cell types, in particular, renal cells and cells of the innate and adaptive immune systems (9, 12-15, 17, 18). Moreover, NFAT5 has been shown to regulate expression of a number of cytokines, growth factors, and surface receptors in lymphocytes and macrophages in a tonicity-independent manner (9, 12, 14). Finally, recent work has implicated NFAT5 in the induction of pathogenic T helper 17 (TH17) lymphocytes in a murine model of autoimmunity (31, 32), raising the intriguing possibility that abnormalities in components of the osmoadaptation pathway might result in human disease.
To confirm that haploinsufficiency of NFAT5 resulted in reduced expression in the patient's cells, PBMCs isolated from the patient and an unrelated healthy control were subjected to hypertonic culture conditions. Levels of NFAT5 protein and mRNA were assessed by immunoblotting and real-time quantitative PCR. We observed a 6-fold reduction in NFAT5 protein (Fig. 2B) and a 5.3-fold reduction in NFAT5 mRNA levels (Fig. 2C) in the patient's cells compared to control cells.
We investigated NFAT5 status in three additional patients with AIE using copy number SNP arrays and targeted re-sequencing. Although the arrays did not demonstrate NFAT5 deletion (Fig. 3), NFAT5 sequencing revealed a deficit in heterozygous variants, suggesting a loss of heterozygosity, or uniparental disomy (UPD), in all of the patients. All four AIE patients had an F statistic, the expected heterozygosity within a population, greater than 0.8, whereas 0 of 7 of the unaffected controls had an F statistic that surpassed this threshold (Fig. 4). In addition, we considered an additional population control and examined 78 Caucasian individuals from the 1000 Genomes Project. We measured the F statistic using the 451 SNPs for which the genotypes were available. Only 10/78 of the population controls had such a high F (p=2.6 × 10-17). UPD occurs when both homologues of a chromosomal segment are inherited from only one parent. UPD can cause disease if an individual inherits two abnormal copies of a gene from the parent of origin, resulting in an altered level of gene expression (33, 34). Aberrant hypermethylation of the NFAT5 promoter was not detected in the proband, nor in the additional patients with AIE (Supplemental Fig. 1). Nonetheless, AIE patients exhibited a significantly higher incidence of UPD than our measured controls and the population controls, raising the possibility that inheritance of NFAT5 abnormalities from one parent might influence disease penetrance.
Figure 3.
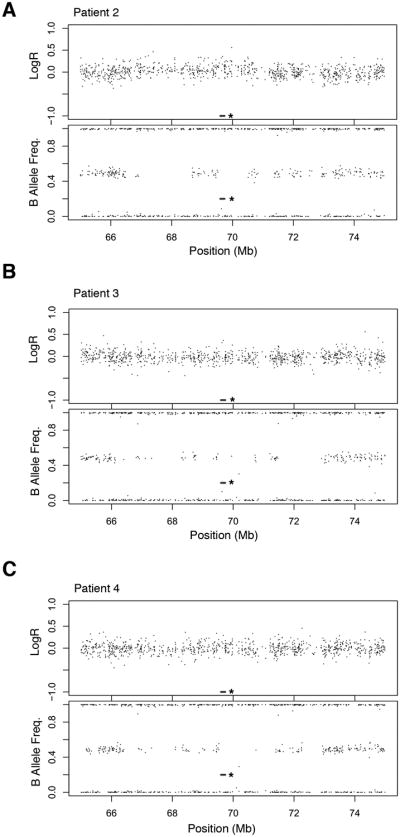
Copy number status of NFAT5. The log ratio (upper panel) and B allele frequency (lower panel) of the Infinium HumanCore BeadChip array located ∼10Mb around the NFAT5 gene (marked by an asterisk) are displayed for three individual patients with autoimmune enterocolopathy.
Figure 4.

Higher incidence of uniparental disomy in cases of autoimmune enterocolopathy compared to controls. An interval of 139 kb was sequenced using 16 long range PCR amplicons in 11 individuals: 4 cases affected with autoimmune enterocolopathy (“cases,” individuals 1-4) and 7 unaffected controls (“controls,” individuals 5-11). F statistic was calculated in the cases and control samples using patient genotypes and 451 SNPs present in dbSNP.
NFAT5 deficiency is associated with immunologic deficits in T lymphocyte function
To provide a mechanistic link between NFAT5 deficiency in the proband and the abnormalities observed in his immune system, we tested whether inhibition of NFAT5 function in immune cells would recapitulate the patient's deficits. Because prior studies have demonstrated that NFAT5 deficiency in proliferating murine T lymphocytes impairs their survival capacity in hypertonic conditions (16, 18), we first tested the viability of the patient's PBMCs and observed reduced survival in hypertonic culture conditions (Fig. 5A) relative to control cells.
Figure 5.

Dysregulated T lymphocyte responses linked to NFAT5 deficiency. (A) Survival capacity of proliferating PBMCs isolated from the proband and 13 healthy controls. PBMCs were activated with anti-CD3 and anti-CD28 antibodies and cultured in isotonic (280 mOsm/kg) or hypertonic (420 mOsm/kg) media; viability was quantified using flow cytometry based on forward and side scatter properties. Survival is expressed as % cell death in hypertonic media, normalized to isotonic media (% live lymphocytes in isotonic culture - % live lymphocytes in hypertonic culture / % live lymphocytes in isotonic culture). (B-E) Jurkat cells were retrovirally transduced with control or dominant negative NFAT5 (DN NFAT5) constructs (B, D) or control or NFAT5 shRNA constructs (C, E). Cells were rested for 2 days, then cultured in either isotonic or hypertonic media for an additional 5 days. Cells expressing constructs were detected on the basis of GFP positivity. Cell survival was calculated as in (A). Cells were stained with Annexin V and propidium iodide (PI) to quantify apoptotic and dead cells using flow cytometry (C, E). (F) Intracellular TNFα production from gated CD8+ T cells was measured by flow cytometry after stimulation of PBMCs from the proband and 25 healthy controls for 24 hours with anti-CD3 and anti-CD28 antibodies. Representative dot plots (left) and scatter plots representing the percent TNFα+ of CD8+ T cells from all individuals analyzed are shown. Error bars represent standard error of the mean (SEM). (G) Intracellular TNFα production from gated CD8+ T cells retrovirally transduced with control or NFAT5 DN constructs, measured by flow cytometry after stimulation for 2 days with anti-CD3 and anti-CD28 antibodies. (H) Intracellular TNFα production from gated CD8+ T cells expressing control or DN NFAT5 constructs, derived from bone marrow chimeric mice, following stimulation with PMA and ionomycin. Chimeric mice were generated by adoptively transferring bone marrow cells transduced with control or DN NFAT5 constructs into lethally irradiated RAG1-deficient mice. Error bars represent SEM. ** p < 0.01,*** p < 0.001. Results shown are representative of at least three independent experiments.
To provide evidence that this effect might be due to NFAT5 deficiency, we used a dominant negative NFAT5 (NFAT5 DN) construct, which has been shown to interfere with dimerization, thereby reducing NFAT5 transcriptional activity by approximately 75% (14). Using a retroviral approach, we expressed the NFAT5 DN or control construct in Jurkat cells, a human T lymphocyte cell line, and observed that proliferating cells expressing the NFAT5 DN construct exhibited a survival defect in hypertonic conditions (Fig. 5B) similar to that displayed by the patient's PBMCs. As a second, confirmatory method to measure cell viability, we stained the cells for Annexin V and propidium iodide (PI), with similar results (Fig. 5D).
As a confirmatory approach, we generated a short hairpin RNA (shRNA) construct that reduced NFAT5 expression by approximately 50% (Supplemental Fig. 2). Jurkat cells were transduced with retrovirus expressing control shRNA or shRNA targeting the NFAT5 gene and subsequently cultured in isotonic or hypertonic media. We observed that proliferating cells with reduced expression of NFAT5 exhibited decreased viability in hypertonic conditions (Fig. 5C and 5E, Supplemental Fig. 2).
To determine whether NFAT5 deficiency might be responsible for decreased production of TNFα by the patient's CD8+ T cells (Table I and Fig. 5F), we expressed the control or DN NFAT5 constructs in PBMCs isolated from healthy controls. CD8+ T cells expressing the dominant negative NFAT5 construct exhibited reduced production of TNFα (Fig. 5G), analogous to the defect observed in the patient's CD8+ T cells. To provide further evidence that NFAT5 regulates T lymphocyte cytokine production, we generated bone marrow chimeras in which CD8+ T cells have diminished NFAT5 function. Bone marrow cells from wild-type mice were transduced with control or dominant negative NFAT5 constructs and adoptively transferred into lethally irradiated RAG1-deficient mice. After reconstitution, CD8+ T cells from the blood were analyzed for intracellular cytokine production. We observed reduced TNFα production in CD8+ T cells expressing the dominant negative NFAT5 construct compared to those cells expressing the control construct (Fig. 5H). Taken together, these human and murine data are consistent with prior evidence that TNFα is a direct target of NFAT5 (9, 14) and suggest that NFAT5 deficiency may underlie the impaired functional capabilities of the proband's T lymphocytes.
NFAT5 deficiency may be associated with a reduction in NK cells
We next investigated whether NFAT5 deficiency might be related to the finding of reduced NK cells in the patient (Table I and Fig. 6A). Because a reduction in the number of NK cells had not been previously reported in NFAT5-deficient mice, we analyzed NK cells from wild type, NFAT5-heterozygous, and NFAT5-null mice. The percentages and absolute numbers of CD122+CD49b+ NK cells were reduced in a dose-dependent manner, with a modest reduction observed in NFAT5-heterozygous mice and a more pronounced reduction in NFAT5-null mice (Fig. 6B). These results suggested that NFAT5 deficiency might contribute to the development and/or survival of NK cells, although the underlying mechanism remains unknown and it remains possible that other factors may be involved.
Figure 6.

Decreased natural killer cell frequency in absence of NFAT5. (A) NK cell frequencies in the proband and 25 healthy controls, assessed by % CD16+CD56+ events gated on lymphocytes. (B) NK cell frequencies in the spleen of wild-type (Wt/Wt), NFAT5-heterozygous (Wt/Nfat5-), and NFAT5-null (Nfat5-/Nfat5-) mice were assessed by % CD49b+CD122+ events gated on lymphocytes (left panel). The total NK cell numbers in each mouse genotype were quantified (right panel). Error bars represent SEM; * p < 0.05, *** p < 0.001.
NFAT5 expression is reduced in patients with inflammatory bowel disease
To determine whether the link between reduced NFAT5 expression and autoimmunity might be generalizable to other immune-mediated diseases, we examined NFAT5 mRNA expression in intestinal tissue biopsies from patients with inflammatory bowel disease (IBD). Crohn's disease (CD) and ulcerative colitis (UC), which together comprise IBD, are believed to result from an aberrant immune response to commensal gut microbes, leading to chronic intestinal inflammation. Compared to healthy controls, we observed that NFAT5 mRNA expression was significantly reduced in patients with active UC and CD (Fig. 7), raising the possibility that NFAT5 and other components of the osmoadaptation pathway may be dysregulated in IBD.
Figure 7.

Reduced NFAT5 mRNA expression in patients with active inflammatory bowel disease. Total RNA was extracted from rectal biopsies obtained from 8 healthy controls, 7 patients with active ulcerative colitis (UC) and 6 patients with active Crohn's disease (CD). Relative expression was calculated by ddCt method using the average Ct of CANX and RPLP0 as a reference genes and normalizing to the average NFAT5 expression of the healthy controls. Error bars represent SEM; *** p < 0.001.
Discussion
In the current study, we describe a patient with a previously unrecognized primary immunodeficiency syndrome affecting components of innate and adaptive immunity, predominantly manifesting as autoimmune enterocolopathy. Although we cannot completely rule out the contribution of other genes found in the patient's deleted segment, our studies provide the first evidence linking this syndrome with a deficiency in NFAT5, which has not previously been associated with disease in mice or humans.
Several groups have shown in mice that an important consequence of NFAT5 deficiency is impaired lymphocyte proliferation and survival in hypertonic culture conditions (16, 18). The critical need for osmoadaptive mechanisms in lymphocytes is underscored by the finding that lymphoid tissues are hyperosmotic relative to blood (18). Thus, because lymphocytes encounter microbes within hypertonic lymphoid tissues, these cells must be capable of osmoadaptation in order to proliferate and orchestrate anti-microbial functions. Our observation that lymphocytes from the proband exhibited a reduced ability to survive in hypertonic conditions ex vivo suggests that these cells may also be functionally impaired in vivo in the setting of microbial challenges. This impairment may be more pronounced in the gut microenvironment, where rapid shifts in tonicity due to fluid absorption combined with constant microbial stimuli may explain, in part, the predominance of gastrointestinal manifestations in the proband (35).
Functional defects observed within different types of immune cells in the proband support the contention that NFAT5 deficiency may indeed confer a state of immunodeficiency. First, CD8+ T lymphocytes from the patient exhibited reduced degranulation and production of the pro-inflammatory cytokine TNFα, consistent with prior evidence that TNFα is a downstream target of NFAT5 (9, 14). Second, NK cells, innate immune cells with important roles in anti-viral and anti-tumor immunity, were reduced in the proband and in NFAT-deficient mice. These results raise the possibility that NFAT5 may influence development and/or survival of NK cells, though the precise mechanisms will require additional investigation and other factors are likely to be contributory. Taken together, these findings suggest a role for NFAT5 in innate and adaptive immune responses against microbial pathogens.
The clinical significance and etiology of the findings within the patient's B lymphocyte compartment remains unclear. Certain developing B cell subsets appeared to be dysregulated in the patient, raising the possibility that B cell activating factor, a regulator of B cell proliferation and differentiation, a known gene target of NFAT5 (36), might be involved. However, the patient's BAFF levels and BAFF receptor levels were normal, suggesting that the dysregulation of the patient's B cell subsets may be linked to NFAT5 via an alternate mechanism, or unrelated to NFAT5 deficiency altogether. Moreover, the patient exhibited an IgG2 subclass deficiency, which has been associated with an increased susceptibility to sinopulmonary infections as well as certain autoimmune disorders (37-39). However, the presence of protective titers of antibodies to microbes against which he was vaccinated seemed to suggest functional humoral immunity, and contrasts with the finding of reduced antigen-specific antibodies in immunized NFAT5-heterozygous mice (18).
The patient exhibited a defective proliferative response against Candida albicans and tetanus toxoid, but not against mitogens, suggesting an antigen-specific deficiency. The patient had been previously vaccinated against tetanus and had likely been exposed to Candida, ubiquitously found in the environment, arguing against lack of primary exposure to these antigens as the underlying mechanism. Another possibility might be a defect in the ability of the proband to form memory T cells, although the presence of normal percentages of memory T cells makes this possibility less likely. Finally, the patient's lymphocytes might exhibit a defect in their ability to be activated by antigen and undergo proliferation. However, the precise mechanism underlying the patient's defect in antigen-specific proliferation remains unknown.
Our findings thus provide the first evidence that genetic abnormalities in NFAT5 might be associated with AIE. However, we did not find the identical genetic deletion in the AIE patients that were evaluated, making it unlikely that abnormalities in NFAT5 underlie all cases of AIE. Nonetheless, we observed that NFAT5 deletion in the proband was associated with defects in lymphocyte function, and inhibition of NFAT5 in normal murine and human cells recapitulated those deficits. Together with the finding of reduced NFAT5 gene expression in the intestinal mucosa of patients with IBD, these results suggest that NFAT5 and other components of the osmoadaptation pathway may play a previously unappreciated role in a variety of human immune-mediated diseases and highlight the need for additional studies.
While the association between primary immunodeficiency and autoimmunity has long been appreciated, the molecular mechanisms underlying this association are not yet understood. It has been suggested that defects in one component of immunity result in inadequate control of microbes, leading to a persistent and pathologic state of activation within a second arm of immunity that ultimately triggers an autoimmune response (1, 2). Autoimmune manifestations are often treated with immunosuppressive medications, but if the initial triggering event is indeed immunodeficiency, an alternative therapeutic approach might be to correct the immune component that is defective. Taken together, our findings suggest that patients presenting with unexplained infections and/or autoimmune manifestations should consider comprehensive genetic and immunologic evaluation that may reveal new insights into the mechanisms underlying their conditions.
Supplementary Material
Acknowledgments
We thank members of the Chang laboratory for helpful discussions.
This work was supported by the National Institutes of Health Grants (R01DK061769 to S.E.C.; R01CA157885 to J.D.B.; R01CA181572 and R01CA72851 to A.G; DK093507, DP2OD008469, and R01AI095277 to J.T.C.); the Howard Hughes Medical Institute (Physician-Scientist Early Career Award to J.T.C.); the Spanish government (SAF2009-08066 and SAF2012-36535 to C.L.-R.; BFU2008-01070 and SAF2011-24268 to J.A.); the European Union (MCIRG516308 to C.L.-R.); and the San Diego Clinical and Translational Research Institute (UL1RR031980).
Abbreviations used in this article
- NFAT5
nuclear factor of activated T cells 5
- CVID
common variable immunodeficiency
- AIE
autoimmune enteropathy
- PBMC
peripheral blood mononuclear cell
- TNFα
tumor necrosis factor, alpha
- UPD
uniparental disomy
- IBD
inflammatory bowel disease
- CD
Crohn's disease
- UC
ulcerative colitis
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Grammatikos AP, Tsokos GC. Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus. Trends Mol Med. 2012;18:101. doi: 10.1016/j.molmed.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verbsky JW, Chatila TA. T-regulatory cells in primary immune deficiencies. Curr Opin Allergy Clin Immunol. 2011;11:539. doi: 10.1097/ACI.0b013e32834cb8fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen FS, Cooper MD, Wedgwood RJ. The primary immunodeficiencies. N Engl J Med. 1995;333:431. doi: 10.1056/NEJM199508173330707. [DOI] [PubMed] [Google Scholar]
- 4.Etzioni A. Immune deficiency and autoimmunity. Autoimmun Rev. 2003;2:364. doi: 10.1016/s1568-9972(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 5.Akram S, Murray JA, Pardi DS, Alexander GL, Schaffner JA, Russo PA, Abraham SC. Adult autoimmune enteropathy: Mayo Clinic Rochester experience. Clin Gastroenterol Hepatol. 2007;5:1282. doi: 10.1016/j.cgh.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catassi C, Fabiani E, Spagnuolo MI, Barera G, Guarino A. Severe and protracted diarrhea: results of the 3-year SIGEP multicenter survey. Working Group of the Italian Society of Pediatric Gastroenterology and Hepatology (SIGEP) J Pediatr Gastroenterol Nutr. 1999;29:63. doi: 10.1097/00005176-199907000-00016. [DOI] [PubMed] [Google Scholar]
- 7.Unsworth DJ, Walker-Smith JA. Autoimmunity in diarrhoeal disease. J Pediatr Gastroenterol Nutr. 1985;4:375. doi: 10.1097/00005176-198506000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Singhi AD, Goyal A, Davison JM, Regueiro MD, Roche RL, Ranganathan S. Pediatric autoimmune enteropathy: an entity frequently associated with immunodeficiency disorders. Mod Pathol. 2014;27:543. doi: 10.1038/modpathol.2013.150. [DOI] [PubMed] [Google Scholar]
- 9.Buxade M, Lunazzi G, Minguillon J, Iborra S, Berga-Bolanos R, Del Val M, Aramburu J, Lopez-Rodriguez C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J Exp Med. 2012;209:379. doi: 10.1084/jem.20111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drews-Elger K, Ortells MC, Rao A, Lopez-Rodriguez C, Aramburu J. The transcription factor NFAT5 is required for cyclin expression and cell cycle progression in cells exposed to hypertonic stress. PLoS One. 2009;4:e5245. doi: 10.1371/journal.pone.0005245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esensten JH, Tsytsykova AV, Lopez-Rodriguez C, Ligeiro FA, Rao A, Goldfeld AE. NFAT5 binds to the TNF promoter distinctly from NFATp, c, 3 and 4, and activates TNF transcription during hypertonic stress alone. Nucleic Acids Res. 2005;33:3845. doi: 10.1093/nar/gki701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Rodriguez C, Aramburu J, Rakeman AS, Rao A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc Natl Acad Sci U S A. 1999;96:7214. doi: 10.1073/pnas.96.13.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci U S A. 1999;96:2538. doi: 10.1073/pnas.96.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Rodriguez C, Aramburu J, Jin L, Rakeman AS, Michino M, Rao A. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity. 2001;15:47. doi: 10.1016/s1074-7613(01)00165-0. [DOI] [PubMed] [Google Scholar]
- 15.Trama J, Go WY, Ho SN. The osmoprotective function of the NFAT5 transcription factor in T cell development and activation. J Immunol. 2002;169:5477. doi: 10.4049/jimmunol.169.10.5477. [DOI] [PubMed] [Google Scholar]
- 16.Berga-Bolanos R, Drews-Elger K, Aramburu J, Lopez-Rodriguez C. NFAT5 regulates T lymphocyte homeostasis and CD24-dependent T cell expansion under pathologic hypernatremia. J Immunol. 2010;185:6624. doi: 10.4049/jimmunol.1001232. [DOI] [PubMed] [Google Scholar]
- 17.Trama J, Lu Q, Hawley RG, Ho SN. The NFAT-related protein NFATL1 (TonEBP/NFAT5) is induced upon T cell activation in a calcineurin-dependent manner. J Immunol. 2000;165:4884. doi: 10.4049/jimmunol.165.9.4884. [DOI] [PubMed] [Google Scholar]
- 18.Go WY, Liu X, Roti MA, Liu F, Ho SN. NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc Natl Acad Sci U S A. 2004;101:10673. doi: 10.1073/pnas.0403139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Rodriguez C, Antos CL, Shelton JM, Richardson JA, Lin F, Novobrantseva TI, Bronson RT, Igarashi P, Rao A, Olson EN. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc Natl Acad Sci U S A. 2004;101:2392. doi: 10.1073/pnas.0308703100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goel A, Xicola RM, Nguyen TP, Doyle BJ, Sohn VR, Bandipalliam P, Rozek LS, Reyes J, Cordero C, Balaguer F, Castells A, Jover R, Andreu M, Syngal S, Boland CR, Llor X. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010;138:1854. doi: 10.1053/j.gastro.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, Xu Q, Hengartner MO, Elledge SJ, Hannon GJ, Lowe SW. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41:733. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 24.Bamias G, Goukos D, Laoudi E, Balla IG, Siakavellas SI, Daikos GL, Ladas SD. Comparative study of candidate housekeeping genes for quantification of target gene messenger RNA expression by real-time PCR in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2840. doi: 10.1097/01.MIB.0000435440.22484.e8. [DOI] [PubMed] [Google Scholar]
- 25.Hamer HM, Jonkers DM, Vanhoutvin SA, Troost FJ, Rijkers G, de Bruine A, Bast A, Venema K, Brummer RJ. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin Nutr. 2010;29:738. doi: 10.1016/j.clnu.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanz I, Wei C, Lee FE, Anolik J. Phenotypic and functional heterogeneity of human memory B cells. Semin Immunol. 2008;20:67. doi: 10.1016/j.smim.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubinsky MC, Lamothe S, Yang HY, Targan SR, Sinnett D, Theoret Y, Seidman EG. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705. doi: 10.1016/s0016-5085(00)70140-5. [DOI] [PubMed] [Google Scholar]
- 29.Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology. 2006;130:1047. doi: 10.1053/j.gastro.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 30.Wright S, Sanders DS, Lobo AJ, Lennard L. Clinical significance of azathioprine active metabolite concentrations in inflammatory bowel disease. Gut. 2004;53:1123. doi: 10.1136/gut.2003.032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol Cell. 2011;103:303. doi: 10.1042/BC20110013. [DOI] [PubMed] [Google Scholar]
- 34.Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O'Brien WE, Beaudet AL. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet. 1988;42:217. [PMC free article] [PubMed] [Google Scholar]
- 35.Schilli R, Breuer RI, Klein F, Dunn K, Gnaedinger A, Bernstein J, Paige M, Kaufman M. Comparison of the composition of faecal fluid in Crohn's disease and ulcerative colitis. Gut. 1982;23:326. doi: 10.1136/gut.23.4.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kino T, Takatori H, Manoli I, Wang Y, Tiulpakov A, Blackman MR, Su YA, Chrousos GP, DeCherney AH, Segars JH. Brx mediates the response of lymphocytes to osmotic stress through the activation of NFAT5. Sci Signal. 2009;2:ra5. doi: 10.1126/scisignal.2000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oxelius VA. Immunoglobulin G (IgG) subclasses and human disease. Am J Med. 1984;76:7. doi: 10.1016/0002-9343(84)90314-0. [DOI] [PubMed] [Google Scholar]
- 38.Shackelford PG, Polmar SH, Mayus JL, Johnson WL, Corry JM, Nahm MH. Spectrum of IgG2 subclass deficiency in children with recurrent infections: prospective study. J Pediatr. 1986;108:647. doi: 10.1016/s0022-3476(86)81035-6. [DOI] [PubMed] [Google Scholar]
- 39.Siber GR, Schur PH, Aisenberg AC, Weitzman SA, Schiffman G. Correlation between serum IgG-2 concentrations and the antibody response to bacterial polysaccharide antigens. N Engl J Med. 1980;303:178. doi: 10.1056/NEJM198007243030402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.