

Abstract
Background
Between- and within-person variation in DNA methylation levels are important parameters to be considered in epigenome-wide association studies. Temporal change is one source of within-person variation in DNA methylation that has been linked to aging and disease.
Methods
We analyzed CpG-site-specific intra-individual variation and short-term temporal trend in leukocyte DNA methylation among 24 healthy Chinese women, with blood samples drawn at study entry and after 9 months. Illumina HumanMethylation450 BeadChip was used to measure methylation. Intraclass correlation coefficients (ICC) and trend estimates were summarized by genomic location and probe type.
Results
The median ICC was 0.36 across nonsex chromosomes and 0.80 on the X chromosome. There was little difference in ICC profiles by genomic region and probe type. Among CpG loci with high variability between participants, over 99% had ICC > 0.8. Statistically significant trend was observed in 10.9% CpG loci before adjustment for cell type composition and in 3.4% loci after adjustment.
Conclusions
For CpG loci differentially methylated across subjects, methylation levels can be reliably assessed with one blood sample. More samples per subject are needed for low-variability and unmethylated loci. Temporal changes are largely driven by changes in cell type composition of blood samples, but temporal trend unrelated to cell types is detected in a small percentage of CpG sites.
Keywords: methylation, intra-individual variation, epigenetic drift, effect attenuation
Introduction
Epigenetic modifications such as DNA methylation of cytosine residues at CpG dinucleotides have been extensively examined for their potential association with disease and aging in humans. DNA methylation marks are stable indicators of tissue lineage, thus different tissues have fundamentally different methylation patterns. DNA methylation profile is established at neonatal stage and undergoes rapid change at the early age and further modifications throughout lifespan. Changes of methylation patterns have been seen in normal human aging process and aberrant methylation changes in tissues such as blood have been linked to a number of diseases including cancer (1–5). A link has also been established between age-related and cancer-related methylation changes. Important cancer related genes become hypermethylated during aging, including key developmental genes and those encoding the estrogen receptor, insulin growth factor and E-cadherin (1, 6). Further, the acquisition of methylation changes during aging could underlie the development of age-related pathological conditions (7).
Research into the role of DNA methylation in disease is affected by variability in measured methylation levels, which can be attributed to a number of sources: differences among study participants due to demographic, environmental and genetic factors; random and systematic variation between samples taken from the same individual, which may result from differences in tissue sample composition or timing of sample collection; technical variability due to measurement error and limited precision of analytic tools. The relative proportion of between- and within-person variability in the total variability is typically described by the intraclass correlation coefficient (ICC). The ICC varies between 0 and 1, with high (low) proportion of within-person variation represented by values close to 0 (respectively, 1). Low values of ICC signify lower data reliability and may affect validity of analysis results. A number of studies (8–11) have analyzed within-person variability of different blood biomarkers; however, we are unaware of any reports on that in DNA methylation, although the importance of this aspect for epigenome-wide association studies has been recognized (12).
In the present study of leukocyte DNA methylation among 24 healthy women, we examined CpG-site-specific intra-individual variation in DNA methylation and analyze short-term temporal changes in DNA methylation using Illumina HumanMethylation450 BeadChip (13). This array has been reported to have low technical variability and high reproducibility (13), although a number of issues have been pointed out (14). The BeadChip contains 485,577 loci that include 482,421 (99.35%) CpG dinucleotides, 3,091 (0.64%) CNG targets, and 65 (0.01%) single nucleotide polymorphism (SNP) sites. The array covers 96% of known CpG islands (CGI) and 99% of NCBI Reference Sequence (RefSeq) genes, with an average of 17 CpG sites per gene distributed across the upstream of the transcription start sites (TSS)1500, TSS200, 5′ untranslated regions (UTR), first exon, gene body, and the 3′UTR. Because the function of DNA methylation seems to vary by functional location and by proximity to CGIs (15), we also sought to describe the distribution of high-low variability CpG sites across functional locations in the genome, and identify whether such changes are more likely to occur in specific types of genomic regions. To assess a potential effect of X chromosome inactivation on methylation variability, separate analyses were conducted for X chromosome CpG sites.
Materials and Methods
Study population and sample collection
Study participants were drawn from the Shanghai Physical Activity (SPA) study subcohort of the Shanghai Women’s Health Study (SWHS). SWHS a prospective cohort study of 74,943 women aged 40–70 years who were recruited from 7 communities in Shanghai, China, between 1997 and 2000 (16). SPA subcohort of SWHS comprises 300 women from two communities (17), who provided blood samples at study entry and after 9 months. Twenty-four women from the SPA subcohort were randomly selected for the present analysis.
For each consented participant, a 10-ml blood sample was drawn into an EDTA vacutainer tube and kept in a portable Styrofoam box with ice packs at 0–4°C. Within 6 hours of collection, samples were processed and separated into 2-ml aliquots of plasma (4), buffy coat (2), and red blood cells (2). Samples were then stored at −80°C until DNA extraction.
Laboratory analysis
DNA extraction and bisulfite conversion
DNA was extracted from buffy coats using a QIAamp® DNA Blood Mini Kit (Qiagen Inc, Valencia, CA). The quality and quantity of extracted DNA was examined by NanoDrop-2000 spectrometer (Thermo Scientific, Wilmington, DE). Bisulfite conversion of 500 ng of DNA was performed on each sample according to manufacturer’s recommendations for the HumanMethylation450 BeadChip using the EZ DNA Methylation kit (Zymo Research, Irvine, CA). The treatment protocol included 16 cycles of denaturing at 95°C for 30 sec, incubation at 50°C for 60 min, and holding at 4°C for a minimum of 2 hours.
Illumina methylation platform
Four μl of bisulfite-converted DNA was hybridized onto and analyzed using HumanMethylation450 BeadChip (Illumina, San Diego, CA). Hybridization protocol consisted of a whole genome amplification step followed by enzymatic endpoint fragmentation, precipitation and resuspension. Resuspended samples were hybridized onto the BeadChip for 16 hours at 48°C. After hybridization, unhybridized and non-specifically hybridized DNA was washed away, followed by single nucleotide extension using the hybridized bisulfite-treated DNA as a template. The Illumina iScan SQ scanner was used to create images of the single arrays. Image intensities were extracted using GenomeStudio (v.2011.1) Methylation module (v.1.9.0) software.
Data quality assessment and pre-processing
Data normalization was performed in GenomeStudio using ‘Background Subtraction’ and ‘Normalization to Internal Controls’ methods and has been described elsewhere (18). Briefly, background subtraction values were derived from built-in negative control bead signals and subtracted from probe intensities. Normalization was performed using internal control probe pairs designed to target the same region within housekeeping genes. The methylation score for each CpG site was represented by a β-value, calculated according to normalized probe fluorescence intensity ratios between methylated and unmethylated signals and varying between 0 (fully unmethylated) and 1 (fully methylated). Data quality control (QC) analyses performed on β-values included principal component analysis, to assess potential batch effects, and sample histograms for signal distributions (data not shown). Probes with detection p-value above 0.05 were excluded.
Statistical analysis
Statistical analyses were performed using methylation levels represented by M-values, computed as logit of β-values: M = log2(β/(1-β)). Because the distribution of M-values is closer to normality, they are widely used as a measure of DNA methylation in association studies (19). For every CpG locus, we estimated variance components that correspond to within-person and between-person variation using a mixed model with study participants as random effects:
ICCs were then computed as . To summarize patterns of variability by functional genomic location, by type of probe and for known differentially methylated regions (DMR), we computed the percentage of CpG loci with high (>0.8), midrange (0.5 – 0.8) and low (<0.5) ICC for each of these groups. The estimated between- and within-person variance components were converted to the β-scale as follows:
where is the mean M-value across all study subjects. For 14,248 CpG loci with very low mean methylation, ICCs could not be estimated and were set to 0, as the worst possible case. The short-term temporal trend in DNA methylation across all CpG loci was examined using a mixed model with time since study entry (years) as a fixed effect, and study participants as random effects:
To assess the influence of quantile normalization on ICC and trend, all models were fit with untransformed and quantile normalized M-values. All analyses were adjusted for cell type composition using the cell mixture deconvolution method of Houseman et al. (20). which establishes DNA methylation signature panels for each cell type and uses constrained optimization to map methylation profiles of interest onto the signature panel and to predict proportions of cell types in a blood sample.
A number of recent reports define meaningful difference (14) in methylation as Δβ value (a measure of average difference in methylation level at a particular CpG site between study subjects) being above a certain threshold. A threshold of 0.2 on the β-scale is commonly used. To assess the impact of intra-individual variation on such high-variance CpG loci, we summarized ICC and trend by genomic location and probe type for high-variance (Δβ ≥ 0.2), mid-variance (0.1 ≤ Δβ < 0.2) and low-variance loci (Δβ < 0.1). We estimated the Δβ value for every CpG site as twice the between-person standard error: . All analyses were conducted using R 3.0.3 software and Bioconductor package nlme 3.1–117.
Results
The median age at study entry among the 24 study participants was 54.5 years (range: 46.5 – 68.8 years). Across the 483,880 CpG loci that passed data QC, the ICCs ranged from 0 to 0.999, with a median of 0.37. For the majority of loci, the within-person variance component tended to be smaller than and uncorrelated with the between-person component (Figure 1). The ranges of and were 0 – 0.42 and 0 – 0.36, respectively.
Figure 1.
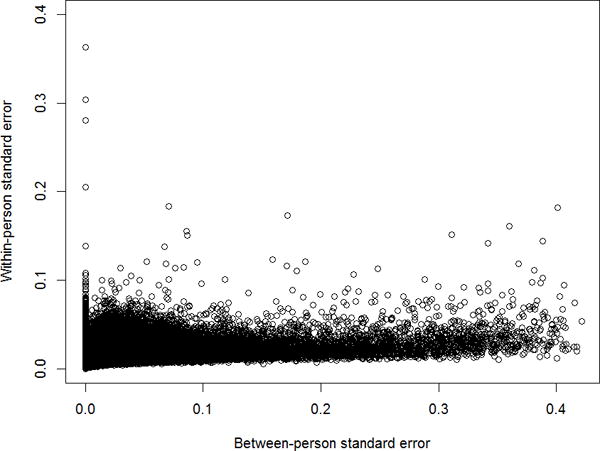
Scatter plot of within- vs. between-person standard error (β-scale), all CpG sites.
Table 1 lists proportions of low, mid and high-ICC loci by genomic location, probe type and locus variability for nonsex chromosomes. The median ICC across all CpG loci was 0.36 (interquartile range (IQR): 0.13 – 0.63). Over 64% loci had low ICC, while 23% had mid-range ICC, and 13% had high ICC. Among the low-variability loci, the proportions of low and mid-range ICC were comparable, while only 8.1% loci exhibited high ICC. On the other hand, over 90% of moderate-variability loci (0.1 ≤ Δβ < 0.2) and over 99% of high-variability loci (Δβ ≥ 0.2) had high ICC, while <1% loci in these two groups had low ICC. This latter result was observed across all genomic locations.
Table 1.
Summary of intraclass correlation coefficients (β-scale) of CpG locus methylation, by genomic location and probe type, nonsex chromosomes.1
| Δβ < 0.1
|
0.1 ≤ Δβ < 0.2
|
Δβ ≥ 0.2
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location/probe type | ICC
|
Percent loci by ICC range
|
Percent loci by ICC range
|
Percent loci by ICC range
|
Percent loci by ICC range
|
||||||||||||||
| N | Median | IQR | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | ||
| All CpG loci | 472665 | 0.36 | 0.13 | 0.63 | 64.1 | 23.0 | 12.9 | 446677 | 67.8 | 24.1 | 8.1 | 20851 | 0.2 | 5.6 | 94.2 | 5137 | 0.0 | 0.4 | 99.5 |
| Enhancer | 101663 | 0.39 | 0.15 | 0.66 | 61.1 | 24.4 | 14.5 | 95364 | 65.1 | 25.7 | 9.2 | 5110 | 0.2 | 5.7 | 94.1 | 1189 | 0.0 | 0.4 | 99.6 |
| Known DMR2 | 36053 | 0.49 | 0.24 | 0.72 | 51.5 | 31.8 | 16.7 | 33802 | 54.9 | 33.6 | 11.5 | 1965 | 0.1 | 5.4 | 94.5 | 286 | 0.0 | 0.7 | 99.3 |
| Location with respect to CpG island: | |||||||||||||||||||
| CpG Island | 145571 | 0.34 | 0.12 | 0.59 | 67.6 | 21.8 | 10.6 | 139254 | 70.7 | 22.7 | 6.6 | 5157 | 0.2 | 3.0 | 96.8 | 1160 | 0.0 | 0.6 | 99.4 |
| Shore3 | 108839 | 0.43 | 0.18 | 0.71 | 57.2 | 25.4 | 17.4 | 101091 | 61.6 | 26.9 | 11.5 | 6623 | 0.2 | 6.2 | 93.6 | 1125 | 0.0 | 0.4 | 99.6 |
| Shelf3 | 45790 | 0.34 | 0.12 | 0.59 | 67.1 | 22.1 | 10.7 | 43614 | 70.5 | 22.9 | 6.6 | 1591 | 0.4 | 8.7 | 90.9 | 585 | 0.0 | 0.0 | 100.0 |
| Functional location: | |||||||||||||||||||
| TSS1500 | 81267 | 0.36 | 0.13 | 0.62 | 64.5 | 23.1 | 12.4 | 77265 | 67.8 | 24.0 | 8.2 | 3433 | 0.2 | 7.0 | 92.8 | 569 | 0.0 | 0.5 | 99.5 |
| TSS200 | 60005 | 0.29 | 0.09 | 0.53 | 73.0 | 19.1 | 7.8 | 58235 | 75.3 | 19.6 | 5.2 | 1464 | 0.2 | 6.1 | 93.7 | 306 | 0.0 | 1.3 | 98.7 |
| 5′UTR | 62975 | 0.32 | 0.11 | 0.56 | 69.5 | 21.6 | 8.9 | 60820 | 72.0 | 22.2 | 5.9 | 1794 | 0.1 | 6.4 | 93.6 | 361 | 0.0 | 0.6 | 99.4 |
| 1st Exon | 37656 | 0.33 | 0.11 | 0.57 | 69.4 | 21.4 | 9.2 | 36406 | 71.8 | 22.0 | 6.2 | 1079 | 0.2 | 3.6 | 96.2 | 171 | 0.0 | 0.6 | 99.4 |
| Body | 172068 | 0.35 | 0.13 | 0.60 | 65.8 | 22.4 | 11.8 | 163309 | 69.3 | 23.3 | 7.3 | 6813 | 0.2 | 5.4 | 94.3 | 1946 | 0.1 | 0.4 | 99.6 |
| 3′UTR | 19174 | 0.32 | 0.10 | 0.56 | 69.4 | 20.8 | 9.8 | 18381 | 72.4 | 21.5 | 6.2 | 603 | 0.8 | 7.1 | 92.0 | 190 | 0.0 | 0.0 | 100.0 |
| Location by probe type: | |||||||||||||||||||
| Infinium I | 132039 | 0.37 | 0.13 | 0.62 | 64.0 | 24.7 | 11.3 | 124892 | 67.6 | 25.8 | 6.6 | 5640 | 0.4 | 6.9 | 92.7 | 1507 | 0.1 | 0.9 | 99.1 |
| Infinium II | 340626 | 0.36 | 0.13 | 0.63 | 64.1 | 22.3 | 13.5 | 321785 | 67.9 | 23.4 | 8.7 | 15211 | 0.2 | 5.1 | 94.7 | 3630 | 0.0 | 0.3 | 99.7 |
All estimates are adjusted for cell-type composition. Δβ is estimated as twice the between-person standard error (β-scale).
DMR: differentially methylated region; TSS: transcription start site; UTR: untranslated region.
Shores: up to 2 kb from CpG islands; shelves: from 2 to 4 kb from CpG islands.
Among the CpG loci in known DMR (13), ICCs tended to be higher than across the entire genome, with median 0.49 (IQR: 0.24 – 0.72), and 51.5%, 31.8% and 16.7% loci exhibiting low, mid-range and high ICC, respectively. Across loci from CGIs, shores and shelves, the median ICC ranged from 0.34 to 0.43, with the proportion of high-ICC loci highest in CpG shores (overall and among low-variability sites) and CGIs (moderate-variability sites). The ICC profiles across different functional locations were similar, with median ICC between 0.29 and 0.36, and the percentage of high-ICC loci 7.8% – 12.4% overall, 5.2% – 8.2% among low-variability sites and 92.0% – 96.2% among moderate-variability sites. There was also little difference in ICC profiles by probe type. CpG loci with methylation measured by Infinium I probes, compared to Infinium II probes, had lower proportion of high-ICC loci overall (11.3% vs. 13.5%), among low-variability sites (6.6% vs. 8.7%) and among moderate-variability sites (92.7% vs. 94.7%).
The distribution of ICCs on the X chromosome (Table 2) differed substantially from that on nonsex chromosomes, with the median ICC of 0.80 (IQR: 0.60–0.89) overall and nearly half of the X chromosome CpG loci having high ICC. The main difference from nonsex chromosomes in the distribution of ICCs was found among low-variability sites, where 27.8–51.1% loci had high ICC. Of all genomic locations, lineage-defining DMRs appeared the most stable, with 61.6% loci having high ICC. CGIs and shores and functional locations on the promoter side of gene coding regions contained more high-ICC loci than CGI shelves, gene body and 3′UTR locations. The proportion of high-ICC loci measured by Infinium II probes was somewhat higher than that among Infinium I probes. ICC estimates without cell type composition adjustment were very similar (Supplementary Tables S1–S2).
Table 2.
Summary of intraclass correlation coefficients (β-scale) of CpG locus methylation, by genomic location and probe type, X chromosome.1
| Δβ < 0.1
|
0.1 ≤ Δβ < 0.2
|
Δβ ≥ 0.2
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location/probe type | ICC
|
Percent loci by ICC range
|
Percent loci by ICC range
|
Percent loci by ICC range
|
Percent loci by ICC range
|
||||||||||||||
| N | Median | IQR | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | N | 0 – 0.5 | 0.5 – 0.8 | 0.8 – 1 | ||
| All CpG loci | 11215 | 0.80 | 0.60 | 0.89 | 18.5 | 31.8 | 49.7 | 9014 | 23.0 | 38.4 | 38.6 | 2161 | 0.1 | 5.0 | 95.0 | 40 | 0.0 | 2.5 | 97.5 |
| Enhancer | 629 | 0.84 | 0.71 | 0.91 | 9.9 | 31.5 | 58.7 | 478 | 13.0 | 40.2 | 46.9 | 149 | 0.0 | 4.0 | 96.0 | 2 | 0.0 | 0.0 | 100.0 |
| Known DMR2 | 1231 | 0.85 | 0.72 | 0.91 | 10.6 | 27.8 | 61.6 | 936 | 14.0 | 34.9 | 51.1 | 290 | 0.0 | 5.2 | 94.8 | 5 | 0.0 | 0.0 | 100.0 |
| Location with respect to CpG island: | |||||||||||||||||||
| CpG Island | 4272 | 0.82 | 0.67 | 0.90 | 13.2 | 32.0 | 54.8 | 3391 | 16.6 | 38.9 | 44.4 | 867 | 0.0 | 5.2 | 94.8 | 14 | 0.0 | 0.0 | 100.0 |
| Shore3 | 2837 | 0.82 | 0.63 | 0.90 | 17.3 | 29.7 | 53.0 | 2176 | 22.5 | 37.2 | 40.3 | 649 | 0.3 | 5.1 | 94.6 | 12 | 0.0 | 0.0 | 100.0 |
| Shelf3 | 1138 | 0.71 | 0.46 | 0.85 | 28.0 | 36.3 | 35.7 | 1003 | 31.8 | 40.4 | 27.8 | 130 | 0.0 | 6.2 | 93.8 | 5 | 0.0 | 0.0 | 100.0 |
| Functional location: | |||||||||||||||||||
| TSS1500 | 2777 | 0.80 | 0.60 | 0.90 | 18.5 | 31.3 | 50.2 | 2211 | 23.2 | 38.4 | 38.4 | 555 | 0.0 | 3.4 | 96.6 | 11 | 0.0 | 0.0 | 100.0 |
| TSS200 | 2441 | 0.82 | 0.67 | 0.90 | 13.5 | 31.4 | 55.1 | 1866 | 17.5 | 39.3 | 43.2 | 564 | 0.4 | 5.9 | 93.8 | 11 | 0.0 | 9.1 | 90.9 |
| 5′UTR | 2401 | 0.82 | 0.67 | 0.90 | 13.2 | 32.1 | 54.7 | 1873 | 16.9 | 39.6 | 43.5 | 522 | 0.0 | 5.6 | 94.4 | 6 | 0.0 | 0.0 | 100.0 |
| 1st Exon | 1635 | 0.83 | 0.67 | 0.90 | 12.3 | 32.9 | 54.8 | 1293 | 15.5 | 40.7 | 43.8 | 340 | 0.0 | 3.5 | 96.5 | 2 | 0.0 | 0.0 | 100.0 |
| Body | 2893 | 0.76 | 0.52 | 0.88 | 23.3 | 32.5 | 44.2 | 2407 | 28.0 | 38.3 | 33.7 | 481 | 0.0 | 3.7 | 96.3 | 5 | 0.0 | 0.0 | 100.0 |
| 3′UTR | 513 | 0.71 | 0.44 | 0.84 | 28.7 | 37.4 | 33.9 | 466 | 31.5 | 40.1 | 28.3 | 46 | 0.0 | 10.9 | 89.1 | 1 | 0.0 | 0.0 | 100.0 |
| Location by probe type: | |||||||||||||||||||
| Infinium I | 3042 | 0.78 | 0.60 | 0.87 | 16.8 | 38.3 | 44.8 | 2444 | 20.9 | 45.7 | 33.4 | 583 | 0.2 | 8.4 | 91.4 | 15 | 0.0 | 6.7 | 93.3 |
| Infinium II | 8173 | 0.81 | 0.60 | 0.90 | 19.1 | 29.4 | 51.5 | 6570 | 23.7 | 35.7 | 40.6 | 1578 | 0.1 | 3.7 | 96.3 | 25 | 0.0 | 0.0 | 100.0 |
All estimates are adjusted for cell-type composition. Δβ is estimated as twice the between-person standard error (β-scale).
DMR: differentially methylated region; TSS: transcription start site; UTR: untranslated region.
Shores: up to 2 kb from CpG islands; shelves: from 2 to 4 kb from CpG islands.
A summary of temporal trend estimates by genomic location, probe type and locus variability is presented in Table 3. Less than 4% CpG loci exhibited a statistically significant trend overall and for most functional locations, with the exception of 5.3% loci with trend for 3′UTR on the X chromosome. There was little difference in the proportion of CpG sites with temporal trend between low-, mid- and high-variability locus groups (data not shown). The proportion of sites with negative trend was somewhat higher in most functional locations. The proportion of loci with positive trend among Infinium II sites on the X chromosome was about 1.5 times that for Infinium I sites. Without adjustment for cell type composition, the proportion of loci with significant negative trend was several times higher across functional locations, 8–17.5% on nonsex chromosomes and 5.2–9.4% on the X chromosome (Supplementary Table S3). We estimated the effect of quantile normalization transformation on ICC profiles and trend estimates of CpG loci. Across all genomic locations and probe types, the application of quantile normalization to DNA methylation data had no noticeable effect on the proportion of high-ICC loci, but lowered the proportion of mid-ICC loci and, correspondingly, increased the proportion of low-ICC loci (Figure 2A). This effect was observed in both nonsex chromosomes and the X chromosome (data not shown). In temporal trend analysis, quantile normalization had the effect of equalizing the proportions with negative and positive trend, and an overall increase in the proportion of loci with trend (Figure 2B).
Table 3.
Summary of temporal trend (β-scale) in CpG site methylation, by genomic location and probe type.1
| Nonsex chromosomes
|
X chromosome
|
|||||
|---|---|---|---|---|---|---|
| Percent loci with significant trend
|
Percent loci with significant trend
|
|||||
| Location/probe type | N | Negative trend | Positive trend | N | Negative trend | Positive trend |
| All CpG loci | 472665 | 1.86 | 1.54 | 11215 | 1.84 | 1.64 |
| Enhancer | 101663 | 1.83 | 1.79 | 629 | 1.75 | 1.91 |
| Known DMR2 | 36053 | 1.91 | 1.29 | 1231 | 1.54 | 1.62 |
| Location with respect to CpG island: | ||||||
| CpG Island | 145571 | 1.91 | 1.31 | 4272 | 2.01 | 1.17 |
| Shore3 | 108839 | 1.93 | 1.50 | 2837 | 1.97 | 1.55 |
| Shelf3 | 45790 | 1.86 | 1.70 | 1138 | 1.85 | 1.76 |
| Functional location: | ||||||
| TSS1500 | 81267 | 1.94 | 1.44 | 2777 | 2.02 | 1.76 |
| TSS200 | 60005 | 1.88 | 1.38 | 2441 | 2.17 | 1.11 |
| 5′UTR | 62975 | 1.88 | 1.42 | 2401 | 2.25 | 1.71 |
| 1st Exon | 37656 | 1.93 | 1.34 | 1635 | 2.32 | 1.59 |
| Body | 172068 | 1.86 | 1.58 | 2893 | 1.69 | 1.56 |
| 3′UTR | 19174 | 1.83 | 1.60 | 513 | 1.95 | 3.31 |
| Location by probe type: | ||||||
| Infinium I | 132039 | 1.55 | 1.59 | 3042 | 1.94 | 1.15 |
| Infinium II | 340626 | 1.99 | 1.52 | 8173 | 1.80 | 1.82 |
All estimates are adjusted for cell-type composition. Δβ is estimated as twice the between-person standard error (β-scale).
DMR: differentially methylated region; TSS: transcription start site; UTR: untranslated region.
Shores: up to 2 kb from CpG islands; shelves: from 2 to 4 kb from CpG islands.
Figure 2.
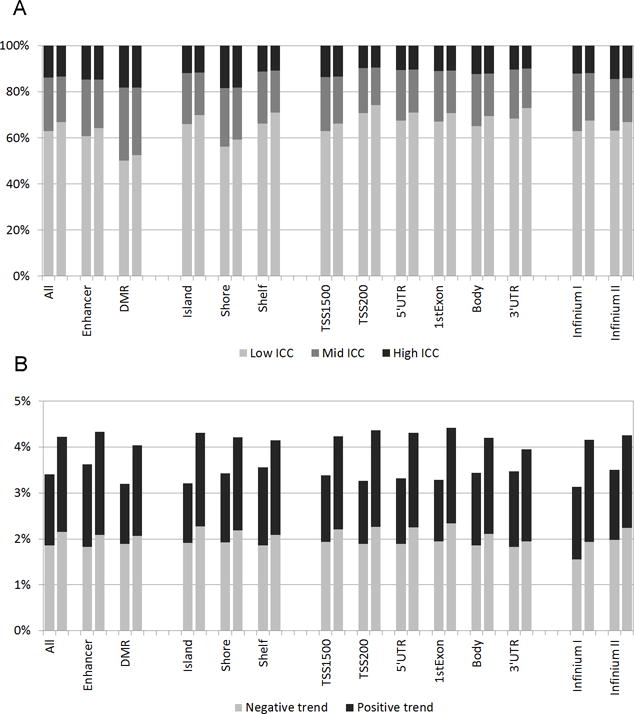
The effect of quantile normalization on ICC distribution and trend estimates, all CpG sites. (A) High, mid and low ICC distribution for untransformed and quantile-normalized methylation levels, by genomic location and probe type. (B) Proportion of CpG loci with significant temporal trend for untransformed and quantile-normalized methylation levels, by genomic location and probe type. All estimates are adjusted for cell type composition. DMR: differentially methylated region; TSS: transcription start site; UTR: untranslated region.
Figure 3 presents a comparison of temporal trend estimates from untransformed and quantile-normalized models, with and without cell type adjustment. The application of quantile normalization alone resulted in the reduction of the temporal effect magnitude, consistent across most loci (Figure 3A). Cell type adjustment alone left temporal effects nearly unaffected (Figure 3B). Compared with cell-type adjusted model, quantile normalization resulted in inconsistent changes in the temporal effect magnitude, with larger changes for some loci and smaller changes for others (Figure 3C,D). Neither cell type adjustment nor quantile normalization, alone or combined, changed the direction of the observed temporal effects.
Figure 3.
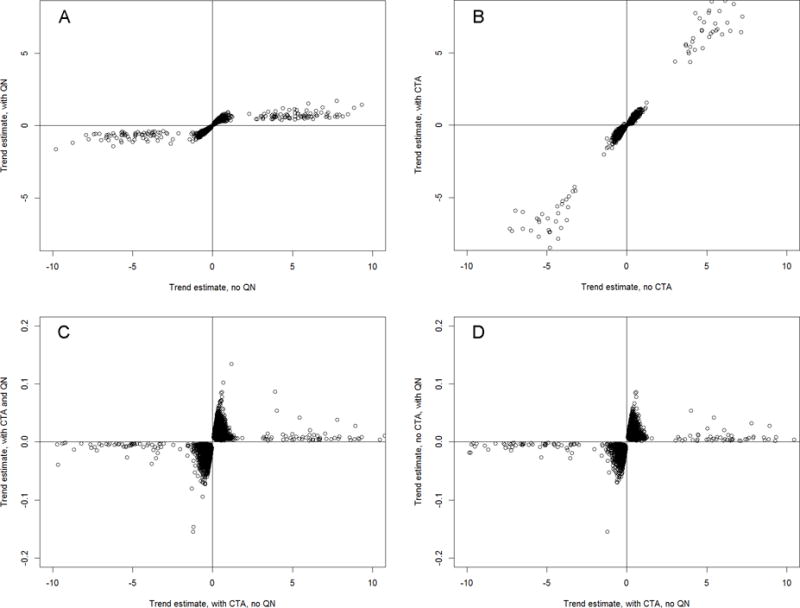
The effect of cell-type composition adjustment and quantile normalization on temporal trend in DNA methylation: scatter plots of trend estimates. (A) Untransformed vs. quantile-normalized data, no cell-type adjustment. (B) Untransformed data: without vs. with cell-type adjustment. (C) Untransformed vs. quantile-normalized data, with cell-type adjustment., (D) Untransformed data with cell-type adjustment vs. quantile-normalized data without cell-type adjustment. All plots show only CpG loci with significant trend in both models. CTA: cell type adjustment; QN: quantile normalization.
Discussion
To our knowledge, this is the first study to look at within- and between-person variability profiles of CpG sites from the HumanMethylation450 BeadChip. Our results show that on average, ICC < 0.5 is common across the genome, and that ICC profiles are similar across all functional locations and probe types. This implies that genomic location at or near CGIs or functional regions has little effect on inter- and intra-individual variability of a CpG locus. We also found that within-person variation tended not to exceed a certain threshold (0.36 for ). Thus, larger between-person variation generally implies a higher ICC. In particular, among CpG loci with between-person variation above the threshold of meaningfulness (Δβ ≥ 0.2), over 99% loci had high ICC. Therefore, for differentially methylated loci with sizeable differences in methylation across subjects, i.e. loci of interest to most association studies, one measurement may be sufficient for a reliable estimate of methylation level.
We note that CpG loci within DMRs had better ICC profiles: even among those with low between-person variation, the percentage of high-ICC loci was higher than in other genomic locations. This observation is expected, as methylation of tissue-specific lineage-defining regions is stable in blood DNA.
The low ICC values may result in substantial attenuation (reduction in magnitude compared to the true value) in estimated parameters, so repeated measurements would be required per study participant to keep attenuation within some acceptable level. The average ICC across all CpG sites in our study was under 0.5, with the largest proportion of low and mid-range ICC values observed among low-variability CpG loci. More than one measurement would be needed for these loci to adequately assess their methylation and to limit attenuation in the estimates; however, because their variability is well below the threshold of meaningful difference, they will likely not be primary targets of an association study.
It should be noted that the low-variability CpG locus group would include most of unmethylated loci with mean methylation close to 0, as such averages can only be attained with low variation. Due to the nature of the logit transportation, this translates to negative M-values of very large magnitude, whereby even small differences in the β-values may translate to M-values that are wide apart. As a result, any estimates for such loci are very unstable and should be treated cautiously. For these loci it may be preferable to use β-values rather than M-values, with appropriate adjustments in analytic methods. For example, beta regression techniques (21) could be used for comparison of DNA methylation between two groups.
We have found that the percentage of high-ICC loci is much larger on the X chromosome, implying more stability in methylation patterns. Prior studies have shown that X chromosome inactivation is accompanied by methylation increase at CGIs and at the promoters of genes silenced by X chromosome inactivation (22). Furthermore, methylated promoter CGIs are usually associated with genes in a stable long-term silenced state (15). Although we were unable to distinguish between active and inactive X chromosomes, our observation of more stable methylation patterns at CGIs and near the start of gene coding regions on the X chromosome appears consistent with the effect of gene silencing in X chromosome inactivation.
In our study we have found that most CpG sites did not exhibit temporal trend that is sufficiently strong to be detected in a small window of <1 year. Among a rather small percentage of sites that did exhibit a trend, negative trend was somewhat more common than positive trend. This finding is in agreement with several prior studies of age-related methylation changes. Using HumanMethylation450 platform on 421 individuals aged 14–94 years, Johansson et al. (23) reported 29% of CpG sites significantly associated with age, of which 60.5% exhibited decrease, and 39.5% increase in methylation. Heyn et al. (24) showed that most of the genome undergoes age-associated hypomethylation. At the same time, a number of studies found that high CpG density promoters of key developmental genes tend to exhibit age-associated hypermethylation, during both early and late stages of life (6, 25, 26). In two longitudinal studies of newborns followed for 1.5–5 years, Martino et al. (26, 27) found clear distinction in methylation profile between samples collected at birth and at subsequent clinic visits. They also observed increase in methylation across all classes of annotated genomic regions, with intergenic regions most likely to undergo such changes. A unique aspect of our study is its longitudinal nature combined with a focus on short-term temporal changes. We have been able to show that in a number of CpG loci, temporal changes in methylation that occur in mature adulthood are detectable over 1-year period of time.
In addressing intra-individual variability in DNA methylation, adjustment for known sources of such variability could improve the ICC. Temporal change may be one such source of within-person variability for CpG loci that exhibit methylation changes over time. Removing a temporal trend may potentially reduce intra-individual variation and improve statistical power. However, because age-related methylation change may be an important contributor to carcinogenesis or other pathogenic process, temporal trend removal may sometimes obscure a true association between DNA methylation and disease risk.
Different cell mixture composition between samples taken from the same individual may also confound measured DNA methylation and contribute to the observed within-person variability. Reinius et al. (28) established that CpG methylation differs between cell types, such as mononuclear cells, granulocytes, natural killer (NK) cells, B-cells, and T-cells. Jacoby et al. (29) analyzed methylation of 58 CpG sites and observed differences in inter-individual variability across blood cell types. We adjusted our analyses for cell type composition using the method of Houseman et al. (20). Koestler et al. (30) further tested this algorithm on data from the Gene Expression Omnibus database, reporting moderate to high agreement between predicted cell type composition and that from complete blood cell counts. In our analyses, there was little difference in ICCs before and after cell type adjustment. It has also been suggested that changes in cell composition of human blood across a person’s lifespan may largely explain age-associated methylation change (1, 31). In our study, we found that a significant temporal trend disappears after cell type adjustment in the majority of CpG sites with such trend (Table 3, Supplementary Table S3). We also observed an age-related decrease in the proportion of CD8+ T-cells (3.5%/year; P=0.015) and B-cells (1.2%/year; P=0.056) and a corresponding increase (4.7%/year; P=0.044) in the proportion of granulocytes (data not shown). Thus, our results support the hypothesis that cell composition change largely accounts for temporal changes in DNA methylation. While two of the aforementioned studies of methylation change did not account for cell type composition of whole blood (23, 25), others either considered cell type composition (27) or focused on specific cell types (6, 24). Thus, associations reported by this latter group of studies are independent of age-related changes in cell composition.
Quantile normalization is a widely used technique to correct for batch effects in DNA methylation and gene expression data. As this technique changes the data, it can affect variability patterns and trends in methylation levels. In our study we have observed that although quantile normalization has little effect on the ICC of differentially methylated CpG sites, it often lowers the ICC of low-variability loci. In addition, it changes the magnitude of the temporal trend estimates for CpG loci with significant trend, but does not change its direction. These observations suggest that for most likely targets of a DNA methylation study, such as differentially methylated loci or those with significant trend, applying quantile normalization as a batch correction technique is unlikely to significantly alter analysis results. However, for studies interested in the magnitude of temporal effects in methylation, this normalization technique can have a profound effect on the results and should be used with caution. Other batch correction techniques should also be considered (32).
Limitations of the present study include its modest sample size, which, nonetheless, is not unusual for epigenome-wide DNA methylation studies to date due to the cost constraint. Second, the absence of samples from men may restrict generalizability of our results. Although a recent study by Lam et al. (33) found only subtle sex differences in methylation of a small subset of CpG loci, one cannot discount the possibility that variability and temporal changes in some CpG loci may differ between men and women. Besides, there is conflicting evidence on the effect of sex on age-related changes in the methylome outside of sex chromosomes (23, 34). Third, in the absence of duplicate contemporaneous samples from the same subject and of technical replicates, it is unclear how much of the detected within-person variation is due to the temporal trend or other non-temporal factors, such as cell fraction differences. Also for this reason, we could not separate technical variability as part of the total variability in DNA methylation. Despite these limitations, we were able to examine variability patterns and detect short-term temporal methylation changes in a substantial number of CpG sites.
In summary, for CpG loci with differences in methylation between people, methylation levels can be reliably assessed with one blood sample; however, more samples and possibly special statistical methods are needed for low-variability and unmethylated loci. The X chromosome exhibits more stable methylation patterns, especially in CGIs and gene promoters, which is consistent with the effects of X chromosome inactivation. Although short-term temporal changes are largely driven by changes in the cell type composition of blood, trend unrelated to cell type was also detected in a small fraction of CpG sites. Further studies are needed to examine whether CpG loci with short-term temporal trend undergo similar methylation changes throughout lifespan and to what extent such changes are related to the onset or progression of disease.
Supplementary Material
Impact.
This study shows that one measurement can reliably assess methylation of differentially methylated CpG loci.
Acknowledgments
We thank Wei Zheng for his leadership and support, Dake Liu, Gong Yang and Charles Matthew for their contribution to the SPA, Regina Courtney and Jie Wu for assistance in DNA extraction, and Hui Cai for data preparation. Sample preparation was performed at the Survey and Biospecimen Shared Resource, Vanderbilt-Ingram Cancer Center.
FINANCIAL SUPPORT: This study was supported by NIH grants and contracts P30 CA068485 (Vanderbilt-Ingram Cancer Center), R37 CA070867 (W. Zheng) and NO2-CP11010-66 (X. Shu).
Footnotes
POTENTIAL CONFLICTS OF INTEREST: None reported
References
- 1.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One. 2009;4:e8274. doi: 10.1371/journal.pone.0008274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011;29:1133–9. doi: 10.1200/JCO.2010.31.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang R, Pfutze K, Zucknick M, Sutter C, Wappenschmidt B, Marme F, et al. DNA methylation array analyses identified breast cancer-associated HYAL2 methylation in peripheral blood. Int J Cancer. 2014 doi: 10.1002/ijc.29205. [DOI] [PubMed] [Google Scholar]
- 4.Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828–37. doi: 10.4161/epi.6.7.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marsit C, Christensen B. Blood-derived DNA methylation markers of cancer risk. Adv Exp Med Biol. 2013;754:233–52. doi: 10.1007/978-1-4419-9967-2_12. [DOI] [PubMed] [Google Scholar]
- 6.Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Otterdijk SD, Mathers JC, Strathdee G. Do age-related changes in DNA methylation play a role in the development of age-related diseases? Biochem Soc Trans. 2013;41:803–7. doi: 10.1042/BST20120358. [DOI] [PubMed] [Google Scholar]
- 8.Block G, Dietrich M, Norkus E, Jensen C, Benowitz NL, Morrow JD, et al. Intraindividual variability of plasma antioxidants, markers of oxidative stress, C-reactive protein, cotinine, and other biomarkers. Epidemiology. 2006;17:404–12. doi: 10.1097/01.ede.0000220655.53323.e9. [DOI] [PubMed] [Google Scholar]
- 9.Tangney CC, Shekelle RB, Raynor W, Gale M, Betz EP. Intra- and interindividual variation in measurements of beta-carotene, retinol, and tocopherols in diet and plasma. Am J Clin Nutr. 1987;45:764–9. doi: 10.1093/ajcn/45.4.764. [DOI] [PubMed] [Google Scholar]
- 10.Shvetsov YB, Hernandez BY, Wong SH, Wilkens LR, Franke AA, Goodman MT. Intraindividual variability in serum micronutrients: effects on reliability of estimated parameters. Epidemiology. 2009;20:36–43. doi: 10.1097/EDE.0b013e318187865e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson JN, Boca SM, Shu XO, Stolzenberg-Solomon RZ, Matthews CE, Hsing AW, et al. Metabolomics in epidemiology: sources of variability in metabolite measurements and implications. Cancer Epidemiol Biomarkers Prev. 2013;22:631–40. doi: 10.1158/1055-9965.EPI-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10:949–55. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 13.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Dedeurwaerder S, Defrance M, Bizet M, Calonne E, Bontempi G, Fuks F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform. 2013 doi: 10.1093/bib/bbt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 16.Zheng W, Chow WH, Yang G, Jin F, Rothman N, Blair A, et al. The Shanghai Women’s Health Study: rationale, study design, and baseline characteristics. Am J Epidemiol. 2005;162:1123–31. doi: 10.1093/aje/kwi322. [DOI] [PubMed] [Google Scholar]
- 17.Peters TM, Moore SC, Xiang YB, Yang G, Shu XO, Ekelund U, et al. Accelerometer-measured physical activity in Chinese adults. Am J Prev Med. 2010;38:583–91. doi: 10.1016/j.amepre.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One. 2013;8:e55761. doi: 10.1371/journal.pone.0055761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari SLP, Pinheiro EC. Improved likelihood inference in beta regression. Journal of Statistical Computation and Simulation. 2012;81:431–43. [Google Scholar]
- 22.Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, et al. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21:1592–600. doi: 10.1101/gr.112680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson A, Enroth S, Gyllensten U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS One. 2013;8:e67378. doi: 10.1371/journal.pone.0067378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109:10522–7. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martino D, Loke YJ, Gordon L, Ollikainen M, Cruickshank MN, Saffery R, et al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14:R42. doi: 10.1186/gb-2013-14-5-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martino DJ, Tulic MK, Gordon L, Hodder M, Richman TR, Metcalfe J, et al. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics. 2011;6:1085–94. doi: 10.4161/epi.6.9.16401. [DOI] [PubMed] [Google Scholar]
- 28.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacoby M, Gohrbandt S, Clausse V, Brons NH, Muller CP. Interindividual variability and co-regulation of DNA methylation differ among blood cell populations. Epigenetics. 2012;7:1421–34. doi: 10.4161/epi.22845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koestler DC, Christensen B, Karagas MR, Marsit CJ, Langevin SM, Kelsey KT, et al. Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics. 2013;8:816–26. doi: 10.4161/epi.25430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Z, Chai HS, Wu Y, White WM, Donkena KV, Klein CJ, et al. Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med Genomics. 2011;4:84. doi: 10.1186/1755-8794-4-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, et al. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17253–60. doi: 10.1073/pnas.1121249109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.