

Abstract
Choroideremia is an outer retinal degeneration with a characteristic clinical appearance that was first described in the nineteenth century. The disorder begins with reduction of night vision and gradually progresses to blindness by middle age. The appearance of the fundus in sufferers is recognizable by the characteristic pale color caused by the loss of the outer retina, retinal-pigmented epithelium, and choroidal vessels, leading to exposure of the underlying sclera. Choroideremia shows X-linked recessive inheritance and the choroideremia gene (CHM) was one of the first to be identified by positional cloning in 1990. Subsequent identification and characterization of the CHM gene, which encodes Rab escort protein 1 (REP1), has led to better comprehension of the disease and enabled advances in genetic diagnosis. Despite several decades of work to understand the exact pathogenesis, no established treatments currently exist to stop or even slow the progression of retinal degeneration in choroideremia. Encouragingly, several specific molecular and clinical features make choroideremia an ideal candidate for treatment with gene therapy. This work describes the considerations and challenges in the development of a new clinical trial using adeno-associated virus (AAV) encoding the CHM gene.
Choroideremia is a degenerative disease of the retina, caused by mutations in the X-linked gene CHM. No treatments currently exist for choroideremia, but a gene therapy clinical trial is in progress.
Choroideremia (CHM; OMIM Phenotype MIM number: 303100; ICD10 code H31.2) is an X-linked, recessive, degenerative disease of the retina. In males, the condition is gradually progressive: This begins with reduced night vision in adolescence and develops with a gradual loss of peripheral vision and blindness in middle age (MacDonald et al. 2009). In contrast to Leber congenital amaurosis and many other visual cycle disorders, choroideremia is not thought to affect eye formation and early visual development. The retinal pigment epithelium (RPE), retina, and choroid do, however, begin to slowly undergo atrophy at an early age. The name of the disease refers to the clinical appearance of atrophy of the choroid, and the appearance of the fundus in sufferers is normally easily recognizable because of the characteristic pale color resulting from exposure of the underlying white sclera at later stages of the degeneration (Fig. 1). Male patients generally maintain good visual acuity until the degeneration encroaches on the fovea, but underlying changes in the retina can be identified in childhood and are associated with significant reductions in parafoveal retinal sensitivity as measured by psychophysical testing (Jacobson et al. 2006b). Because disease progression is relatively slow and occurs after the period of visual development, patients have largely unaffected vision, particularly in early life. Hence, the majority of patients would likely benefit from successful therapeutic intervention to preserve remaining vision, even at late stages of the disease.
Figure 1.
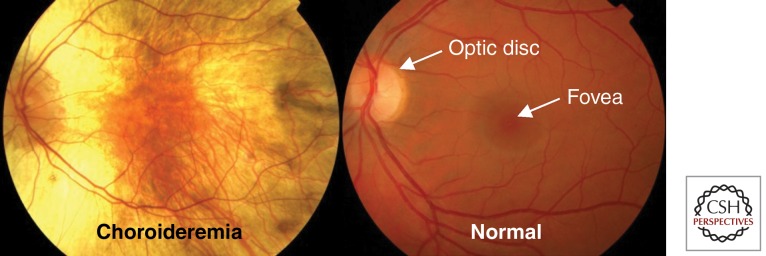
A fundus photograph of a patient with advanced choroideremia. Patient eye (left) compared with a normal eye (right). In the choroideremia eye, there is a residual island of healthy tissue in the central macula and fovea.
When examined, most carrier females show characteristic pigmentary changes of the fundus but these are generally nonprogressive if the X-inactivation occurs late during development—in other words, if there is an even and fine distribution of affected and unaffected RPE cells. These individuals remain predominantly asymptomatic in early years, but may develop reduced night vision in later years. In contrast, X-inactivation very early during development can result in large areas of retina affected by choroideremia caused by clonal expansion of cells expressing only the defective CHM gene (the “good” copy on the other X chromosome having been silenced). These rare, severe cases can lead to blindness in female carriers (Bonilha et al. 2008).
Choroideremia is caused by mutations in the CHM gene (OMIM Gene/Locus MIM number: 300390), which is located on the long arm of the X chromosome (Cremers et al. 1990). CHM encodes Rab escort protein 1 (REP1), which is sometimes also referred to as component A of Rab geranylgeranyl-transferase (Seabra et al. 1992, 1993). This protein is expressed ubiquitously and is found in all cell types and tissues throughout the body. Through its action on Rab GTPases, REP1 plays a role in the fundamental cellular processes of intracellular vesicle trafficking and recycling. Despite about two decades of work to understand the role of REP1 deficiency in the pathogenesis of the disease (e.g., Seabra et al. 1992, 1993, 1995; Larijani et al. 2003; Pylypenko et al. 2003; Tolmachova et al. 2006, 2010), no established treatments currently exist to stop or even slow the progression of retinal degeneration in choroideremia.
Nevertheless, in vivo gene augmentation therapy, in which a working and healthy copy of the gene is introduced into the cells within the body that need it, is an appealing strategy for this currently incurable, blinding disease. The slow rate of degeneration means that there is a long therapeutic window in which to replace the gene in ocular cells before they are lost. In addition, virtually all cases reported so far are functionally null mutations and are often predicted to result in the severe truncation or absence of endogenous REP1 protein (van den Hurk et al. 1997, 2003; McTaggart et al. 2002; Esposito et al. 2011). This is useful because the product of the therapeutic gene will not have to compete with a large pool of mutant protein. This would limit latent dominant negative effects and in theory would mean only a small amount of vector-delivered REP1 might be needed to arrest degeneration. Furthermore, recent reports showing that replacement gene therapy can be safely and successfully used to treat the retinal disease (Leber congenital amaurosis type 2) (Bainbridge et al. 2008; Hauswirth et al. 2008; Maguire et al. 2008), provide encouragement that a similar technique could be applied to choroideremia.
THE HISTORY, PREVALENCE, AND MOLECULAR-GENETIC DIAGNOSIS OF CHOROIDEREMIA
Choroideremia was first described in the nineteenth century by the Austrian ophthalmologist Ludwig Mauthner (1872). Seemingly as a result of the barren appearance of the fundus, Mauthner named the condition “Chorioideremie,” which is thought to be a combination of the ancient Greek “erēmia,” meaning barren land or desert, as a suffix to the stem word “chorioid” a common alternative spelling of “choroid” (also originally from ancient Greek words for skin—“chorion,” and form/type—“eidos”) (Myers 2006). Thus, the disease name means “an area barren of choroid.” This entered English usage as choroideremia, but an alternative spelling, choroideraemia, has been used extensively (e.g., Martin 2010). It is unclear if the addition of an “a” comes from a desire to differentiate the first and second vowel sounds in “erēmia” (which indeed are different Greek letters). Alternatively, it may have been believed that the disease name was derived by adding the prefix “er-” (possibly from the Latin “ir-” meaning not/negation) to the suffix “aemia” (derived from ancient Greek and in usage in British medicine as referring to the state or condition of the blood), thereby meaning an observation of “no blood in the choroid.” Whatever the case, the preferred modern English spelling is Choroideremia, as the “ae” diphthong, seen increasingly as archaic, is being replaced by a simple “e.”
Originally, the disease was thought of as a congenital absence of the majority of the choroid, probably caused by a developmental disorder (Mauthner 1872). In fact, Theodor Leber saw the same case and classified it as one of retinitis pigmentosa with extreme atrophy of the choroid (reported by Wolf 1930). Although closer to the truth, Leber’s analysis seems to have been rejected at the time, and the idea of the disease being caused by a congenital (and largely stationary) absence of the choroid predominated for several decades. After more cases were studied, it became clear that the stationary nature of the disease was questionable and the idea was flatly rejected by Pameyer et al. (1960) who said that choroideremia should be considered as an “abiotrophic process of the ‘tapetum nigrum’ (a reference to the RPE) with a secondary progressive dystrophy of the choriocapillary layer and outer retinal layers” and were clear that “the hypothesis of a dysplasia of the choroid as a primary cause of the fundus alterations must be rejected.” Even though choroideremia is now accepted as being a progressive degenerative disease, the idea that degeneration of the choroid is the primary cause still lingers in some medical definitions (e.g., Myers 2006). We now know that the name choroideremia, although an apt description of the fundus appearance of atrophy of the choroid at later stages of the disease, does not reflect the etiology of the disease very well. This is an important consideration when developing therapeutic strategies (see below).
Choroideremia has a prevalence of approximately 1:50,000 in people of European descent, with the highest prevalence in northern Finland (Sankila et al. 1992). There are thought to be more than 500 affected male patients in the United Kingdom and around 3000 throughout Europe (MacDonald et al. 2009). Thus, although relatively rare, a significant patient population exists for this blinding disease, which lacks any major treatment options.
The characteristic appearance of the fundus can be used to first identify choroideremia clinically. A family history consistent with an X-linked recessive disease can help rule out other diseases with a similar appearance (e.g., gyrate atrophy, Oliver-McFarlane syndrome, bifocal chorio-retinal atrophy). Mutations that cause choroideremia invariably lead to a complete absence of the REP1 protein. Because of this, it has been suggested that clinical diagnosis of choroideremia can be confirmed by the absence of REP1 protein in peripheral blood samples in an immunoblot analysis with anti-REP1 antibody (MacDonald et al. 1998). However, genetic testing and molecular diagnosis of a pathogenic mutation in the CHM gene should be used to support the clinical diagnosis. It is possible to order routine sequencing of CHM via health service approved diagnostic laboratories in many countries, including the UK, although in some cases a more thorough examination of the genomic DNA or the mRNA must be performed to detect the molecular cause of CHM (Furgoch et al. 2014).
THE EXPRESSION AND FUNCTION OF REP1 PROTEIN
In many inherited retinal dystrophies, the protein product of the causative gene directly contributes to a process that is essential for the light-sensitive function of the eye (such as phototransduction in the retina or the visual cycle in the RPE). This is not the case in choroideremia. Instead, the gene product, REP1, is involved in lipid modification of Rab GTPases, members of the Ras superfamily of monomeric G proteins (Fig. 2) (Seabra et al. 1992, 1995; Pylypenko et al. 2003). This modification, known as prenylation, involves the covalent attachment of one or more prenyl groups (geranylgeranyl or farnesyl) to carboxy-terminal cysteines of a Rab protein. REP1 binds newly synthesized, unprenylated Rabs and presents them to a catalytic Rab geranylgeranyltransferase (GGTase) subunit for the geranylgeranyl transfer reaction. REP1 also helps escort prenylated Rabs to their specific destination membrane by binding the hydrophobic, insoluble prenyl groups and carrying them through the cytoplasm (Pylypenko et al. 2003). Rabs themselves have an important function in intracellular vesicle trafficking, for which the addition of a prenyl moiety “lipid anchor” is essential (Seabra et al. 1995). Thus, in choroideremia, the affected gene plays a role in fundamental cellular processes.
Figure 2.

A schematic of the cellular function of Rab escort protein (REP1). REP1 binds Rabs (member of the Ras subfamily of small GTPases, ∼70 members) as they are made by ribosomes (R) on the endoplasmic reticulum (ER). REP1 present newly synthesized, unprenylated Rabs to a catalytic Rab geranylgeranyltransferase (GGTase) subunit. Rabs undergo prenylation, which involves the covalent attachment of one or more hydrophobic prenyl groups (geranylgeranyl, GG shown here) to carboxy-terminal cysteine residues. REP1 also escorts prenylated Rabs to specific destination membranes in the Golgi apparatus (GA) and of diverse cellular vesicles (Vesicle, represents various cellular vesicles, e.g., lysosomes or transport vesicles).
Within the eye, there are a number of processes reliant on Rab protein function. In the photoreceptor, proteins destined for the outer segment, such as rhodopsin, travel from the Golgi apparatus to the connecting cilium via vesicular transport that is regulated by Rab8 and Rab6. In the RPE, intracellular trafficking controls the phagocytosis and degradation of disk membranes shed from the apical tips of photoreceptor outer segments. It is believed that Rab proteins function during phagocytosis by the RPE, possibly through an effector-mediated interaction with myosin VIIA, which is required for phagosome transport (Gibbs et al. 2003).
Although choroideremia is almost exclusively a disease affecting the eye, REP1 is expressed in all cells and tissues throughout the body. As an additional complexity, mammals (including man) also produce REP2, a protein very similar to REP1, with an (amino acid sequence 75% identical, 90% similar to REP1) (Cremers et al. 1994). REP2 is encoded by the gene CHML (choroideremia-like), which resides on chromosome 1 and is thought to have arisen from a retrogene insertion of the REP1 mRNA transcript sometime during vertebrate evolution (as evidenced by the lack of introns). REP2, like REP1, is expressed ubiquitously. Thus, loss of REP1 function can in theory be compensated for by the action of REP2, at least in cells outside the eye.
It is thought, therefore, that the phenotype of choroideremia is not so much dictated by the cells in which REP1 is normally expressed, but by those in which REP2 is unable to compensate for its function, for whatever reason. It has been shown that some Rabs, such as Rab27a, are preferentially prenylated by REP1 (Larijani et al. 2003). Rab27a has a key role in the retina (Tolmachova et al. 1999), and its affinity toward REP1 and REP2 proteins is similar. However, the Rab27a-REP1 and Rab27a-REP2 complexes have different affinities for the Rab GGTase enzyme, which might explain why REP2 is unable to support Rab27a prenylation in the retina of choroideremia patients over the long term (Larijani et al. 2003).
INVESTIGATING THE PATHOLOGY OF REP1 DEFICIENCY USING LABORATORY ANIMAL MODELS
To investigate the pathology of choroideremia, researchers have attempted to develop laboratory animal models of the disease. However, the global ablation of CHM gene function appears to be more severe in animal models than in humans. In the zebrafish, a common nonmammalian vertebrate model organism, mutation of the chm gene leading to a completely nonfunctional (null) allele manifests as an extremely severe systemic illness resulting in early lethality at postnatal day 6 (Starr et al. 2004; Moosajee et al. 2009). This may be explained by the fact that chml/Rep2 is not present in the zebrafish genome (Moosajee et al. 2009). In mice, despite the presence of the Chml gene, the genetic deletion of Chm causes embryonic lethality in hemizygous male mice (Chmnull/Y), caused by abnormalities in extraembryonic tissues such as the placenta and yolk sac (Shi et al. 2004). Interestingly, carrier female mice (Chmnull/+) are viable and do show progressive retinal degeneration (van den Hurk et al. 1997; Tolmachova et al. 2006). Thus, the Chmnull/+ female mouse seems to best model the phenotype of affected hemizygous male patients. The reasons for this are not completely clear, but subtle species differences in RabGGT compatibility with REP2-Rab complexes and differences in X-inactivation may partially explain why the sex-linked phenotype of choroideremia differs substantially between species.
An important question in choroideremia pathology has been whether one cell type/tissue layer acts as a primary site of degeneration (which then leads to the demise of the other cell types/tissue layers) or if the disease appears in multiple cell types/tissue layers independently and/or simultaneously. The answer dictates which cell types need to be targeted by for gene replacement therapy (and which can or should be avoided). The question is complicated by the fact that the three affected eye layers (choroid, photoreceptors, and RPE) are highly interdependent. For example, genetically normal photoreceptors will degenerate secondary to loss of the RPE by any cause (for instance, in myopic and dry age-related macular degeneration), and the morphology and distribution of RPE cells will change in response to photoreceptor loss (as in many forms of retinitis pigmentosa, for example).
Therefore, more complex animal models have been used in an attempt to dissect the intricate pathologic changes that occur during the choroideremia degenerative process. Krock et al. (2007) used the chm zebrafish model to create genetically mosaic mutant/wild-type eyes (by transplanting cells in blastula stage embryos) and found that REP1 appears to be critical for the pigment epithelium but not photoreceptors. In the mouse, a conditional knockout allele (ChmFlox) has been made that allows for the selective ablation of the Chm gene in a particular cell/tissue type (Tolmachova et al. 2006). When Chm was selectively ablated in either the neurosensory retina or the RPE, it was found that different subsets of Rabs were underprenylated, implying that both layers have an intrinsic prenylation defect (Tolmachova et al. 2006). In further work, Chm was selectively ablated in photoreceptor cells or the RPE, resulting in independent degeneration of both layers (Tolmachova et al. 2010). Although neither condition could initiate significant degeneration of the other layer, the inherent and cell-autonomous need for REP1 function in the photoreceptors and RPE means that both layers could be considered primary sites of the disease (Tolmachova et al. 2010). Furthermore, when ablation of Chm in the RPE and photoreceptors was combined in the same mouse line, the phenotype was greater than the sum of the effects observed by ablating Chm in each cell type separately (Tolmachova et al. 2010). This highlights once again that the adjacent cell layers in the eye influence each other and that these interactions are important factors that can determine the degenerative process in choroideremia.
Overall, the animal models indicate that REP1 deficiency causes cell-autonomous problems in the eye and that both the RPE and retina/photoreceptors can be considered primary sites of the disease. It is logical therefore to propose that future gene therapy approaches for choroideremia should use viral vectors that target both photoreceptors and RPE.
FURTHER CONSIDERATIONS FOR CHOROIDEREMIA PATHOLOGY BASED ON HUMAN DATA
Caution is needed when extrapolating observations from laboratory models directly to humans, because of potential species differences as mentioned above. It is therefore crucial to use human data wherever possible to identify the retinal layers that are wholly dependent on REP1. This information can come from multiple sources, using a range of techniques.
In human female carriers, skewed X-inactivation across the retinal layers can leave affected clones of photoreceptors against normal pigment epithelial cells, and histological postmortem specimens suggest that photoreceptors degenerate independently of the RPE (Flannery et al. 1990; Syed et al. 2001; Bonilha et al. 2008). There is also evidence from optical coherence tomography scans of male choroideremia patients that the photoreceptor layer thins in advance of degeneration of the RPE (Jacobson et al. 2006b).
This data confirm the logic of designing a REP1-expressing vector that targets both the photoreceptor layer and the RPE, but further refinement might also be necessary. Because the early stages of choroideremia manifest as loss of night vision, with central, cone-mediated vision preserved until very late stages of the disease, it seems reasonable to assume that rods are affected significantly more than cones. For reasons that are not completely clear, cones are known to be dependent on surrounding rods, and any genetic condition that causes rod death will also lead to complete loss of cones in a centripetal pattern as a secondary phenomenon. In a postmortem analysis of the retina from an X-linked carrier female, apparently normal-looking, REP1-deficient cones could be identified within regions containing REP1 expressing rods (Syed et al. 2001). The differential expression of REP1 in these two photoreceptor subclasses results from differential X-inactivation (Reese et al. 1995) and provides further evidence that cones may exist without REP1 when surrounded by rods. It therefore seems reasonable to assume that cone death may occur as a phenomenon secondary to loss of rods and RPE, rather than a dependency on REP1. Hence, from a safety perspective, it might actually be better to avoid transduction of cone cells as much as possible.
It might seem ironic that despite the characteristic and defining atrophy of the choroid in choroideremia, the relative importance of REP1 deficiency in the choroidal layers has not been dealt with comprehensively in the available animal models. Fortunately, human data and observations exist that are more illuminating. Studies from postmortem carrier females show no focal loss or abnormalities of the choroid except underlying areas of severe retinal degeneration (Syed et al. 2001), which suggests that the choroidal vascular system is not dependent on REP1. Similarly, choroideremia is not known to affect blood vessels elsewhere in the body. It is therefore likely that choroidal atrophy occurs secondary to loss of the overlying RPE. In other retinal conditions, the choroid undergoes progressive atrophy many years after subretinal surgery if the pigment epithelium is damaged, leading to a gradual and focal unmasking of scleral autofluorescence, which is similar to the progression of choroideremia (MacLaren et al. 2005). Despite this, a slow primary degeneration of the choroid cannot be excluded completely.
CHOOSING A GENE THERAPY VECTOR
For gene replacement therapy to work, the genetic material (DNA/RNA) needs a delivery system (vector), and it is common in clinical trials to use a modified virus to carry the gene into cells. Adeno-associated virus (AAV) is currently a popular vector for clinical trials, particularly for ocular gene therapy, but is it applicable for use in choroideremia?
An important issue is the amount of genetic material that can be placed into each viral particle. This is known as the packaging capacity or limit, and for AAV vectors it is thought to be <5 kb of single-stranded DNA (Wu et al. 2010). Although this capacity is considered to be small, and a potential limitation of using AAV, it is more than sufficient to include the full coding sequence of REP1 (at 1.9 kb), while still allowing space for a choice of promoters and other regulatory elements.
Another consideration is whether the viral vector can effectively target and transfer DNA to (transduce) the specific cell types and tissues that need to be treated. There are many different AAV subtypes, each with slightly different DNA sequences and capsid proteins on their outer shell (serotypes). These serotypes have different cellular tropism; they preferentially target specific cell types within a given host species. Many AAV vectors target neurons effectively, but only AAV serotype 2 has been shown to transduce both photoreceptors and the RPE after subretinal injection in nonhuman primates (Bennett et al. 1999; Jacobson et al. 2006a). AAV2 therefore appears to be a good choice, as rod photoreceptors and RPE cells both need to be targeted for REP1 replacement in choroideremia.
In the photoreceptor layer, AAV2 appears to predominantly target rods rather than cones (Bennett et al. 1999). This is also beneficial because cone photoreceptors do not intrinsically suffer in the absence of REP1 in choroideremia and therefore, from a safety perspective, it might be better to avoid transduction of cone cells as much as possible. However, avoiding cone transduction completely by using AAV2 cannot be guaranteed in the degenerating retina. For instance, in the absence of rods, viral particles are likely to have greater access to cone cells. There may also be promoter-serotype interactions. AAV5, for example, does not transduce macaque primate cones effectively with the CMV promoter (Lotery et al. 2003), but it is effective in cones of the squirrel monkey when using a cone-specific promoter (Mancuso et al. 2007). It is impossible to precisely predict whether AAV2 will limit photoreceptor gene expression exclusively to rod photoreceptors in any given human retinal degeneration; however, it is probably safe to say that AAV2 will target rods more effectively than cones in humans. The tropism of AAV2 for the deeper vascular layers of the choroid is relatively unexplored in animal models and is therefore hard to predict in humans. It seems unlikely that AAV2 will be able to bypass the combined barriers of the RPE and Bruch’s membrane, but any possible transduction and treatment of choroidal layers could only be beneficial. As REP1 is ubiquitously expressed, the unlikely event of a more widespread transduction pattern of the gene therapy virus might not be considered a major cause for concern, even in cells and tissue outside the eye.
AAV2 seems a suitable choice for use in choroideremia gene therapy for reasons beyond those described above. Although it is not currently known to cause disease, AAV naturally infects humans (and some other primate species). Consequently, antibodies against capsid antigens of wildtype AAVs can be detected in a significant percentage of humans (Mingozzi et al. 2007). Despite this, there are extensive safety data in primates (Jacobson et al. 2006a) and from recent RPE65 gene therapy trials for (LCA2) (Bainbridge et al. 2008; Hauswirth et al. 2008; Maguire et al. 2008) that show subretinal administration of AAV2 does not cause significant immunogenicity, inflammation, or toxicity. In addition, data from dog models of LCA2 treated with AAV2.RPE65 show that the treated dogs maintain vision without reduction for several years, and even over a decade in some cases (Bennicelli et al. 2008; Cideciyan et al. 2013). In humans, clinical trials have shown that visual gains following gene transfer using AAV2.RPE65 are sustained vision for at least 3 yr (Jacobson et al. 2012; Cideciyan et al. 2013; Testa et al. 2013). Clearly, the lifelong effects in humans will not be known for some time, but these data suggest that expression from the AAV2 vector is sustained, and early loss of therapeutic transgene expression through gene silencing, such as by methylation of the promoter DNA, is not a significant issue. It should be noted, however, that follow-up results from one study (Bainbridge et al. 2008) were never published and, although a maze navigation test in the one reported patient looked impressive, close inspection of the data shows that a similarly dramatic improvement in maze navigation time was seen in the control eye as well. Because RPE65 patients dark-adapt with great difficulty, variation in pretest dark adaptation is a possible explanation for the alleged improvement in vision, as this would also benefit the control eye. In any case, the absence of any follow-up data or further analysis of visual function in this patient raises the possibility that the reported improvement in vision may have been an artifact of testing. It is also unclear why, after 5 yr, there are no reported results for the other nine patients who underwent gene therapy in this trial. The results may have been unspectacular, perhaps owing to the choice of an unsuitable promoter (MacLaren 2009).
Taken together, AAV2 displays tropism for the correct cell types needed, is capable of packaging the full REP1 coding sequence and has been shown to be safe and effective in ocular gene therapy trials for a different eye disease (LCA2) up to 3 yr after treatment. For these reasons, AAV2 was selected for use in a CHM-replacement gene therapy strategy for choroideremia. As AAV2 is such a compelling choice, other types of virus, such as lentiviruses and adenoviruses, were not considered.
DESIGNING AN EFFICIENT AAV2-REP1 EXPRESSION CASSETTE
Because REP1 is ubiquitously expressed, there was little point in using its 5′ upstream promoter sequence to drive regulated transgene expression. Instead, a well-characterized ubiquitous promoter, capable of driving sustained, high levels of expression, was chosen: a hybrid of the human cytomegalovirus (CMV) upstream enhancer with the chicken β-actin promoter. This promoter, variously termed CBA or CAG, has been used extensively in preclinical studies. The original CBA promoter construct was generated in Japan (Miyazaki et al. 1989; Niwa et al. 1991) and first used successfully in AAV in vivo by Mark Sands in the USA (Daly et al. 1999). A later study showed that an AAV vector with the CBA promoter administered via portal vein injection yielded 9.5-fold higher expression compared to an AAV vector with the EF1α promoter and 137-fold higher expression compared to an AAV vector with the CMV promoter/enhancer, and the CBA-mediated expression was sustained for at least a year (Xu et al. 2001). CBA promoters have provided highly effective transduction in retinal gene therapy clinical trials (Hauswirth et al. 2008; Maguire et al. 2008) and appear to sustain expression for over a decade in dogs (Bennicelli et al. 2008; Cideciyan et al. 2013) and at least 3 yr in humans (Jacobson et al. 2012; Cideciyan et al. 2013; Testa et al. 2013).
Other sequences in the transgene expression cassette can be used to improve expression of the therapeutic gene. The inclusion of a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) downstream from the gene coding sequence enhanced AAV transgene expression in the rat brain (Paterna et al. 2000; Klein et al. 2002), in human embryonic kidney cells (HEK293), and human skin fibroblasts in vitro by up to 10-fold (Loeb et al. 1999). Although such an element has not been used previously in retinal gene therapy trials, the same WPRE sequence was approved by the United States Food and Drug Administration and, when used in the AAV2-GAD vector for gene therapy of Parkinson’s disease, did not lead to any severe adverse events (Kaplitt et al. 2007). Importantly, this WPRE has been modified to ablate the translation initiation site of the viral X antigen, expression of which has previously been linked to increased tumor susceptibility in the liver of mice (Flajolet et al. 1998). The choice of polyadenylation signal sequence type can also be used to enhance gene expression. Bovine growth hormone polyA sequence has been shown to yield threefold higher expression levels than do other polyA sequences such as SV40 and human collagen polyA, and this increased expression is largely independent of the type of upstream promoter or transgene (Pfarr et al. 1986).
Thus, AAV transgene expression levels can be maximized by a combination of an optimized promoter and polyA signal, along with the inclusion of the WPRE sequence. This allows a lower overall dose of AAV vector to achieve the same therapeutic effect, which is likely to help minimize the immune response to viral proteins in patients. One might argue that this could also be achieved by using other AAV serotypes, such as AAV8, that appear to produce more efficient expression, especially in photoreceptors (Lebherz et al. 2008). However, as long-term data on safety and sustained expression are currently only available for AAV2, it is preferential to optimize the expression cassette rather than switch capsids.
AAV expression cassettes must be flanked by identical inverted terminal repeat sequences. These are part of the genome of wild-type AAV and are important in packaging the single-stranded DNA into virions and also in somehow protecting and maintaining the unpackaged DNA in an episomal state in transduced cells. Most inverted terminal repeat sequences used for clinical trials are derived from the genome of AAV2, regardless of which capsid serotype they are packaged into. The full annotation AAV2/2 is used to indicate that AAV2 ITRs have been used to package the expression cassette into the AAV2 serotype.
The final necessary component of the cassette is, of course, the cDNA sequence encoding human REP1. Fortunately, this has previously been fully cloned and characterized (Cremers et al. 1990; van Bokhoven et al. 1994). Overall, each AAV virion would contain a single stranded DNA sequence of ∼4.2 kb in length comprising a CMV enhancer/CBA hybrid promoter, human REP1 cDNA, WPRE, and bovine growth hormone polyadenylation sequence (BGH-polyA). This would all be flanked by inverted terminal repeat sequences from the wild-type AAV2 genome. The structural organization of the AAV-REP1 vector is shown in Fig. 3.
Figure 3.
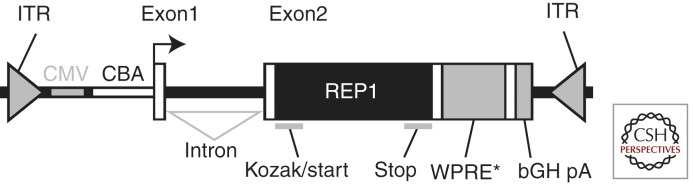
Design of the AAV2/2-REP1 vector used for choroideremia gene therapy. ITR, AAV2 inverted terminal repeats; CMV, cytomegalovirus enhancer element; CBA, chicken β actin promoter; Exon1, chicken β-actin exon 1; Intron, hybrid chicken β-actin and rabbit β-globin intron; Exon2, rabbit β-globin exon 3 fragment (creates an artificial splice site) and human REP1 cDNA (REP1) containing Kozak consensus sequence and start codon initiating translation (Kozak/start) and stop codon terminating translation (stop); WPRE*, modified woodchuck hepatitis virus posttranscriptional regulatory element; bGH pA, bovine growth hormone polyadenylation signal.
PRECLINICAL TESTING OF THE VECTOR
To translate seamlessly and safely from the laboratory to a clinical trial, the AAV2 expression cassette was produced using protocols that had previously been scaled up to good manufacturing practice grade, approved by the United States Food and Drug Association and used safely in patients in another trial (Kaplitt et al. 2007). All preclinical work was conducted using AAV2-REP1 that had been packaged and purified to a research grade standard, but was otherwise identical to the good manufacturing practice-grade vector that could be used in a choroideremia clinical trial (Tolmachova et al. 2013).
Preclinical testing of the gene therapy expression cassette also involved creation of a green fluorescent protein (GFP) reporter vector. In the reporter vector, the coding sequence of GFP replaced the REP1/Chm cDNA, but otherwise, the structure and sequence were identical. Subretinal injection of AAV2-GFP reporter vector in mice confirmed targeting of the transgene to RPE and a high level of expression in photoreceptors (Tolmachova et al. 2013). Furthermore, in vitro treatment of explants of human retina in culture with the AAV2-GFP vector confirmed that the gene therapy expression cassette and capsid type were capable of transducing photoreceptors and driving a high level of transgene expression (Tolmachova et al. 2013).
Importantly, the AAV2-REP1 vector was able to produce therapeutic protein in cultured fibroblasts of choroideremia patients. By examining prenylation activity in these transduced cells, correct function of the REP1 transgene product could be confirmed (Tolmachova et al. 2013). Subretinal injection of AAV2-REP1 in wild type mice did not significantly impair retinal function (as assessed by electroretinography), and thus no obvious toxic effects are evident following overexpression of REP1 (Tolmachova et al. 2013). When subretinal injection of AAV2-REP1 was used to treat Chmnull/+ mice, a dose dependent improvement/preservation of retinal function was observed after several months in treated eyes compared to paired, sham-injected control eyes (Tolmachova et al. 2013). Although large animal models have been used in the preclinical assessment of gene therapy for retinal disease such as LCA2 (Bennicelli et al. 2008; Cideciyan et al. 2013) and achromatopsia (Komaromy et al. 2010), no such large animal model exists for choroideremia. Thus, using a range of techniques and experiments, preclinical work confirmed the effectiveness and safety of the AAV2-REP1 gene therapy vector.
THE INITIATION OF A GENE THERAPY CLINICAL TRIAL FOR CHOROIDEREMIA
The necessary funding and regulatory and ethical approvals were obtained to embark on a gene therapy trial for choroideremia (ClinicalTrials.gov Identifier: NCT01461213). After a period of enrolment and baseline testing, the first treatment was given in October 2011. In all, the clinical trial will assess the effects of the AAV2.REP1 vector in 12 male patients with a clinical phenotype of choroideremia and a confirmed molecular diagnosis of a null mutation in the gene encoding REP1.
Choroideremia presents a number of challenges for gene therapy. Treatment must be performed when retinal structure relatively intact (i.e., before major cell loss) and when “good” foveal vision is still retained. The potential risks are significant, as delivery of the vector involves detachment of the fovea in a patient with good visual acuity, although if no form of treatment is given, vision will eventually be lost because of the ongoing degeneration. Although there is no preclinical evidence that REP1 is toxic, and it is present in virtually all nucleated cells in nonchoroideremia patients, it is possible that REP1 (over)expression might have a detrimental effect on retinal cells, in particular functional photoreceptors. In addition, this is the first gene therapy trial for retinal degeneration that specifically targets photoreceptor cells (in trials for RPE65-LCA2, the RPE cells were the primary target).
To ensure that photoreceptor cells are not compromised, the trial involves an escalation of the vector dose from a low baseline level. The starting dose of AAV2 (1010 genome particles) is at the lower end of the range (1010–1012) that has been shown to be safe in current AAV2 ocular gene therapy trials (Bainbridge et al. 2008; Hauswirth et al. 2008; Maguire et al. 2008). Because of this, reactions to AAV capsid protein are unlikely, even allowing for a large number of empty vector particles. The low initial dose also addresses the concern that, because the REP1 protein will be expressed against a null background, there is a theoretical risk of immune reaction to a new protein. The starting dose will be used in the first group of six patients and, if no serious adverse effects or reactions are seen, this will then be increased to a dose of AAV2 that is 10-fold higher (1011 genome particles) in the second group of six patients.
The primary objective of the study is to assess the safety and tolerability of the AAV2-REP1 vector. This will be performed by monitoring for evidence of ocular inflammation or detrimental effects to visual function in patients for 24 mo after administration. Changes in the treated eye can be compared to baseline measurements and to the fellow untreated eye. Gene transfer to the photoreceptors and RPE cells might enable the cells to survive long-term. The secondary endpoint of the study is to identify any therapeutic benefit of subretinal injection of AAV2-REP1 vector. As choroideremia is a progressive disease, this would be evidenced by a decrease in both loss of visual function and retinal degeneration in the treated eye compared to the control eye at 24 mo after gene delivery. Ophthalmological tests used to assess this will include microperimetry, optical coherence tomography scans and fundus autofluorescence imaging.
Although it is not expected that any anatomical parameter will show an improvement posttreatment compared to baseline, it is possible that there will be some recovery and restoration of vision when measured by the functional tests. These gains might be detected by microperimetry testing. Although male patients generally maintain good visual acuity until the degeneration encroaches on the fovea, significant deficits in parafoveal retinal sensitivity can be seen in more-discriminating tests (Jacobson et al. 2006b).
If the primary and secondary objectives are met, follow-up clinical trials will be initiated using the same vector at the efficacious dose. Beyond the 24 mo of formal testing, patients will continue long-term follow-up, with further assessments taking place annually as part of normal National Health Service care. This will continue for the lifetime of the patient, and the data will continue to be analyzed by members of the study group to determine if AAV2 transduction of the human retina can result in lifelong gene expression.
CONCLUDING REMARKS
More than 130 yr since the first description of choroideremia, a clinical trial involving gene therapy to treat the disease has begun. The conception and design of the clinical trial has been built on several decades of research focusing on the causes and pathology of the disease. Recent advances in the use of viral vectors for ocular gene therapy have also been invaluable. The trial is the first time photoreceptors have been specifically targeted for treatment in retinal degeneration. It is also the first time treatment of nondegenerated foveas with normal visual acuity has been attempted. Preclinical data on the testing of the AAV2-REP1 gene therapy vector have been encouraging, and the results of the human trial are awaited with great anticipation (MacLaren et al. 2014)
ACKNOWLEDGMENTS
The authors’ work is supported by the National Institute for Health Research (NIHR) Biomedical Research Centres at the Oxford University Hospitals and Moorfields Eye Hospital NHS Trusts. Additional funding is provided by the Health Foundation, Fight for Sight, the Lanvern Foundation, the Special Trustees of Moorfields Eye Hospital, the Royal College of Surgeons of Edinburgh. The CHM gene therapy clinical trial is funded by the Health Innovation Challenge Fund, a parallel funding partnership between the Wellcome Trust and the United Kingdom Department of Health.
Footnotes
Editors: Eric A. Pierce, Richard H. Masland, and Joan W. Miller
Additional Perspectives on Retinal Disorders: Genetic Approaches to Diagnosis and Treatment available at www.perspectivesinmedicine.org
REFERENCES
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, et al. 2008. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358: 2231–2239. [DOI] [PubMed] [Google Scholar]
- Bennett J, Maguire AM, Cideciyan AV, Schnell M, Glover E, Anand V, Aleman TS, Chirmule N, Gupta AR, Huang Y, et al. 1999. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc Natl Acad Sci 96: 9920–9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennicelli J, Wright JF, Komaromy A, Jacobs JB, Hauck B, Zelenaia O, Mingozzi F, Hui D, Chung D, Rex TS, et al. 2008. Reversal of blindness in animal models of Leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther 16: 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL, Trzupek KM, Li Y, Francis PJ, Hollyfield JG, Rayborn ME, Smaoui N, Weleber RG 2008. Choroideremia: Analysis of the retina from a female symptomatic carrier. Ophthalmic Genet 29: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, Roman AJ, Olivares MB, Schwartz SB, Komaromy AM, et al. 2013. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci 110: E517–E525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers FP, van de Pol DJ, van Kerkhoff LP, Wieringa B, Ropers HH 1990. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 347: 674–677. [DOI] [PubMed] [Google Scholar]
- Cremers FP, Armstrong SA, Seabra MC, Brown MS, Goldstein JL 1994. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J Biol Chem 269: 2111–2117. [PubMed] [Google Scholar]
- Daly TM, Okuyama T, Vogler C, Haskins ME, Muzyczka N, Sands MS 1999. Neonatal intramuscular injection with recombinant adeno-associated virus results in prolonged beta-glucuronidase expression in situ and correction of liver pathology in mucopolysaccharidosis type VII mice. Hum Gene Ther 10: 85–94. [DOI] [PubMed] [Google Scholar]
- Esposito G, De Falco F, Tinto N, Testa F, Vitagliano L, Tandurella IC, Iannone L, Rossi S, Rinaldi E, Simonelli F, et al. 2011. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat 32: 1460–1469. [DOI] [PubMed] [Google Scholar]
- Flajolet M, Tiollais P, Buendia MA, Fourel G 1998. Woodchuck hepatitis virus enhancer I and enhancer II are both involved in N-myc2 activation in woodchuck liver tumors. J Virol 72: 6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery JG, Bird AC, Farber DB, Weleber RG, Bok D 1990. A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Scie 31: 229–236. [PubMed] [Google Scholar]
- Furgoch MJ, Mewes-Ares J, Radziwon A, MacDonald IM 2014. Molecular genetic diagnostic techniques in choroideremia. Mol Vis 20: 535–544. [PMC free article] [PubMed] [Google Scholar]
- Gibbs D, Kitamoto J, Williams DS 2003. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci 100: 6481–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, et al. 2008. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum Gene Ther 19: 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Boye SL, Aleman TS, Conlon TJ, Zeiss CJ, Roman AJ, Cideciyan AV, Schwartz SB, Komaromy AM, Doobrajh M, et al. 2006a. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in Leber congenital amaurosis. Hum Gene Ther 17: 845–858. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Sumaroka A, Aleman TS, Schwartz SB, Windsor EA, Roman AJ, Stone EM, MacDonald IM 2006b. Remodeling of the human retina in choroideremia: Rab escort protein 1 (REP-1) mutations. Invest Ophthalmol Vis Sci 47: 4113–4120. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, et al. 2012. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: Safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130: 9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, et al. 2007. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 369: 2097–2105. [DOI] [PubMed] [Google Scholar]
- Klein RL, Hamby ME, Gong Y, Hirko AC, Wang S, Hughes JA, King MA, Meyer EM 2002. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp Neurol 176: 66–74. [DOI] [PubMed] [Google Scholar]
- Komaromy AM, Alexander JJ, Rowlan JS, Garcia MM, Chiodo VA, Kaya A, Tanaka JC, Acland GM, Hauswirth WW, Aguirre GD 2010. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet 19: 2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock BL, Bilotta J, Perkins BD 2007. Noncell-autonomous photoreceptor degeneration in a zebrafish model of choroideremia. Proc Natl Acad Sci 104: 4600–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larijani B, Hume AN, Tarafder AK, Seabra MC 2003. Multiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseases. J Biol Chem 278: 46798–46804. [DOI] [PubMed] [Google Scholar]
- Lebherz C, Maguire A, Tang W, Bennett J, Wilson JM 2008. Novel AAV serotypes for improved ocular gene transfer. J Gene Med 10: 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ 1999. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: Implications for gene therapy. Hum Gene Ther 10: 2295–2305. [DOI] [PubMed] [Google Scholar]
- Lotery AJ, Yang GS, Mullins RF, Russell SR, Schmidt M, Stone EM, Lindbloom JD, Chiorini JA, Kotin RM, Davidson BL 2003. Adeno-associated virus type 5: Transduction efficiency and cell-type specificity in the primate retina. Hum Gene Ther 14: 1663–1671. [DOI] [PubMed] [Google Scholar]
- MacDonald IM, Mah DY, Ho YK, Lewis RA, Seabra MC 1998. A practical diagnostic test for choroideremia. Ophthalmology 105: 1637–1640. [DOI] [PubMed] [Google Scholar]
- MacDonald IM, Russell L, Chan CC 2009. Choroideremia: New findings from ocular pathology and review of recent literature. Surv Ophthalmol 54: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren RE 2009. An analysis of retinal gene therapy clinical trials. Curr Opin Mol Ther 11: 540–546. [PMC free article] [PubMed] [Google Scholar]
- MacLaren RE, Bird AC, Sathia PJ, Aylward GW 2005. Long-term results of submacular surgery combined with macular translocation of the retinal pigment epithelium in neovascular age-related macular degeneration. Ophthalmology 112: 2081–2087. [DOI] [PubMed] [Google Scholar]
- MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FPM, Black GCM, et al. 2014. Initial observations in patients undergoing retinal gene therapy for choroideremia. Lancet (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, et al. 2008. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358: 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso K, Hendrickson AE, Connor TB Jr, Mauck MC, Kinsella JJ, Hauswirth WW, Neitz J, Neitz M 2007. Recombinant adeno-associated virus targets passenger gene expression to cones in primate retina. J Opt Soc Am A 24: 1411–1416. [DOI] [PubMed] [Google Scholar]
- Martin EA 2010. Concise medical dictionary. Oxford University Press, Oxford. [Google Scholar]
- Mauthner L 1872. Ophthalmologische. Mittheilungen: 2. Ein fall von chorioideremie. in Berichte des naturwissenschaftlichen-medizinischen Verein Innsbruck, pp. 191–197 Medical and Scientific Association Innsbruck. [Google Scholar]
- McTaggart KE, Tran M, Mah DY, Lai SW, Nesslinger NJ, MacDonald IM 2002. Mutational analysis of patients with the diagnosis of choroideremia. Hum Mutat 20: 189–196. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, Ragni MV, Manno CS, Sommer J, Jiang H, et al. 2007. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat Med 13: 419–422. [DOI] [PubMed] [Google Scholar]
- Miyazaki J, Takaki S, Araki K, Tashiro F, Tominaga A, Takatsu K, Yamamura K 1989. Expression vector system based on the chicken β-actin promoter directs efficient production of interleukin-5. Gene 79: 269–277. [DOI] [PubMed] [Google Scholar]
- Moosajee M, Tulloch M, Baron RA, Gregory-Evans CY, Pereira-Leal JB, Seabra MC 2009. Single choroideremia gene in nonmammalian vertebrates explains early embryonic lethality of the zebrafish model of choroideremia. Invest Ophthalmol Vis Sci 50: 3009–3016. [DOI] [PubMed] [Google Scholar]
- Myers T 2006. Mosby’s dictionary of medicine, nursing & health professions. Elsevier, London. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108: 193–199. [DOI] [PubMed] [Google Scholar]
- Pameyer JK, Waardenburg PJ, Henkes HE 1960. Choroid eremia. Br J Ophthalmol 44: 724–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterna JC, Moccetti T, Mura A, Feldon J, Bueler H 2000. Influence of promoter and WHV post-transcriptional regulatory element on AAV-mediated transgene expression in the rat brain. Gene Ther 7: 1304–1311. [DOI] [PubMed] [Google Scholar]
- Pfarr DS, Rieser LA, Woychik RP, Rottman FM, Rosenberg M, Reff ME 1986. Differential effects of polyadenylation regions on gene expression in mammalian cells. DNA 5: 115–122. [DOI] [PubMed] [Google Scholar]
- Pylypenko O, Rak A, Reents R, Niculae A, Sidorovitch V, Cioaca MD, Bessolitsyna E, Thoma NH, Waldmann H, Schlichting I, et al. 2003. Structure of Rab escort protein-1 in complex with Rab geranylgeranyltransferase. Mol Cell 11: 483–494. [DOI] [PubMed] [Google Scholar]
- Reese BE, Harvey AR, Tan SS 1995. Radial and tangential dispersion patterns in the mouse retina are cell-class specific. Proc Natl Acad Sci 92: 2494–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankila EM, Tolvanen R, van den Hurk JA, Cremers FP, de la Chapelle A 1992. Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nat Genet 1: 109–113. [DOI] [PubMed] [Google Scholar]
- Seabra MC, Brown MS, Slaughter CA, Sudhof TC, Goldstein JL 1992. Purification of component A of Rab geranylgeranyl transferase: Possible identity with the choroideremia gene product. Cell 70: 1049–1057. [DOI] [PubMed] [Google Scholar]
- Seabra MC, Brown MS, Goldstein JL 1993. Retinal degeneration in choroideremia: Deficiency of Rab geranylgeranyl transferase. Science 259: 377–381. [DOI] [PubMed] [Google Scholar]
- Seabra MC, Ho YK, Anant JS 1995. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 270: 24420–24427. [DOI] [PubMed] [Google Scholar]
- Shi W, van den Hurk JA, Alamo-Bethencourt V, Mayer W, Winkens HJ, Ropers HH, Cremers FP, Fundele R 2004. Choroideremia gene product affects trophoblast development and vascularization in mouse extra-embryonic tissues. Dev Biol 272: 53–65. [DOI] [PubMed] [Google Scholar]
- Starr CJ, Kappler JA, Chan DK, Kollmar R, Hudspeth AJ 2004. Mutation of the zebrafish choroideremia gene encoding Rab escort protein 1 devastates hair cells. Proc Natl Acad Sci 101: 2572–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed N, Smith JE, John SK, Seabra MC, Aguirre GD, Milam AH 2001. Evaluation of retinal photoreceptors and pigment epithelium in a female carrier of choroideremia. Ophthalmology 108: 711–720. [DOI] [PubMed] [Google Scholar]
- Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, Banfi S, Surace EM, Sun J, Acerra C, et al. 2013. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital amaurosis type 2. Ophthalmology 120: 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachova T, Ramalho JS, Anant JS, Schultz RA, Huxley CM, Seabra MC 1999. Cloning, mapping and characterization of the human RAB27A gene. Gene 239: 109–116. [DOI] [PubMed] [Google Scholar]
- Tolmachova T, Anders R, Abrink M, Bugeon L, Dallman MJ, Futter CE, Ramalho JS, Tonagel F, Tanimoto N, Seeliger MW, et al. 2006. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest 116: 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachova T, Wavre-Shapton ST, Barnard AR, MacLaren RE, Futter CE, Seabra MC 2010. Retinal pigment epithelium defects accelerate photoreceptor degeneration in cell type-specific knockout mouse models of choroideremia. Invest Ophthalmol Vis Sci 51: 4913–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachova T, Tolmachov OE, Barnard AR, de Silva SR, Lipinski DM, Walker NJ, Maclaren RE, Seabra MC 2013. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med 91: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, van den Hurk JA, Bogerd L, Philippe C, Gilgenkrantz S, de Jong P, Ropers HH, Cremers FP 1994. Cloning and characterization of the human choroideremia gene. Hum Mol Genet 3: 1041–1046. [DOI] [PubMed] [Google Scholar]
- van den Hurk JA, Hendriks W, van de Pol DJ, Oerlemans F, Jaissle G, Ruther K, Kohler K, Hartmann J, Zrenner E, van Bokhoven H, et al. 1997. Mouse choroideremia gene mutation causes photoreceptor cell degeneration and is not transmitted through the female germline. Hum Mol Genet 6: 851–858. [DOI] [PubMed] [Google Scholar]
- van den Hurk JA, van de Pol DJ, Wissinger B, van Driel MA, Hoefsloot LH, de Wijs IJ, van den Born LI, Heckenlively JR, Brunner HG, Zrenner E, et al. 2003. Novel types of mutation in the choroideremia (CHM) gene: A full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum Genet 113: 268–275. [DOI] [PubMed] [Google Scholar]
- Wolf S 1930. Choroideremia. Arch Ophthalmol 3: 80–87. [Google Scholar]
- Wu Z, Yang H, Colosi P 2010. Effect of genome size on AAV vector packaging. Mol Ther 18: 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Daly T, Gao C, Flotte TR, Song S, Byrne BJ, Sands MS, Parker Ponder K 2001. CMV-β-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1α promoter and results in therapeutic levels of human factor X in mice. Hum Gene Ther 12: 563–573. [DOI] [PubMed] [Google Scholar]