

Abstract
Telomere maintenance is a highly coordinated process, and its misregulation is linked to cancer as well as telomere-shortening syndromes. Recent studies have shown that the TEL-patch – a cluster of amino acids on the surface of the shelterin component TPP1 – is necessary for the recruitment of telomerase to the telomere in human cells. However, there has been only basic biochemical analysis of the role of TPP1 in the telomerase recruitment process. Here we develop an in vitro assay to quantitatively measure the contribution of the TEL-patch to telomerase recruitment – binding and extension of the first telomeric repeat. We also demonstrate that the TEL-patch contributes to the translocation step of the telomerase reaction. Finally, our quantitative observations indicate that the TEL-patch stabilizes the association between telomerase and telomeric DNA substrates, providing a molecular explanation for its contributions to telomerase recruitment and action.
Keywords: telomere, telomerase recruitment, DNA substrate-competition, TPP1 TEL-patch, processivity
Introduction
Telomeres are specialized nucleoprotein caps found at linear chromosomal termini. Telomeres are essential for genomic integrity. Telomerase is the enzyme responsible for the synthesis of telomeric DNA repeats [1], which are in turn bound by protective telomeric protein complexes [2]. In humans, telomerase recruitment and its subsequent enzymatic action at the telomere during late S/G2 phase of the cell cycle are essential for both germ cells and stem cells to retain proliferative capacity [3]. In dividing somatic cells, in which telomerase is not expressed, telomeres progressively erode as a result of incomplete 3′ end-replication and the cells enter replicative senescence [4]. Escape from senescence results in critically short telomeres and apoptosis [5]. However, on occasion, cells evade apoptosis by reactivating telomerase, which results in renewed telomere length maintenance and cellular immortalization, a hallmark of 80-95% of cancers [5, 6]. In contrast, telomerase or telomere maintenance deficiencies in proliferative cell populations cause organ failure in a class of telomere- shortening diseases, examples of which include dyskeratosis congenita (DC), idiopathic pulmonary fibrosis, and aplastic anemia (AA) [7, 8]. Thus, the proper regulation of telomerase is critical to telomere homeostasis.
Telomerase is a ribonucleoprotein complex that is minimally comprised of a templating Telomerase RNA (TR) and the enzymatically active Telomerase Reverse Transcriptase (TERT) [9, 10]. Human TERT (hTERT) has an N-terminal domain (TEN), Telomerase RNA-binding domain (TRBD), and conserved Reverse Transcriptase domain (RT) and C-terminal extension (polymerase thumb) [2, 11]. The 450 nt hTR contains the canonical pseudoknot-template domain and a partially conserved three-way junction (CR4/5), which are bound by hTERT and necessary for enzymatic activity [12, 13]. During telomeric DNA synthesis, single-stranded DNA anneals to the hTR RNA template and the RT domain of TERT catalyzes the sequential addition of nucleotides to the 3′ end of the DNA. When TERT reaches the end of the RNA template, the telomeric DNA repositions on the template, enabling another repeat to be added. The addition of multiple repeats by telomerase, prior to dissociating from the telomeric substrate, is known as repeat addition processivity (RAP). The TEN-domain, RT domain, and CTE of hTERT have been proposed to interact with the telomeric DNA substrate to promote RAP [14-18], as has the TRBD in Tetrahymena TERT [19]. Outside of the active site, hTR also contains a scaRNA domain that is recognized by dyskerin and a CAB box motif that is recognized by TCAB1 to promote Cajal body localization [20-22].
Human telomerase is sequestered in Cajal bodies for most of the cell cycle through an interaction with TCAB1 [23, 24]. During S-phase, telomerase is recruited to telomeres through an interaction with shelterin component TPP1 [25, 26]. We find it useful to distinguish “recruitment” of telomerase from simple binding, with recruitment designating preferential localization of telomerase to its site of biological activity (e.g., telomeres vs. internal chromatin or telomeres vs. Cajal bodies) in a manner that is competent for telomere extension. Proper recruitment of telomerase to the telomere is crucial to maintaining telomere length. The number of active telomerase molecules per cell roughly equals the number of telomere ends during S-phase (∼240 in HEK 293 and HeLa cells) [27]. Furthermore, telomeres are thought to shorten by 50-100 bp per cell division [28], while telomerase is thought to extend each telomere by about 60 nt per cycle [29]. Thus, a precarious balance between telomere erosion and telomerase action exists, and small changes in telomerase activity or recruitment may have significant impacts on telomere length in cells [30].
Telomerase and shelterin work synergistically to promote telomere homeostasis. Shelterin is the six protein complex responsible for protecting telomeres and regulating telomerase action [2, 31]. Loss of the double-stranded DNA-binding proteins, TRF1 and TRF2, results in activation of the DNA-damage response in mice [32, 33]. The single-stranded DNA binding protein POT1 serves to restrict telomerase access to telomeric substrates both in vivo and in vitro [34, 35]. TIN2 and TPP1 bridge the double-stranded and single-stranded binding proteins within shelterin. Additionally, TIN2 is necessary for the recruitment of TPP1 to shelterin [25]. TPP1, which also associates with POT1, is required for the recruitment of telomerase to telomeres in vivo [25, 26]. In particular, the acidic TEL-patch found on the surface OB-domain of TPP1 is both necessary and sufficient to recruit telomerase [36-39] through a direct interaction with the TEN-domain of hTERT [40].
In addition to recruiting telomerase, the TPP1-POT1 complex is a processivity factor for telomerase because the binding of TPP1-POT1 to primers in direct telomerase extension assays stimulates RAP [41]. TPP1-POT1 interacts with telomerase to stimulate processivity through at least two mechanisms: (i) decreasing the rate of primer dissociation from the enzyme, and (ii) increasing the apparent rate of translocation and efficiency [42]. Mutations to the TEL-patch of TPP1 also decrease TPP1-POT1 RAP stimulation of telomerase [36]. Moreover, RAP stimulation and in vivo recruitment defects of TPP1 TEL-patch mutants can be rescued by a compensatory charge-swap mutation in the TEN-domain of hTERT [40]. Collectively, experimental evidence suggests that TPP1-POT1 RAP stimulation and telomerase recruitment are manifestations of the same direct interaction between telomerase and TPP1.
To better understand the contributions of the TEL-patch to telomerase recruitment, we have developed a novel in vitro substrate competition assay. Using this assay, we show that the TEL-patch participates in the preferential extension of TPP1-POT1-bound substrates and that mutation of the TEL- patch results in less efficient substrate usage by telomerase in vitro. In addition, we show that mutation to the TEL-patch reduces the apparent translocation rate, decreases the efficiency of translocation, and increases the rate of primer DNA dissociation from actively synthesizing telomerase.
Results
The TEL-patch on TPP1 promotes the translocation of human telomerase
Mutations in the TEL-patch impact the ability of TPP1 to stimulate RAP by telomerase in vitro [36], suggesting that the TEL-patch interacts with telomerase during catalysis. To understand TEL-patch contributions in stimulating telomerase RAP, we compared wild-type TPP1 and a previously described TPP1 TEL-patch mutant E169A;E171A (EE mutant) [36] in a number of in vitro telomerase assays. Assays were used to query various steps in the telomerase catalytic cycle (Fig. 1a).
Fig. 1.
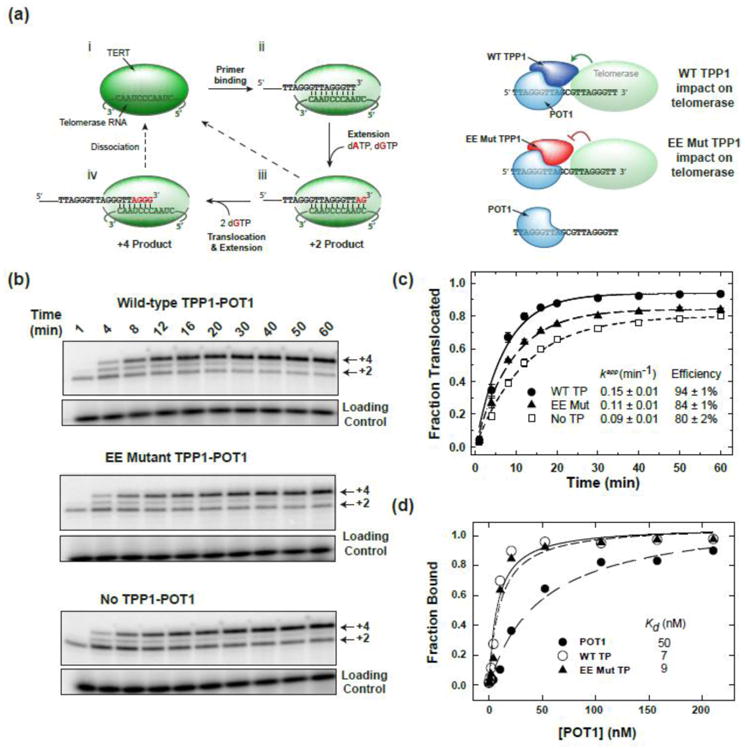
Mutations in the TEL-patch adversely impact telomerase translocation. (a) (Left) the human telomerase catalytic cycle. i) Telomerase is a ribonucleoprotein complex that contains an internal template Telomerase RNA (TER) which is utilized by Telomerase Reverse Transcriptase (TERT) to synthesize telomeric repeats ii) upon DNA substrate binding. iii) Nucleotides are sequentially added to the 3′ end of the substrate until the end of the internal RNA template is reached. iv) The primer is next repositioned on the RNA template (translocation) and a subsequent round of repeat addition ensues. Additionally, the substrate or product can dissociate from the enzyme during any step of the cycle, although dissociation coincides most often with the translocation step in vitro. (Right) POT1 complexed with TPP1 binds the DNA primer; wild-type TPP1 with an intact TEL-patch engages with TERT.
(b) Translocation rate assay for primer alone, primer bound by wild-type TPP1-POT1, or primer bound by E169A;E171A (EE mutant) TPP1-POT1. Reaction time (min) noted above gel, and +2 and +4 products denoted on the side of the gel correspond to products shown in (a). Precipitation and loading control shown below each translocation panel.
(c) Fraction translocated was calculated as the sum of counts in the +3 and +4 products divided by the total counts and plotted as a function of time for primer alone (open squares), wild-type TPP1 (closed circles), and E169A;E171A TPP1 (closed triangles). TP signifies TPP1-POT1. Translocation data were fit to the equation y = A(1-e(−kt)) where k (or kapp) represents the rate constant for a single round of translocation plus subsequent nucleotide incorporation and A (the horizontal limit) represents the maximum efficiency. The efficiency and rate constants for each condition are noted. The averaged data are plotted ± standard deviation (n = 2).
(d) Equilibrium binding of POT1 (closed circles) and complexes wild-type TPP1–POT1 (open circles) and E169A;E171A mutant TPP1–POT (triangles) to the primer a5TT. Native gel-shift assay data were fit to a one-site binding equation by non-linear regression to obtain equilibrium dissociation constants (Kd). One replicate was carried out as our values matched previous measurements.
Wild-type TPP1 was previously shown to impact both the translocation rate and the efficiency of translocation [42]. We hypothesized that mutations in the TEL-patch would decrease RAP stimulation by impacting translocation, and we tested this with a single-turnover translocation experiment [42-44]. Wild-type TPP1-POT1 or EE mutant TPP1-POT1 was complexed with primer and pre-bound to telomerase. The translocation rate was measured by initiating telomerase extension by adding only dATP and dGTP (dTTP was omitted) and monitoring the fraction of product formation before (+2 products) and after translocation (+3,+4 products) (Fig. 1b). We note that the “translocation rate” that we measure relies on translocation as well as nucleotide incorporation to determine the fraction translocated and yields a complex rate constant that may not be solely dependent on primer repositioning. A single translocation event (Fig. 1a; steps iii and iv) was observed because dTTP was absent and an excess of chase primer was added simultaneously with the dNTPs to prevent dissociated substrates from rebinding telomerase.
TPP1-POT1 increased both the translocation rate and efficiency of translocation compared to primer alone. The apparent rate constant for primer alone was 0.09 ± 0.01 min-1, in agreement with previous measurement [42]. Having wild-type TPP1-POT1 bound to the primer increased the apparent rate constant to 0.15 ± 0.01 min-1, while the TEL-Patch mutant TPP1-POT1 retained partial activity (0.11 ± 0.01 min-1) (Fig. 1c). In addition, the overall efficiency of translocation differed between the samples. In the case of primer alone, only 80% of the incorporated dGTP was present in the +4 product, while 20% remained un-translocated. Addition of TEL-patch or wild-type POT-TPP1 resulted in incremental increases in efficiency to 84% and 94%, respectively (Fig. 1c). We verified that differences observed in the translocation assay result in increased telomerase RAP, using a full extension assay with dTTP (Fig. S1a,b). The full extension assay was carried out under similar conditions; however, the concentration of Mg2+ was titrated to account for overall higher dNTP concentration. Collectively, these results confirm that the TEL-patch contributes to the apparent translocation rate and increases the efficiency of translocation.
To ensure that our TEL-patch EE mutant TPP1 was active in interacting with POT1 and correctly folded, we determined the Kd of both wild-type and mutant TPP1-POT1 complexes for DNA by electrophoretic mobility shift assay. TPP1 is known to enhance the affinity of POT1 for DNA [41], and mutation to the TEL-patch should not disrupt POT1 binding [36]. POT1 alone bound the DNA with a Kd of 50 nM, and the addition of either wild-type or mutant TPP1 resulted in increased affinity to 7 and 9 nM, respectively (Fig. 1d), consistent with previous results [36, 41]. These results indicate that the TEL-patch EE mutant TPP1 is competent to form a complex with POT1 and thereby increase its affinity for DNA.
Developing an in vitro assay for telomerase recruitment to a telomere
The TEL-patch of TPP1 was previously shown to interact with the TEN-domain of TERT, and it is crucial for telomerase recruitment to telomeres in vivo [36, 37, 40]. We postulated that telomerase should be preferentially recruited (i.e., binding and extension) to TPP1-POT1-bound substrates with a wild-type TEL-patch. To test this hypothesis, we developed a competition assay with two competing substrates (Fig. 2a). Two primers with slightly different lengths (38 and 44 nt) and the same molar concentration were used in the competition. One primer was complexed with wild-type TPP1-POT1 and the other with the EE mutant TPP1-POT1 (Fig. S2a). The competition was initiated when the two substrates were simultaneously added to a low concentration of telomerase in buffer containing dATP and dGTP (dTTP was again omitted). As the primers were slightly longer than the primers that are typically used in direct extension assays, we verified that the extension products were telomerase specific by treating the telomerase with RNase A; this eliminated extension of both primers under the competition conditions (Fig. S2b). In the competition, the two substrates act as competitive inhibitors of one another [45] – i.e., a single active site can accommodate and catalyze product formation of only one substrate at a time. Thus, the ratio of the initial velocities of product formation for the two substrates gives the ratio of their specificity constants (kcat/Km)substrate1/(kcat/Km)substrate2 [45].
Fig. 2.
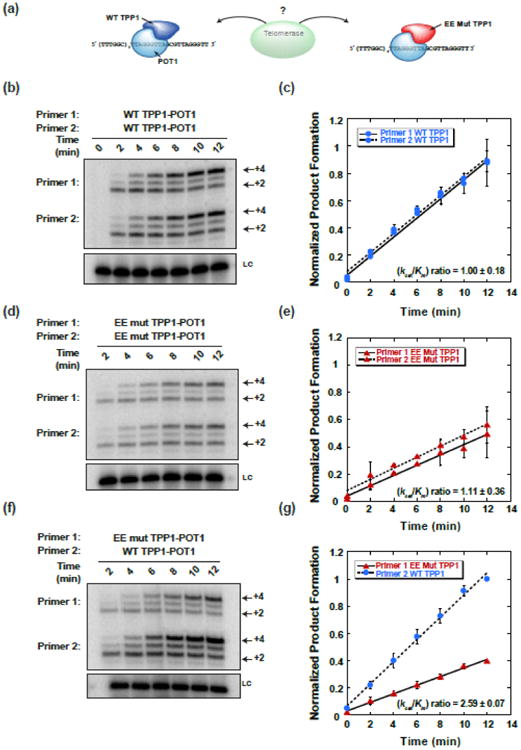
The TEL-patch contributes to substrate recruitment and extension by telomerase. (a) In vitro telomerase competition assays between two primers: a 38 nt primer bound with wild-type TPP1-POT1 and a 44 nt primer bound by TEL-patch mutant TPP1-POT1, cartoon depicts competition shown in panel f and quantitated in panel g. (b) Competition between primer 1 and primer 2 each pre-bound to wild-type (WT) TPP1-POT1. (c) Initial velocity plot for the competition shown in b. Blue circles indicate wild-type TPP1-POT1. (d) Primer 1 and primer 2 each pre-bound to E169A;E171A (EE mut) TPP1-POT1. (e) Initial velocity plot for the competition shown in d. Red triangles indicate E169A;E171A TEL-patch mutant TPP1-POT1. (f) Primer 1 pre-bound to EE mut TPP1-POT1 and primer 2 pre-bound to WT mut TPP1-POT1. (g) Initial velocity plot for competition shown in f, colors and symbols are the same as c,d. Time (min) after simultaneously mixing telomerase and both substrates denoted above gels, and +2 and +4 products indicated on the right side of the gels. Precipitation and loading control (LC) shown below each competition panel. Product formation was calculated by summing the counts of the +2, +3, and +4 products, normalized to the loading control and expressed as a fraction of the total counts incorporated for primer bound by WT TPP1-POT1 at 12 min for each replicate. The average normalized product formation for each competition was plotted as a function of time and fit by linear regression, and the initial velocity of product formation was determined by the slope of the line. Error bars represent the standard deviation in normalized product formation at each time point for the replicates (n = 2). Throughout this work, kcat/Km ratios calculated using the equation: (kcat/Km)primer2/(kcat/Km)primer1 = (νprimer2/νprimer1)*([primer1]/[primer2]). Final concentration of each primer was 100 nM.
We first verified that measured differences in the initial velocity of product formation between the two TPP1-POT1-bound substrates were due to mutations in the TEL-patch rather than other intrinsic differences (e.g. primer length or secondary structure). Both primers were bound either to wild-type TPP1-POT1 or to EE mutant TPP1-POT1 and used in telomerase competitions (Fig. 2b-e). The difference in initial velocity of product formation was negligible when both primers were bound by the same TPP1- POT1 complex (Fig. 2c,e). Competing primers bound to wild-type TPP1-POT1 had a specificity constant ratio of 1.00 ± 0.18 and primers bound by EE mutant TPP1-POT1 had a ratio of 1.11 ± 0.36, consistent with equivalent primers having an expected ratio of 1. Collectively, these data demonstrate that primers of slightly different lengths bind to and are extended by telomerase with equivalent efficiency when associated with the same TPP1-POT1 complex. Additionally, the total velocity of product formation (i.e. vprimer1 + vprimer2) increased when both primers were bound to wild-type TPP1-POT1 compared to both primers bound to TEL-patch mutant TPP1-POT1 (Fig. 2c,e).
Next we carried out a competition between a primer bound to wild-type TPP1-POT1 and a second primer bound to EE mutant TPP1-POT1 (Fig. 2f,g). The initial velocity and specificity constant ratio for the wild-type over mutant TPP1-POT1-primer was 2.59 ± 0.07. The higher initial velocity for the primer bound by wild-type TPP1-POT1 was retained at multiple primer concentrations (Fig. S2c-h). The specificity constant ratio appeared to increase at lower primer concentrations. In addition, there was a lag in the initial velocity of product formation especially noticeable at 20 and 10 nM primer concentrations (Fig. S2f,h), consistent with the association of primer with telomerase contributing to the observed rate.
As a control, a second competition in which the opposite shorter primer was bound to TEL-patch mutant TPP1-POT1 gave a specificity ratio of 1.52 ± 0.03 (Fig. S3a,b). Preferential extension of the longer primer bound by the wild-type TPP1-POT1 was also apparent at 40 nM primer concentration (Fig. S3c,d). Ideally, the specificity constant ratio should be independent of which primer is bound to which protein complex, but this was not the case. For example, 100 nM primer concentrations gave specificity constant ratios of 2.59 ± 0.01 when the shorter primer was bound to WT TPP1-POT1 and 1.52 ± 0.03 when the longer primer was bound to WT TPP1-POT1. The difference likely reflects the contributions of the primers themselves in addition to the contributions of the wild-type or mutant TEL-patch. Despite the differences in specificity constant ratios, the main conclusion is that when either primer was complexed with wild-type TPP1-POT1 it was preferentially bound and extended by telomerase at multiple primer concentrations. For analysis of how this assay relates to telomerase recruitment, see the Discussion.
Our experimental design would underestimate the difference between mutant and wild-type TPP1 if the proteins exchanged between the primers during the course of the experiment. We therefore tested how TPP1 exchange impacted our competition experiment. One primer bound to wild-type TPP1-POT1 and a second primer bound to EE mutant TPP1-POT1 were pre-mixed for varying amounts of time prior to a fixed length of competition extension by telomerase (Fig. 3a-c). Plotting the product ratio versus time in Fig. 3c revealed that approximately 50% of the TPP1 exchanged after 30 min (note: 20 min of pre-equilibration followed by 10 min of extension). The experiment also revealed that after 12 min (the last time point in our competition assay) between 6 and 21% of the TPP1 appeared to have exchanged (i.e., after 2 min of pre-equilibration and 10 min of telomerase extension the product ratio was 21%, which was 6% higher than the starting product ratio of 15% with no pre-equilibration, Fig. 3c). Thus, the TPP1-POT1 complexes bind stably enough to primers that protein exchange has minimal effect on the competition assays.
Fig. 3.
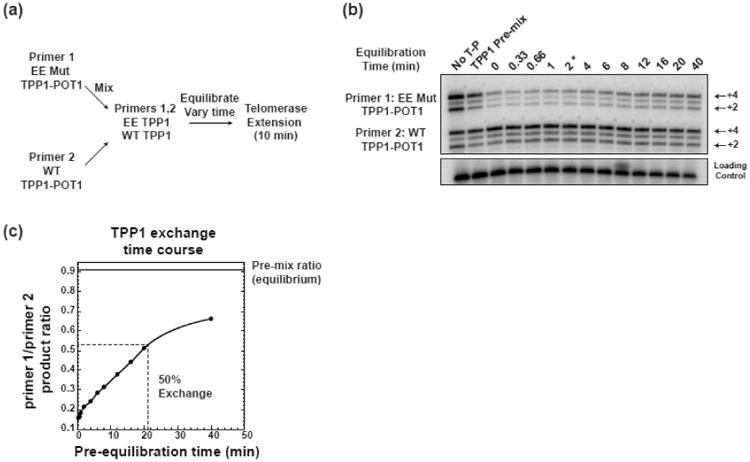
TPP1-POT1 remains stably associated with primers during competition assay. (a) Flow chart of TPP1 equilibration experiment. Primer 1 was bound to TEL-patch mutant TPP1-POT1 (EE mut) and in a separate tube primer 2 was bound by wild-type TPP-POT1. The two primers were mixed and allowed to pre-equilibrate for different amounts of time to allow TPP1 (or TPP1-POT1) to exchange between the primers. Primers were then extended for 10 min.
(b) TPP1 exchange equilibration experiment. Primer 1 was bound to TEL-patch mutant TPP1-POT1 (EE mut) and primer 2 was bound by wild-type TPP-POT1, as noted on left side of gel. No T-P, control experiment consisting of a competition between primer 1 and primer 2 with no TPP1-POT1 present. TPP1 Pre-mix, control experiment in which primers 1 and 2 were first mixed and then added to a mixture of wild-type TPP1-POT1 and E169A;E171A TPP1-POT1. Primer pre-equilibration time above gel; note that products visualized on gel were extended by telomerase for 10 min in addition to the time noted above the gel. Asterisk above 2 min pre-equilibration time corresponds to the final time point (12 min) in the initial velocity competitions shown in Figure 2 (i.e. 2 min of TPP1 pre-equilibration + 10 min telomerase extension = 12 min of TPP1 exchange). Products corresponding to +2 and +4 additions indicated on the right side of the gel. Precipitation and loading control (LC) shown below competition panel.
(c) Plot of the product ratio versus TPP1 pre-equilibration time. Product ratios were calculated by summing the +2, +3, +4 products for each primer and then dividing the total counts of primer 1 by the total counts of primer 2. The ratio of the TPP1-premix control is shown as a horizontal line at the top of the plot, and the pre-equilibration time (min) at which TPP1 has undergone 50% exchange is denoted with a dashed line, taking the starting product ratio as 15% and the pre-mix control ratio of 91% as the point at which equilibrium is reached.
Mutations in the TEL-patch of TPP1 induce faster dissociation of cycling telomerase from DNA substrates
Wild-type TPP1-POT1 was previously shown to slow the rate of primer dissociation from actively cycling telomerase [42]. We hypothesized that mutations in the TEL-patch may destabilize the interaction between TPP1 and telomerase, and thereby increase primer dissociation. To examine this hypothesis, we measured the primer dissociation rate from telomerase engaged in telomeric repeat synthesis [42]. In this experiment, primers were pre-bound to telomerase and repeat synthesis was initiated by addition of cold dATP, dGTP, and dTTP. At time zero, a large excess of 3′-phosphorylated chase primer was added to the reaction to prevent telomerase from re-associating with active primers. Telomerase extension was carried out for varying lengths of time in the presence of cold nucleotides, and products were pulse labeled with α-32P-dGTP prior to reaction termination.
We measured the dissociation rates for primer alone, primer bound by wild-type TPP1-POT1, and primer bound by EE mutant TPP1-POT1. Primers without TPP1-POT1 underwent biphasic dissociation from telomerase - an initial rapid dissociation followed by slower dissociation (Fig. 4a,b), consistent with previous observations [42]. In the absence of TPP1-POT1, primers dissociated with an estimated t1/2Apparent ∼1 min. In contrast, primers bound with wild-type TPP1-POT1 dissociated at a much slower rate, with t1/2Apparent of ∼6.5 min (Fig. 4a,b). The 6.5-fold decrease in the dissociation rate in the presence of wild-type TPP1-POT1 is consistent with previous results [42]. When primers were bound by EE mutant TPP1-POT1, the dissociation rate increased compared to primers bound by wild-type TPP1- POT1; the t1/2Apparent was ∼3.5 min. These results indicate the following mechanism for TEL-patch-dependent RAP stimulation: the TEL-patch stabilizes the interaction between primer-POT1-TPP1 and telomerase during active repeat synthesis, and thus increases the efficiency of telomerase translocation.
Fig. 4.
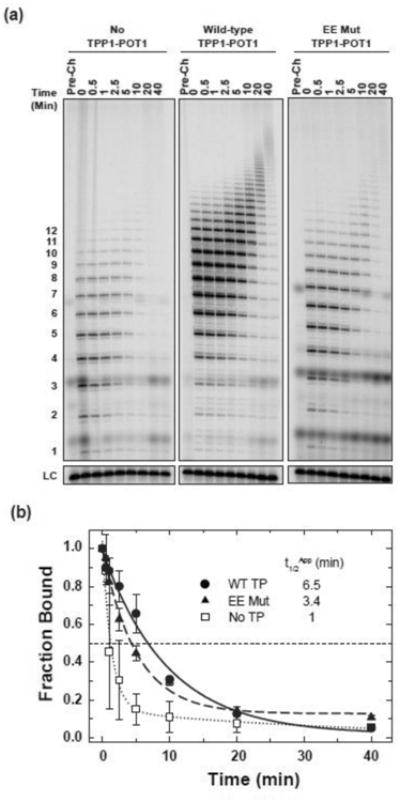
Mutations in the TEL-patch increase primer dissociation from extending telomerase.
(a) Cycling telomerase off-rate assays for free primer (left panel, No TPP1-POT1), primer bound by wild-type TPP1-POT1 (middle panel), or primer bound by E169A;E171A (right panel, EE Mut TPP1-POT1). “Pre-Ch” indicates pre-chase control samples in which 3′-phosphorylated primer was added to the telomerase prior to addition of the substrate primer. Time (min) of the chase denoted above gel; number of telomeric repeats indicated on left side of gel. Precipitation and loading control (LC) shown below each off-rate panel. The diffuse bands found near repeats 1, 3, and 7 are the result of contaminants present in the 32P-α-dGTP. The intensity of these spurious bands was excluded from the calculation of the fraction of primer bound.
(b) The total counts (TC) incorporated in each lane at time n were expressed as fraction of the counts incorporated at time zero, TC(t = n)/TC(t = 0), and plotted versus time. The data were fit to a double exponential. Values of t1/2Apparent were estimated by determining the time at which half of the primer dissociated, represented by the dashed line. Fits for free primer (No TP, open square), wild-type TPP1-POT1 (WT TP, closed circle), and E169A/E171A mutant TPP1-POT1 (EE TP, closed triangle). Averaged data were plotted ± standard deviation (n = 2).
Discussion
Telomerase-telomere interactions are vital to homeostatic telomere maintenance. Here we demonstrate that the TEL-patch contributes to telomerase-substrate interaction at multiple points during the telomerase catalytic cycle including increased binding and extension for the first repeat synthesized, increased translocation, and decreased dissociation. We present a first step towards reconstituting an in vitro telomerase recruitment assay, and validate the dependence of single-repeat synthesis on the TEL-patch of TPP1.
Telomerase recruitment can be measured independently of RAP stimulation in vitro
The crucial role for the TEL-patch in telomerase recruitment has been well established by a variety of cell-based experimental approaches [36-38, 40]. Evidence for TEL-patch-dependent telomerase recruitment in vitro is less direct, and has been inferred from measurements of TPP1-POT1 RAP stimulation of telomerase or TPP1-dependent telomerase pull-down [36, 40, 46]. The relationship between the RAP stimulation assay and telomerase recruitment is unclear, because the interaction being measured could occur after telomerase binds to the primer. The pull-down assay provides some measure of the efficiency of telomerase recruitment in vitro, but the rate constants or equilibrium constants that determine the pull-down efficiency are unknown. Our in vitro competition assay utilizes substrates that simultaneously compete for the telomerase active site and subsequent extension, so it provides the ratio of specificity constants (kcat/Km) for the two substrates. Our data indicate that the TEL-patch contributes to the synthesis of the first repeat of TPP1-POT1-bound primers, with the E169A;E171A mutation within TPP1 resulting in an approximately two-fold decrease in the initial velocity and specificity constant of telomerase for mutant TPP1. We note that our assay does not rely on the measurement of TPP1-POT1 stimulation of telomerase RAP. Furthermore, any enzyme turnover in this assay is distributive rather than processive (i.e., after the +4 incorporation telomerase could in theory dissociate and act on another substrate), because an unextendable chase primer is not added to the competition assay as it was in the translocation assay. Finally, our competition assay strengthens the evidence that the same molecular interaction between the TEL-patch of TPP1 and hTERT mediates both RAP stimulation and recruitment.
One advantage of using the telomerase reaction rather than binding to measure recruitment is that only productive enzyme-TP-primer complexes are counted. A disadvantage, however, is that the assay does not separate binding from the nucleotide addition steps. We acknowledge that the measurement of initial velocity in our competition experiment includes nucleotide additions +1 to +4 and the translocation step, resulting in the measurement of a complex rate constant. One possibility is that both wild-type and EE mutant TPP1-POT1-bound primers associate with telomerase equally well, and the TEL-patch slows dissociation during some other step that is rate-limiting (such as a nucleotide addition), dictating the difference in specificity constants (Fig 2). For example, dGTP incorporation may be rate-limiting in our experiment as its concentration is below Km [27] (although the dGTP concentration used in our experiments is roughly physiological [47, 48]). Although our assay does not isolate the initial substrate binding step, the lag in the initial velocity of product formation seen when the primer concentration is reduced to 10 or 20 nM suggests that substrate association likely contributes to the measured rates (Fig. S2c-h). Future work will be required to measure the individual rate constants of each step in the synthesis of a single repeat to definitively determine the extent to which the TEL-patch contributes to substrate binding to increase the efficiency of +1 nucleotide addition.
The two-fold stimulating effect of the TEL-patch on first repeat addition measured in vitro seems insufficient to explain the essentially on-off switch in telomerase recruitment seen upon TEL-patch mutation in vivo [36-38, 40]. One possible explanation is that telomerase may need to interact with multiple telomere-bound TPP1 proteins in order to locate the 3′ overhang. In this case, a small deficiency in binding during each association could result in a large additive recruitment defect. It is also likely that recruitment is further enhanced by additional factors in vivo. Possibilities include post- translational modification of TPP1 and/or TERT, trafficking of telomerase to the telomere by other proteins, or further stabilization of an initial TPP1-TERT interaction by additional components. The in vitro competition assay developed here should be useful in validating candidate telomerase recruitment factors in the future.
The TEL-patch stabilizes the telomerase-TPP1 interaction during processive telomeric synthesis
Processive repeat synthesis by telomerase requires translocation. During translocation, the telomeric DNA substrate must dissociate and subsequently reposition on the complementary RNA template [49]. Repeat addition is a dynamic process, and several factors contribute to telomerase- substrate association, translocation, and processivity. Interactions between the telomeric substrate and the TEN-domain provide stability throughout the catalytic cycle [14-16]. Processive synthesis is facilitated by multiple portions of the RT domain on hTERT, including the insertion in the fingers domain, motif 3, and the CTE [17, 18]. hTR, the telomerase RNA subunit, also makes direct contributions to translocation and processivity [50, 51]. However, despite the evolutionary tuning of telomerase to synthesize multiple repeats processively, translocation is an inefficient step and primers frequently dissociate [49].
A number of studies indicate that TPP1 is a processivity factor for human telomerase during active telomeric synthesis [2, 41, 42, 52]. TPP1 mediates its role as a processivity factor at least in part by reducing the telomerase-substrate off-rate [42]. Our data demonstrate that the TEL-patch of TPP1 contributes to enzyme-substrate stability during processive telomeric synthesis, resulting in longer products. Moreover, the TEL-patch of TPP1 decreases the dissociation of telomeric DNA from telomerase by approximately two-fold (Fig. 4b). Increased RAP was previously shown to correlate with decreased primer dissociation in a gain-of-function Tetrahymena TERT mutant [53]. In addition, we show that the interaction between telomerase and the TEL-patch of TPP1 stimulates the increase of both the apparent rate of translocation and the overall translocation efficiency. Translocation efficiency was shown to correlate with RAP for a panel of hTERT mutants [44]. Our results demonstrate that TPP1- POT1-dependent increases in translocation efficiency also correlate with increased telomerase RAP stimulation.
There are a number of explanations for how TPP1-POT1 might influence telomerase-substrate interaction to act as a processivity factor. First, TPP1-POT1 might contribute to the interaction solely by influencing the single-stranded DNA conformation in order to make it more accessible to telomerase; but our data and previous studies argue against this. Heterologous telomerase-TPP1-POT1 mixing experiments showed that non-cognate TPP1-POT1 complexes do not stimulate processivity (i.e. human TPP1-POT1 does not stimulate mouse or medaka telomerases, all of which synthesize the same telomeric repeat sequence), indicating that the TPP1-POT1 complex specifically interacts with its cognate TERT [52]. Furthermore, our data indicate that EE mutant and wild-type TPP1 interact with POT1-DNA equally well (Fig. 1d), but binding and extension, translocation, and dissociation are compromised in the presence of TEL-patch mutant TPP1. Second, TPP1 may act as an allosteric regulator or activator of telomerase by inducing conformational changes in telomerase making it more competent for processive elongation. Finally, TPP1 may directly interact with telomerase to stabilize the primer during key steps of the catalytic cycle, acting as an anchor point to provide an additional contact between telomerase and the telomere [42]. Currently our data do not distinguish between the allosteric and anchor mechanisms for TEL-patch stimulation of processive telomerase synthesis. However, dissociation rate experiments suggest that TPP1-POT1 actively interacts with telomerase during processive synthesis, not transiently at the initiation of synthesis (this study and [42]).
In our current recruitment model, the TEN-domain of hTERT directly engages the TEL-patch of TPP1 to scan along the ∼ 50-100 TPP1-POT1 complexes on a telomere [54] to locate the 3′ overhang. In vivo experiments suggest that the interaction between the TEL-patch and telomerase is necessary and sufficient for telomerase recruitment [2, 37-40]. Our competition data confirm that the TEL-patch-telomerase interaction contributes to the synthesis of the first telomeric repeat. Once telomerase engages in synthesis, the TEL-patch of TPP1 directly contacts the TEN-domain of hTERT to either induce a conformational change in telomerase or merely stabilize telomerase-telomere association, resulting in processive synthesis. Previous works suggest that multiple surfaces on TPP1 may contact telomerase [36, 38, 46], and TPP1 may make TEL-patch independent contributions to telomerase activation or processivity [38]. TPP1 may also directly interact with the primer, when complexed with POT1, to increase processivity [55, 56]. As mutation to the TEL-patch did not completely abrogate the stimulatory effects of TPP1 in our experiments, it is possible that TPP1 may contribute to translocation efficiency and substrate dissociation in additional manners. Therefore, our current model does not discount the possibility of multiple TPP1 contributions to the activation, processivity enhancement, and regulation of telomerase. Further elucidation of the contributions and mechanism by which TPP1 contributes to telomerase action is merited.
Deficiencies in telomerase recruitment lead to telomere-shortening disorders
Mutations in hTERT that result in telomerase recruitment defects are thought to cause telomere shortening diseases. The IPF-associated mutation in hTERT V144M [57] has deficiencies in RAP stimulation by wild-type TPP1-POT1 [40], as well as in vivo telomere localization [37]. Recently a mutation in the TEL-patch of TPP1 was shown to associate with aplastic anemia and with Hoyeraal- Hreidarsson syndrome (a severe form of DC) [58, 59]. Patients with ΔK170 TPP1 allele had significantly reduced telomere lengths; furthermore, ΔK170 TPP1 reduced telomerase recruitment in cell-based assays and reduced RAP stimulation in direct telomerase extension assays [58, 59]. These studies strongly suggest that telomerase recruitment is specifically compromised by deletion of K170 in the TEL-patch. K170 is immediately adjacent to the mutated residues in TPP1 used in this study. Based on our data, it is tempting to speculate that K170 disrupts the TPP1-hTERT interface to increase telomerase dissociation during synthesis, in addition to the previously shown recruitment defects [58, 59].
In summary, we have shown that the TEL-patch amino acid cluster of TPP1 stabilizes telomerase on telomeric DNA throughout the catalytic cycle. We have developed a novel telomerase competition assay and demonstrate that the telomerase interacts directly with the TEL-patch during synthesis of the first repeat on telomeric DNA substrates. Our in vitro competition assay presents a facile way to directly test the impacts of new TPP1 mutants, identify critical residues at the POT1-TPP1 interface, or test the impacts of additional recruitment factors. We also show that the TEL-patch increases the rate of product formation, improves translocation efficiency, and reduces telomeric DNA dissociation during active telomerase synthesis. Thus, TPP1 makes multiple contributions to telomerase action. Quantitative studies of the TEL-patch-dependent recruitment of telomerase will contribute to further understanding of molecular origins of disease-associated alleles and might be applied in development of new molecular therapies.
Materials and Methods
Super telomerase extract preparation
In order to overexpress and assemble human telomerase, plasmids encoding hTR (pBS-U1-hTR) and N-terminal HA-tagged hTERT (pVan145) were co-transfected into HEK 293T cells. The wild-type telomerase plasmids were a generous gift of J. Lingner (EPFL, Lausanne)[60]. Mutant hTERT plasmids were generated by quick-change mutagenesis (Agilent). Whole cell extracts were prepared after two days of transient transfection using a CHAPS lysis buffer [61]; extracts were snap frozen in liquid nitrogen and stored at -80°C.
Telomerase was isolated from whole cell extracts with slight modifications to a previous purification protocol [60]. In short, 200 IgG Sepharose 6 Fastflow beads (GE Healthcare) were equilibrated in 30 mL of buffer A lacking 1 mM DTT. Buffer A was comprised of 20 mM HEPES- KOH pH 7.9, 2 mM MgCl2, 300 mM KCl, 10 % glycerol (v/v), 1 mM DTT, 1 mM EDTA, 0.1 % Triton X-100 (v/v), 1 mM PMSF. The ionic strength of the extract was adjusted by diluting the extract in an equal volume of 2× buffer A. Telomerase was captured by nutating at 4°C for approximately 4 h. The Sepharose beads were then washed in 50 mL of buffer A. Telomerase was cleaved from the IgG beads with 50 U of AcTEV protease (Life Technologies), in the presence of 200 U of RNasin Plus (Promega), overnight while nutating at 4°C. Following elution, telomerase was frozen in liquid nitrogen and stored at -80°C for future use.
Protein purification
Recombinant human wild-type and mutant TPP1-N constructs, comprised of the OB and POT1 binding domains, were overexpressed in BL21-DE3 cells and purified as previously described [36]. Recombinant human full length POT1 was overexpressed and purified from insect cells as previously described [62]. Protein concentrations were determined by absorbance at 280 nm, and corrections for % active protein were made.
Native gel shift assays
Binding assays were performed as described earlier [62]. POT1, wild-type TPP1-POT1 or E169A;E171A TPP1-POT1 protein complexes were added to 5′-32P-labelled DNA primer a5TT (5′ TTAGGGTTAGCGTTAGGGTT 3′) and incubated for 30 min at room temperature. Binding was carried out in buffer containing 50 mM Tris–HCl pH 8.0, 100 mM NaCl, and 1 mM DTT. Reactions were then loaded onto a native 1× TBE 8–20% polyacrylamide gradient gel (Life Technologies), run in the cold room (4°C) at 200 V for 1 h and dried.
Telomerase full extension assay
Telomerase extension assays were carried out with minor modifications [52]. Reactions were carried out in 1× reaction buffer: 50 mM Tris pH 8.0, 30 mM KCL, 0.1 to 1 mM MgCl2, 1 mM spermidine, and 5 mM β-mercaptoethanol. Final nucleotide concentrations in the assay were 500 μM dATP, dTTP, and 3.23 μM dGTP. The final dGTP concentration included 0.33 μM α- 32P-dGTP (Perkin Elmer). Reactions were initiated by addition of S4A5TT primer (5′(TTTGGC)4TTAGGGTTAGCGTTAGGGTT 3′, 100 nM final), and extension was allowed to proceed for 120 min at 4°C. Reactions were stopped in five volumes of a solution containing 3.6 M ammonium acetate, 20 μg of glycogen, and a 5′ 32P-labled oligo loading control. Reactions were then precipitated with 0.5 mL of 100% ethanol and then washed one time with 1 mL 70% ethanol. The reaction products were then resuspended in equal parts H2O and 2× formamide loading dye (0.5× TBE, 93.5% Formamide, 30 mM EDTA, 0.5% xylene cyanol and bromophenol blue). Telomerase extension products were electrophoresed on a 10% agarose denaturing gel (1× TBE, 7 M urea) at a constant power of 90 watts for approximately 1.5 h. Gels were visualized by phosphoimagery and then quantitated using ImageQuant TL (GE Healthcare).
Translocation assays
Translocation assays were carried out as previously described [42], with minor modifications. POT1 and TPP1 were incubated with 100 nM S4A5TT primer for 30 min at room temperature. TPP1- POT1 bound primers were then bound to telomerase in 1× reaction buffer, lacking dNTPs, for 20 min at 4°C. At time zero, the reactions were initiated by the simultaneous addition of dNTPs and a competitive 3′ phosphorylated chase primer at a final concentration of 1 μM. The chase primer was included to prevent telomerase from re-engaging after substrate dissociation [42]. Reactions contained 100 nM primer, 250 nM POT1, 125 nM TPP1, 50 mM Tris pH 8.0, 30 mM KCL, 0.1 mM MgCl2, 1 mM spermidine, and 5 mM β-mercaptoethanol. Final nucleotide concentrations in the assay were 500 μM dATP and 3.23 μM dGTP; dTTP was omitted from the reaction. The final dGTP concentration included 0.33 μM α-32P-dGTP. Reactions were carried out at 4°C. Reactions were stopped by removing aliquots of the reaction at various time points and quenching in stop solution described above for full extension assays. The fraction translocated was calculated by summing the counts (C) in the +3 and +4 bands, and dividing by the total counts; i.e., fraction translocated = (C+3 + C+4)/(C+2 + C+3 + C+4). The fraction of translocated product was plotted as a function of time and fit to a single exponential using SigmaPlot (Systat).
Competition assays
Competition assays were performed by simultaneously initiating a telomerase reaction with two substrates, in this case two primers of different length. Primer 1 (S4A5TT) in 1× reaction buffer was incubated in with POT1 and TPP1 (either wild-type or mutant TPP1) for 30 min at room temperature, followed by 20 min at 4°C. Primer 2 (S3A5TT = 5′-(TTTGGC)3TTAGGGTTAGCGTTAGGGTT-3′) was incubated under identical conditions in a separate tube with POT1 and either mutant or wild-type TPP1. While primers were complexed with TPP1-POT1, reactions containing telomerase, buffer, and dNTPs were assembled and cooled to 4°C for 20 min. Reactions were initiated by simultaneously adding primer 1 and primer 2 to the telomerase in buffer and nucleotides at 4°C. Competition reactions were stopped by removing aliquots of the reaction at various time points and quenching in the stop solution described above for full extension assays. Competition assays were carried under the buffer and nucleotide conditions as described for the translocation assay. The final concentration of each primer was 100 nM, 500 nM POT1, and 250 nM wild-type TPP1 and 250 nM mutant TPP1. For competitions carried out at lower primer concentrations, the POT1-TPP1 concentrations were also lowered but the ratio of protein to primer remained constant (i.e., 2.5 fold excess of POT1 and 1.25 fold excess of TPP1 over total oligo concentration). Total counts (TC), at each time point, for both primers were normalized to the loading control and expressed as fractions of the product formed for primer complexed with wild-type TPP1- POT1 at the final time point - e.g. Normalized product formation at time n for primer with EE TPP1 = ((TCprimerEE(t = n)/Cload control))\((TCprimerWT(t = 12 min)/Cload control)). The initial velocities were calculated by plotting the normalized product formation versus time, and determining the slope by linear regression. The error values reported in the text were calculated using the equation: reported error = μ(νprimerWT/νprimerEE)((σνprimerWT/μνprimerWT)2 + (σνprimerEE/μνprimerEE)2)1/2.
Extending primer dissociation rate assays
Dissociation rate assays were carried with slight modifications from those previously described [42]. Primer and 3′-phosphorylated chase primer in 1× buffer were complexed with excess TPP1-POT1 at room temperature in separate tubes. Complexed primer was then allowed to equilibrate with telomerase for five min; next cold dATP, dTTP, and dGTP were added to initiate the telomerase reaction. The reaction proceeded for five min, and at time zero a large-excess of 3′ phosphorylated chase primer was added to prevent substrate primer re-association. After chase addition, aliquots of the reaction were removed at varying time points and pulse labeled with 32P-α-dGTP for five min. Reactions stopped as described for full extension assays. Reactions were carried out at 25°C. Final reaction concentrations were 50 nM A5 primer (5′ TTAGGGTTAGCGTTAGGG 3′), 1 μM 3′ phosphorylated chase primer, 1.2 μM POT1, 1.2 μM TPP1, 50 mM Tris pH 8.0, 150 mM KCl, 1 mM MgCl2, 1 mM spermidine, and 5 mM β- mercaptoethanol. Final nucleotide concentrations in the assay were 500 μM each of dATP and dTTP, and 3.23 μM dGTP. The final dGTP concentration included 0.33 μM α-32P-dGTP. The total counts (TC) for each lane were expressed as a fraction of counts at time zero and plotted as a function of time (i.e. fraction bound = (TC(t = n)/TC(t = 0)), as previously described [42]. Data were fit to a double exponential and t1/2Apparent was taken as the time required for 50% dissociation.
Supplementary Material
Highlights.
The TEL-patch on TPP1 promotes the translocation of human telomerase
The TEL-patch of TPP1 reduces substrate dissociation from telomerase
Telomerase interacts with TPP1 to preferentially bind and extend substrates
Towards an in vitro assay for telomerase recruitment to telomeres
Acknowledgments
We thank Jayakrishnan Nandakumar and Arthur Zaug for generously providing experimental reagents as well as technical advice. We thank Ci Ji Lim for insightful discussions regarding mechanisms of telomerase recruitment and Jens Schmidt for critical feedback and reading of this manuscript. In addition we thank the members of the Cech lab for helpful discussions and suggestions. C.H. is supported by the European Regional Development Fund CZ.1.05/1.1.00/02.0068, CZ.1.07/2.3.00/30.0019 and Czech Science Foundation P205/12/0550. T. R. C. is an investigator of the HHMI. This work was supported by NIH grant R01 GM099705 to T. R. C.
Footnotes
Author contributions: A.B.D., C.H., and T.R.C. designed experiments. A.B.D. and C.H. carried out biochemical assays. A.B.D. and C.H. generated materials. A.B.D., C.H. and T.R.C. wrote the manuscript.
Competing interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–9. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 2.Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to telomeres. Nature reviews Molecular cell biology. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batista LF, Artandi SE. Understanding telomere diseases through analysis of patient-derived iPS cells. Current opinion in genetics & development. 2013;23:526–33. doi: 10.1016/j.gde.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. Journal of molecular biology. 1992;225:951–60. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- 5.Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–57. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- 6.Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Semin Cancer Biol. 2011;21:349–53. doi: 10.1016/j.semcancer.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armanios M, Blackburn EH. The telomere syndromes. Nature reviews Genetics. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holohan B, Wright WE, Shay JW. Cell biology of disease: Telomeropathies: an emerging spectrum disorder. The Journal of cell biology. 2014;205:289–99. doi: 10.1083/jcb.201401012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–7. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 10.Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–7. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–9. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 12.Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–14. doi: 10.1016/s0092-8674(00)80687-x. [DOI] [PubMed] [Google Scholar]
- 13.Tesmer VM, Ford LP, Holt SE, Frank BC, Yi X, Aisner DL, et al. Two inactive fragments of the integral RNA cooperate to assemble active telomerase with the human protein catalytic subunit (hTERT) in vitro. Molecular and cellular biology. 1999;19:6207–16. doi: 10.1128/mcb.19.9.6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurczyluk J, Nouwens AS, Holien JK, Adams TE, Lovrecz GO, Parker MW, et al. Direct involvement of the TEN domain at the active site of human telomerase. Nucleic acids research. 2011;39:1774–88. doi: 10.1093/nar/gkq1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu RA, Collins K. Human telomerase specialization for repeat synthesis by unique handling of primer-template duplex. The EMBO journal. 2014 doi: 10.1002/embj.201387205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robart AR, Collins K. Human telomerase domain interactions capture DNA for TEN domain-dependent processive elongation. Mol Cell. 2011;42:308–18. doi: 10.1016/j.molcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huard S, Moriarty TJ, Autexier C. The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic acids research. 2003;31:4059–70. doi: 10.1093/nar/gkg437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie M, Podlevsky JD, Qi X, Bley CJ, Chen JJ. A novel motif in telomerase reverse transcriptase regulates telomere repeat addition rate and processivity. Nucleic acids research. 2010;38:1982–96. doi: 10.1093/nar/gkp1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finger SN, Bryan TM. Multiple DNA-binding sites in Tetrahymena telomerase. Nucleic acids research. 2008;36:1260–72. doi: 10.1093/nar/gkm866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–5. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 21.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323:644–8. doi: 10.1126/science.1165357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jady BE, Bertrand E, Kiss T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. The Journal of cell biology. 2004;164:647–52. doi: 10.1083/jcb.200310138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jady BE, Richard P, Bertrand E, Kiss T. Cell cycle-dependent recruitment of telomerase RNA and Cajal bodies to human telomeres. Molecular biology of the cell. 2006;17:944–54. doi: 10.1091/mbc.E05-09-0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomlinson RL, Ziegler TD, Supakorndej T, Terns RM, Terns MP. Cell cycle-regulated trafficking of human telomerase to telomeres. Molecular biology of the cell. 2006;17:955–65. doi: 10.1091/mbc.E05-09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, et al. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Molecular and cellular biology. 2010;30:2971–82. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, et al. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature. 2007;445:559–62. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- 27.Xi L, Cech TR. Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic acids research. 2014 doi: 10.1093/nar/gku560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harley CB, Futcher AB, Greider CW. Telomeres Shorten during Aging of Human Fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Abreu E, Kim J, Stadler G, Eskiocak U, Terns MP, et al. Processive and Distributive Extension of Human Telomeres by Telomerase under Homeostatic and Nonequilibrium Conditions. Mol Cell. 2011;42:297–307. doi: 10.1016/j.molcel.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaug AJ, Crary SM, Jesse Fioravanti M, Campbell K, Cech TR. Many disease-associated variants of hTERT retain high telomerase enzymatic activity. Nucleic acids research. 2013;41:8969–78. doi: 10.1093/nar/gkt653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes & development. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 32.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 33.Sfeir A, de Lange T. Removal of Shelterin Reveals the Telomere End-Protection Problem. Science. 2012;336:593–7. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–8. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 35.Lei M, Zaug AJ, Podell ER, Cech TR. Switching human telomerase on and off with hPOT1 protein in vitro. The Journal of biological chemistry. 2005;280:20449–56. doi: 10.1074/jbc.M502212200. [DOI] [PubMed] [Google Scholar]
- 36.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012;492:285–9. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012;150:481–94. doi: 10.1016/j.cell.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sexton AN, Regalado SG, Lai CS, Cost GJ, O'Neil CM, Urnov FD, et al. Genetic and molecular identification of three human TPP1 functions in telomerase action: recruitment, activation, and homeostasis set point regulation. Genes & development. 2014;28:1885–99. doi: 10.1101/gad.246819.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakashima M, Nandakumar J, Sullivan KD, Espinosa JM, Cech TR. Inhibition of telomerase recruitment and cancer cell death. J Biol Chem. 2013;288:33171–80. doi: 10.1074/jbc.M113.518175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt JC, Dalby AB, Cech TR. Identification of human TERT elements necessary for telomerase recruitment to telomeres. eLife. 2014:3. doi: 10.7554/eLife.03563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, et al. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–10. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 42.Latrick CM, Cech TR. POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. The EMBO journal. 2010;29:924–33. doi: 10.1038/emboj.2009.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D'Souza Y, Chu TW, Autexier C. A translocation-defective telomerase with low levels of activity and processivity stabilizes short telomeres and confers immortalization. Molecular biology of the cell. 2013;24:1469–79. doi: 10.1091/mbc.E12-12-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi X, Xie M, Brown AF, Bley CJ, Podlevsky JD, Chen JJ. RNA/DNA hybrid binding affinity determines telomerase template-translocation efficiency. The EMBO journal. 2012;31:150–61. doi: 10.1038/emboj.2011.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fersht A. Enzyme structure and mechanism Reading Eng. San Francisco: W. H. Freeman; 1977. [Google Scholar]
- 46.Sexton AN, Youmans DT, Collins K. Specificity requirements for human telomere protein interaction with telomerase holoenzyme. J Biol Chem. 2012;287:34455–64. doi: 10.1074/jbc.M112.394767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mathews CK. DNA precursor metabolism and genomic stability. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:1300–14. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- 48.Traut TW. Physiological concentrations of purines and pyrimidines. Molecular and cellular biochemistry. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 49.Greider CW. Telomerase is processive. Molecular and cellular biology. 1991;11:4572–80. doi: 10.1128/mcb.11.9.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berman AJ, Akiyama BM, Stone MD, Cech TR. The RNA accordion model for template positioning by telomerase RNA during telomeric DNA synthesis. Nature structural & molecular biology. 2011;18:1371–5. doi: 10.1038/nsmb.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller MC, Collins K. Telomerase recognizes its template by using an adjacent RNA motif. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6585–90. doi: 10.1073/pnas.102024699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaug AJ, Podell ER, Nandakumar J, Cech TR. Functional interaction between telomere protein TPP1 and telomerase. Genes & development. 2010;24:613–22. doi: 10.1101/gad.1881810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bryan TM, Goodrich KJ, Cech TR. A mutant of Tetrahymena telomerase reverse transcriptase with increased processivity. J Biol Chem. 2000;275:24199–207. doi: 10.1074/jbc.M003246200. [DOI] [PubMed] [Google Scholar]
- 54.Takai KK, Hooper S, Blackwood S, Gandhi R, de Lange T. In vivo stoichiometry of shelterin components. J Biol Chem. 2010;285:1457–67. doi: 10.1074/jbc.M109.038026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajavel M, Mullins MR, Taylor DJ. Multiple facets of TPP1 in telomere maintenance. Biochimica et biophysica acta. 2014;1844:1550–9. doi: 10.1016/j.bbapap.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang H, Buncher N, Opresko PL, Myong S. POT1-TPP1 regulates telomeric overhang structural dynamics. Structure. 2012;20:1872–80. doi: 10.1016/j.str.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7552–7. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo Y, Kartawinata M, Li J, Pickett HA, Teo J, Kilo T, et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014 doi: 10.1182/blood-2014-08-596445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kocak H, Ballew BJ, Bisht K, Eggebeen R, Hicks BD, Suman S, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes & development. 2014 doi: 10.1101/gad.248567.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sauerwald A, Sandin S, Cristofari G, Scheres SH, Lingner J, Rhodes D. Structure of active dimeric human telomerase. Nature structural & molecular biology. 2013;20:454–60. doi: 10.1038/nsmb.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cristofari G, Lingner J. Telomere length homeostasis requires that telomerase levels are limiting. The EMBO journal. 2006;25:565–74. doi: 10.1038/sj.emboj.7600952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nature structural & molecular biology. 2004;11:1223–9. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.