

Abstract
Peroxiredoxins were not recognized as a family of enzymes until the 1990s but are now known to be the dominant peroxidases in most organisms. Here, the history and fundamental properties of peroxiredoxins are briefly reviewed, with a special focus on describing how an exquisitely tunable balance between fully folded and locally unfolded conformations plays a large role in peroxiredoxin catalytic properties.
Keywords: Hydrogen peroxide, oxidative stress, redox signaling, chaperone, floodgate hypothesis
The discovery of Peroxiredoxins as nature’s dominant peroxidases
It has long been known that cellular antioxidant enzymes provide protection from reactive oxygen species such as the superoxide radical anion, hydrogen peroxide, and the hydroxyl radical that can cause toxicity through oxidizing nucleic acids, proteins, and lipids. In terms of antioxidant defenses, it is a few peroxidases, like catalase, glutathione peroxidases (Gpxs), and peroxiredoxins (Prxs) that are most important because they have a primary purpose of reducing peroxides. In contrast, for a second group of peroxidases – including the heme peroxidases such as horseradish peroxidase and myeloperoxidase – the primary purpose is to use the peroxide as an oxidizing agent to oxidize a second molecule. This review focuses only on the first of these groups. Among such peroxidases, the two historically best-known ones, catalase and glutathione peroxidase (Gpx), were discovered in 1900 [1] and 1957 [2], respectively, and have been extensively studied. To carry out catalysis, these enzymes have special cofactors, either a heme (catalase) or a seleno-cysteine residue (Gpx), and they are very effective catalysts with second order rate constant kcat/KM values of near 108 M−1 s−1, and 107 M−1 s−1, respectively.
Until quite recently, these two enzyme types were thought to be the major peroxide reducing enzymes protecting cells. The shift began in 1994, when protein sequence comparisons led to the recognition of a third abundant and widespread group of peroxidases [3]. The name peroxidoxins was first proposed for this group, but this quickly morphed to become the currently used peroxiredoxins (Prxs) [4]. The characterized enzymes grouped together in that study included just three, known at the time as thiol-specific antioxidants (TSA) from yeast and from rat, and alkyl hydroperoxide reductase C (AhpC) from Salmonella typhimurium. These enzymes were quite distinct from catalases and Gpxs especially in that they had no special cofactor, but simply used cysteine residues for catalysis. Either one or two Cys residues can be involved in catalysis and based on their roles [5], it was proposed by Wood et al. [6] that they be called the ‘peroxidatic’ Cys or CP and, when present, the ‘resolving’ Cys or CR. As seen in Figure 1, for catalysis the peroxidatic Cys (CP) thiolate reacts directly with peroxide and is converted to a CP-sulfenic acid. In those Prxs having a second ‘resolving’ Cys (CR), it then reacts with the CP-sulfenic acid to form a disulfide. This disulfide is then subsequently reduced by a thiol reductant such as thioredoxin (Trx) to complete the catalytic cycle.
Figure 1.
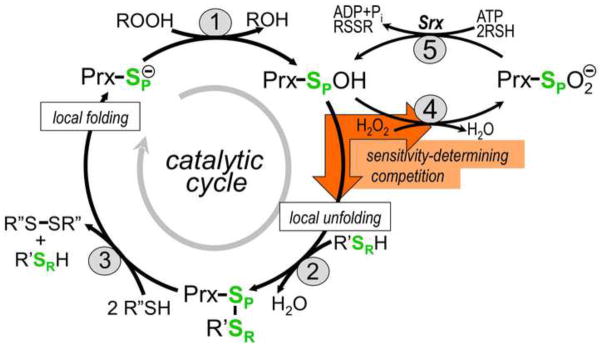
The interplay of five chemical steps and two conformational states in Prx catalysis. The steps are described in the text. The orange block arrows emphasize that the sensitivity of a Prx to hyperoxidation depends on the competition between the hyperoxidation (reaction 4, requiring the FF conformation) and the resolution (reaction 2, requiring the LU conformation.
These enzymes were still for many years thought of as less important than catalase and Gpx because they appeared to be much less efficient catalysts, having reported second-order rate constants of only ~104 M−1 s−1 [7]. However, in an important breakthrough, it was recognized that the ~104 M−1 s−1 value was not reflecting the intrinsic rate of peroxide reduction, but the rate at which the disulfide form of the Prx was reduced by the external reductant added to the assay; after addressing this issue for S. typhimurium AhpC, measurements of the intrinsic limit of the reaction with peroxide yielded a kcat/KM value in excess of 107 M−1 s−1 [8, 9], a range now seen to be typical for Prxs and making it clear that their efficiencies are on par with catalase and Gpx. At the same time, evidence was accumulating that Prxs tend to be much more highly expressed than catalase and Gpxs. For instance in yeast, estimates of protein abundance obtained by GFP-tagging every protein [10] showed that the most abundant Prx was expressed at 50-fold higher levels than the most abundant Gpx, and was 500-fold more abundant than the most abundant catalase (Table 1). In mammals, there are six Prxs (PrxI through VI) and among these, PrxI (cytosolic) and PrxIII (mitochondrial) appear to be the ones relatively highly expressed in all tissue types (Figure 2).
Table 1.
Estimated levels of peroxiredoxins, glutathione peroxidases, and catalases in Saccharomyces cerevisiae. Data based on GFP tagged proteins from [10]. Alternate names of the five Prxs in S. cerevisiae are given.
| enzyme | Estimated molecules/cell | Prx subfamily | Cellular location |
|---|---|---|---|
| cTpxI (Tsa1, YML028W) | 378,000 | Prx1 | cytosol |
| cTpxII (Tsa2, YDR453C) | 4,800 | Prx1 | cytosol |
| cTpxIII (Ahp1p, YLR109W) | 16,200 | Prx5 | cytosol |
| mTpx (Prx1p, YBL064C) | 4,500 | Prx6 | mitochondria |
| nTpx (Dot5p, YIL010W) | 1,840 | PrxQ | nucleus |
| Gpx1 | Low signal | ||
| Gpx2 | 2,000 | ||
| Gpx3 | 8,100 | ||
| Catalase A | 600 | ||
| Catalase T | 600 |
Figure 2.

Relative mRNA expression levels for six mammalian Prxs in bovine tissues. For more detail about the tissue types and experimental details, see the work from which the data were taken [11].
Putting together the high activity of Prxs with their high abundance, it has been estimated that in human cells over 99% of peroxide in the cytosol and over 90% of peroxide in the mitochondria will react with peroxiredoxins rather than other enzymes or small molecule thiols [12, 13]. This sense of the general importance of Prxs is further reinforced by the observation that many organisms, including some human pathogens (reviewed in [14]), have no catalase and/or Gpx enzymes, but they all have Prxs. Taken together, despite their much later discovery than catalase or Gpx, Prxs are now seen as the dominant peroxidases in most living organisms. As Prxs can also show high catalytic reactivity with peroxynitrite, they may also be important for defense against reactive nitrogen species [15].
Basic structural features of Prxs
Prxs are single domain proteins based on a thioredoxin fold that are presumed to have evolved from a thioredoxin-like ancestor [16], and the structural core common to the Prx family contains seven central β-strands that are surrounded by five α-helices. Structures of over 100 Prxs are now known, and they range from about 160 to 220 amino acid residues in length with the longer Prxs, of course, having additional secondary structural elements. Only four positions are highly conserved among all Prxs, and of these three are located in an eight-residue PxxxTxxC motif that makes up the loop preceding helix α2 and the first turn of the helix. The Cys in this segment is the peroxidatic Cys, and the loop leading up to it is called the CP-loop. The Pro and Cys are fully conserved and the Thr position is occasionally filled by a Ser. The fourth conserved position is an Arg residue that is located near the beginning of strand β6. As is further discussed below, these four residues are all close to one another in the fully-folded peroxidatic active site (Figure 3A).
Figure 3.


Views of the Prx active site and Prx1 subfamily decamer. A. The fully-folded active site pocket of a representative Prx is shown with the molecular surface (with grey carbons, red oxygens, blue nitrogens and yellow sulfurs. In the substrate binding pocket is shown an H2O2 molecule (green) as seen in the Michaelis complex of an Aeropyrum pernix Prx [17] and hydrogen bonds (cyan dashed lines) from backbone atoms and the conserved Thr and Arg side chains that stabilize the CP residue and substrate. Also shown are dozens of oxygens (red) representing the positions of water molecules that are bound to various un-liganded FF actives sites (gathered as described in [18]). The water positions clearly define a track along which the substrate oxygens will be stabilized as they move during the reaction. B. Decamer commonly seen for Prx1 subfamily enzymes such as mammalian PrxI, PrxII, PrxIII and PrxIV. Emphasized is how it is constructed of a pentamer of B-type dimers associating through the A-type interfaces.
In terms of quaternary structure, Prxs occur variously as monomers, A-type and B-type dimers, and decameric (or dodecameric) assemblies made of either five (or six) B-type dimers that associate into a ring by interacting via their A-type interfaces (Figure 3B). As defined by Sarma et al. [19], the B- (for β-sheet) type dimers associate via the edges of the β-strands so that the dimer forms a wide 14 stranded β-sheet. In contrast, the A- (for ancient or alternative) type dimers associate mostly via loops at the C-terminal ends of the parallel β-strands 3 through 7 [6, 19]. Two Prx structures have been reported to form octamers [20, 21], but these appear to be artifacts rather than representing true physiological forms [22, 23]. Also, Prx decamers are known in some circumstances – apparently promoted by formation of the hyperoxidized of CP-SO2− residue – to assemble into “high molecular weight” complexes involving stacks of decamers that can have protein-stabilizing chaperone activity [24–26]. Their ability to form these super-assemblies has led them to be considered recently as a useful building block for protein-based nanotechnology (e.g. [27]).
Five main evolutionary subfamilies of Prxs
As information available for Prx structures and sequences has grown, it has been recognized that most Prxs fall into five common subfamilies – Prx1 (or Prx1/AhpC), Prx6, Prx5, Tpx, and PrxQ (or PrxQ/BCP) – and a sixth possible subfamily – AhpE – that is not widely represented so cannot be confidently characterized [28]. Within each subfamily the percent sequence identities tend to be >30% and between subfamilies the identities are typically in the 15–30% range. For a structure-based alignment of sequences from representative family members see Hall et al. [29]. The online PREX database (http://csb.wfu.edu/prex/; [30]) contains much information about Prxs including the subfamily assignment for each known Prx sequence. One must be cautious about inferring anything from the name of a given Prx, because many Prxs have been given a generic name that does not reflect their subfamily. For instance, the Tpx name (short for thiol peroxidase) has been given to many enzymes that are not in the Tpx subfamily (see also examples in [14]). It is worth noting that the Prx1 subfamily enzymes, which are generally decameric, tend to be the ones that are most highly expressed.
The mechanisms of Prx catalysis
As is diagramed in Figure 1, in considering Prx catalytic activities one must consider five main chemical events and two distinct conformational states. The five chemical steps are (1) peroxidation, (2) resolution, (3) recycling, (4) hyperoxidation, and (5) resurrection; and the two conformational states are the “fully folded” (FF) and “locally unfolded” (LU). As far as is known, the resurrection step catalyzed by the enzyme sulfiredoxin (Srx) is only relevant for some Prx1 subfamily enzymes, but as was noted above, in eukaryotes these are often the enzymes that are the most highly expressed. Nevertheless, understanding Prx function involves taking into account the interplay of the chemical steps and the conformational changes.
The fully folded (FF) and locally unfolded (LU) conformational states
As was first described in a structural analysis of S. typhimurium AhpC [6], Prxs can exist in both FF and LU states. The FF state refers to a well-defined and well-conserved [29] active site protein conformation that has its CP thiolate (typically CP residues have pKa values < 7) at the bottom of a pocket that is ready to bind and react with a peroxide substrate (Figure 3A; [17, 18]). In this position, the limited steric accessibility of the CP side chain leads it to be strongly hindered both from being modified by thiol reagents and from reacting with another thiol to form a disulfide. This FF conformation must exist for all Prxs that have substantial peroxidase activity, because it is the conformation that promotes the reaction with peroxide.
In contrast, the LU state refers to a set of conformations that could include a substantial amount of variation both within a given Prx and between Prxs. The essential feature of the LU state is that the active site pocket is no longer formed and the CP side chain is exposed and available for reaction with (or already has reacted with) another thiol to form a disulfide – whether that thiol comes from the Prx itself (as for 2-Cys Prxs) or from another protein or small molecule (as for 1-Cys Prxs). Minimally, LU states have a rearrangement of the PxxxTxxC segment that includes movements of the CP-loop and a partial unwinding of the first turn of the α2 helix. In addition to the many LU conformations, some parameters influencing this transition that may be optimized for each Prx during evolution are the FF
 LU rates of transition and the FF
LU equilibrium constant in the CP-SH, CP-SOH, CP-SO2−, and CP-SO3− forms; in contrast, any form with CP involved in a disulfide bond will not be undergoing an FF
LU equilibrium, but the disulfide bond will effectively lock it into the LU form. For one Prx studied by NMR, the rates of opening and closing were seen to be very rapid, in the range of 1500 s−1 [31].
LU rates of transition and the FF
LU equilibrium constant in the CP-SH, CP-SOH, CP-SO2−, and CP-SO3− forms; in contrast, any form with CP involved in a disulfide bond will not be undergoing an FF
LU equilibrium, but the disulfide bond will effectively lock it into the LU form. For one Prx studied by NMR, the rates of opening and closing were seen to be very rapid, in the range of 1500 s−1 [31].
Another aspect of the FF
LU conformational change is a connection between quaternary structure and conformation that occurs for many members of the Prx1 subfamily that form donut-shaped decamers. For these proteins, the residues near the CP-loop participate in the decamer-building interface and the change in conformation to the LU state weakens these interactions and destabilizes the decamer. This means that unless the protein concentration is very high, when the enzyme becomes locked into the LU state by disulfide formation, the decamers will tend to dissociate into B-type dimers [6]. As the stability of the decamers can vary during evolution, not all subfamily Prx1 enzymes tend to dissociate into dimers (e.g. [32–34]), and the dissociation is of uncertain functional importance.
Peroxidation
The first step in the catalytic cycle is the nucleophilic SN2 attack of the CP-thiolate on the hydrogen peroxide substrate to form CP-SOH and water. As noted above this requires the FF enzyme form which provides a steric and electrostatic environment that enhances catalysis. It lowers the pKa of CP so it is largely deprotonated, yielding a thiolate that reacts at ~20 M−1 s−1 with peroxide [13, 35]. And, beyond this to provide the remaining 105-fold rate enhancement, the FF active site orients and activates the peroxide substrate for the displacement reaction by providing a hydrogen bonding and steric environment that creates an ‘oxygen track’ along which the reaction can take place [18] (Figure 3A). The protein environment geometry appears to be constructed such that the hydrogen bonding interactions will be most linear and strong at the transition state of the reaction (Figure 4). The synergistic hydrogen-bonding interactions of the side chains of the conserved Arg and Thr residues together with the backbone N-H atoms from CP and the preceding residue underscore the extent to which the FF active site is required for this step. Factors directing the specificity of Prxs toward organic peroxides as opposed to H2O2 itself are not well understood, but in some cases those enzymes – such as those from the Tpx and PrxV subfamilies – are seen to have a ‘hydrophobic collar’ around a part of the active site pocket that could make favorable interactions with the alkyl portion of the organic peroxides [18, 36].
Figure 4.

Proposed transition state stabilization by the conserved FF Prx FF active site. In the reaction transition state the S…OA bond is partially formed, the OA…OB bond is partially broken, and the central OA atom is expected to have two lone pairs and its hydrogen in a trigonal planar arrangement. The hydrogen bonds from the environment can be seen to be well-suited to stabilize exactly that structure. Adapted from [18].
Resolution
The resolution step involves a second thiol attacking the CP-SOH to create a disulfide and expel water (or an alcohol for organic peroxide substrates) as a leaving group (Figure 1). This step requires the locally unfolded state so that the CP-SOH group is accessible for attack by the second thiol. Based on typical protein dynamics, the CP-SOH form of the enzyme would be in a rapid equilibrium between the FF and LU conformations, and then the disulfide formation would covalently trap the protein into the LU conformation. A contrasting idea recently suggested for human PrxIV is that the CP-SOH form itself has the FF conformation so destabilized that it is effectively ‘locked’ in the LU form already [37]. As noted above, for so-called ‘2-Cys’ Prxs this second thiol comes from the protein itself, and is called the ‘resolving’ Cys or CR. In the various Prx families the CR residue has been seen to be located in five different places: in helix α2 just five residues after CP, in helix α3, in helix α5, in the C-terminal region of the other chain of a B-type dimer (Figure 5) and between strands β1 and β2 of the other chain of an A-type dimer [38]. For so-called ‘1-Cys’ Prxs this second thiol may be a small molecule thiol such as glutathione.
Figure 5.

Four positions of the resolving thiol in 2-Cys Prxs. Shown is a portion of a ribbon backbone diagram of a Prx1 subfamily B-type dimer in the FF conformation, highlighting side chains of the CP (red) and CR (pale blue from the C-terminus of the second chain). Also mapped to the backbone are the positions of CR in other Prx subfamilies that are in helix α2 (violet; as in some PrxQ subfamily members), helix α3 (green; as in most Tpx and some PrxQ subfamily members), helix α5 (orange; as in some PrxV subfamily members). Not shown is a fifth CR position seen in some PrxV subfamily members that is in the N-terminal region of the second chain of an A-type dimer [38]. The color-coded arrows indicate that forming the disulfide in each of these cases must involve a different kind of local unfolding for the CP and CR to come together.
Recycling
The third step of the normal catalytic cycle is the reductive recycling of the disulfide to reform the CP-thiolate form of the enzyme to be ready for the next catalytic cycle. This reduction is generally rather promiscuous and can be accomplished by a variety of thiol compounds including small molecules such as dithiothreitol. For many Prxs the physiological reductant appears to be thioredoxin, but for some there exists a specific Prx reductase that contains a thioredoxin-like domain. One example of this is the AhpF enzymes in bacteria that specifically reduce the AhpC Prxs [39]. In those recycling reactions that have been studied, the initial attack on the disulfide preferentially occurs on the sulfur from the CR-residue creating a mixed disulfide between the reductant and the CR residue, and freeing the CP-thiol [38, 40–42]. Importantly, the substrate for the recycling reaction is the locally unfolded form of the enzyme (trapped in that conformation by the disulfide bond) and after reduction, the Prx refolds to the fully folded, substrate-ready conformation.
Hyperoxidation
From the CP-SOH form of the enzyme a different reaction can take place instead of the “resolution” reaction described above. This is the hyperoxidation reaction that occurs if a second peroxide substrate binds to the active site pocket and reacts with the CP-SOH (Cys-sulfenic acid) to convert it to CP-SO2− (Cys-sulfinate). This form of the enzyme can no longer move through the catalytic cycle, but is stuck. As has been described [19], this reaction is thought only to occur in the fully folded active site after the OH of the CP-SOH group must rotate away from the substrate binding pocket to make room for the incoming peroxide. It has been recently shown that for human PrxII, this second reaction (CP-SOH to CP-SO2−) is about 1000 times slower than the first reaction [43]. For some Prxs the hyperoxidation process stops at this stage, but for others, by reacting with a third peroxide substrate the CP-SO2− can be further oxidized to become CP-SO3− (Cys-sulfonate) (e.g. [19]).
As is emphasized by the block arrows in Figure 1, the initial hyperoxidation step is in competition with the resolution step that leads to disulfide formation, so that rapid disulfide formation can be thought of as protecting the Prx from becoming hyperoxidized and inactivated. In Prxs for which the resolution step is disfavored and slow – for instance because the local unfolding step is unfavorable – the hyperoxidation will more readily occur. The rate of hyperoxidation (inactivations per catalytic cycle) is roughly linear with peroxide concentration and can be fit to a model for the inactivation kinetics that assumes that the FF and LU conformations are in rapid equilibrium [44]. As was recently described [45], the propensity for hyperoxidation of a Prx can be described as Chyp1%, the peroxide concentration at which 1% of the enzyme becomes inactivated per catalytic cycle. However, important to note is that even for Prxs that are intrinsically highly sensitive to hyperoxidation (i.e. a low Chyp1% value), it may be that in vivo they do not become easily hyperoxidized because the recycling step is slow, which both limits the total number of turnovers per enzyme and leaves the enzyme largely in the protected disulfide form. This seems to be the case for both PrxII in human red blood cells [46, 47] as well as for PrxIV in the endoplasmic reticulum [48].
Hyperoxidation can occur for all Prxs, but it is only some Prxs in subfamily Prx1 that appear to have a feature that was selected during their evolution that makes them particularly sensitive to hyperoxidation [44]. As far as is known, it is only these Prxs that are substrates for sulfiredoxin and can undergo the resurrection reaction described in the next section.
Resurrection
The final reaction included in Figure 1 is the ATP-dependent conversion of the inactive Prx Cys-sulfinate back to a sulfenic acid so its activity is resurrected and the enzyme can continue around the catalytic cycle. The Cys-sulfinate form of Prxs was considered to be irreversibly inactivated until it was reported that there was an ATP-dependent activity in cellular extracts that could reactivate it [49]. Shortly thereafter, using yeast as a hunting ground, the reaction was shown to be catalyzed by the enzyme sulfiredoxin (Srx) [50]. As recently reviewed [51], the reaction involves transfer of the γ-phosphoryl group of ATP to the sulfinate oxygen, followed by attack on the sulfinate sulfur by the Srx thiol to displace a phosphate and form a mixed Srx-Prx oxo-disulfide (i.e. a thiosulfinate). Using designed mutants, a very informative crystal structure of a complex of Prx in a mixed disulfide with Srx has been captured [52]. This structure shows that specificity in this reaction is partly provided by the C-terminal region of the Prx (that becomes disordered during the local unfolding of subfamily Prx1 enzymes) wrapping around the Srx in an intimate embrace. Indeed, as far as is now known, the resurrection reaction only occurs for Prxs from subfamily Prx1, consistent with these being the only group of Prxs for which facile hyperoxidation is a physiologically relevant process.
For what reason has Prx hyperoxidation been selected?
Originally Prx hyperoxidation was seen as an unfortunate by-product of having the reactive Cys-sulfenic acid intermediate necessarily being exposed to peroxide during the catalytic cycle [4, 53], basically along the lines of the old saying that “if you play with fire you’ll get burned.” But with the observations that facile hyperoxidation was not a general Prx property, and was associated with Prx1 subfamily enzymes having certain sequence fingerprints – a ‘GGLG’ segment halfway through the protein, and a ‘YF’ motif in a helix at the C-terminal end of the enzyme – it was proposed that the sensitivity to hyperoxidation was a feature of certain Prxs that had been selected for during the evolution of eukaryotes [44]. The discovery of the ATP-dependent resurrection activity associated with sulfiredoxin made the case even more compelling that the sensitivity to hyperoxidation must have been selected for during evolution. And, while there is still debate over the potential physiological purposes of the facile hyperoxidation of certain family Prx1 subfamily members, there is little doubt that it has been strongly selected, and so must serve one or more positive purposes. As has been summarized [54], roles for which there is some evidence include: the originally proposed role of a peroxide floodgate (Figure 6), that of a triage agent to conserve redox power under conditions of high oxidative stress [55], that of a protein chaperone function that has been observed for high molecular weight assemblies (multiple stacked decamers) of Prxs that are promoted by hyperoxidation [25], and that of an SOS ‘fire alarm’ signal serving as a checkpoint of sorts [56]. These four proposed roles are not exclusive, in that any or all of them may be true, and they are also not exhaustive in that there may be important roles that have not yet been recognized. It is worth noting that the first two of the proposed roles for sensitivity are based on the loss-of-function of the enzyme, and the second two are based on a gain-of-function associated with the hyperoxidized form. Also, worth noting is that among the proposed roles, the hypothesis that sensitive Prxs act as floodgates that allow for local buildup of peroxide concentrations for signaling purposes (Figure 6) is unique in that it is relevant to normal non-stress-related physiology as opposed to conditions of oxidative stress. One concrete example of a mitochondrial Prx acting as floodgate to help regulate corticosteroid biosynthesis was recently described [57].
Figure 6.
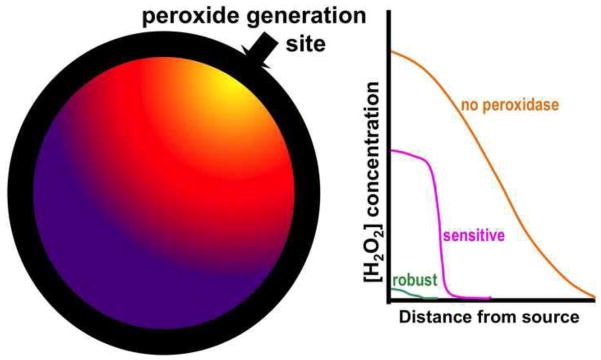
The floodgate hypothesis concept. The left-hand image shows how if peroxide were generated at one site in a cell it would form a radial peroxide gradient from high (yellow) to low (purple). The right hand sketch shows how the peroxide gradient would look in three scenarios: with no peroxidase a simple gradient (orange trace) would be generated; with a high amount of a robust peroxidase, little peroxide would build up (green trace); and with a high amount of a sensitive peroxidase, peroxide would build up locally where the peroxidase was inactivated but not further away where active peroxidase was still present (pink trace). Thus the ‘floodgate’ does not allow the whole cell to be flooded with peroxide, but creates a barrier-less compartmentalization of the peroxide buildup.
The delicate nature of the balances governing Prx catalytic properties
In the first report that certain Prx1 subfamily enzymes had a sensitivity to hyperoxidation that had been selected for during evolution, the sensitivity was found to be correlated with the presence of two sequence fingerprints, a ‘GGLG’ segment halfway through the protein, and a ‘YF’ motif in a helix at the C-terminal end of the enzyme [44]. The C-terminal ‘YF’ motif packed against, and could be seen to stabilize, the FF active site region and mutations altering or removing the C-terminal ‘YF’ motif converted the enzymes to become much more robust [58, 59]. Now, as can be gathered from the above description of the catalytic cycle, it must be understood that sensitivity versus robustness to hyperoxidation is more complicated; it is not an ‘either/or’ phenomenon, but is a continuum with the relative stabilities of the FF and LU states being a crucially important factor governing Prx functionality [60]; Figure 7). If a Prx has an FF active site that is too stable then it will carry out the peroxidation step readily, but the resolution step will be slow and the enzyme will thus be very sensitive to hyperoxidation. In contrast, if a Prx has an FF active site that is not stable enough, then the peroxidation step will not work well but it will be protected against hyperoxidation. This implies that in general Prxs would be expected have their FF/LU equilibria delicately balanced, and it should be expected that single mutations at a variety of places might easily shift properties fairly strongly one way or the other, i.e. toward being more sensitive to hyperoxidation and a more potent peroxidase, or more robust to hyperoxidation but also less potent as a peroxidase ([60]; Figure 7).
Figure 7.

How FF
LU thermodynamics impact Prx functionality. The series of violet to red curves are steps of 1.36 kcal/mol changes in the relative stability of the FF and LU conformations, and the number with each curve gives the equilibrium [FF]/[LU] ratio that would yield. Striking is how comparing the blue to the yellow curve, just a 2.72 kcal/mol change in relative stability alters the fraction of protein in the FF state from ~10% to ~90% and would result in a roughly 10-fold increase in peroxidase activity and a 100-fold increase in susceptibility to hyperoxidation. Yet a further 2.72 kcal/mol change in relative stability (to the red curve) would increase peroxidase activity only another 10%, but increase susceptibility to hyperoxidation by another 100-fold. Adapted from [60].
This delicate balance has been most well documented in Prx1 subfamily enzymes, such as human PrxII and PrxIII and Salmonella typhimurium AhpC. The PrxII and PrxIII enzymes are forms that are naturally rather sensitive to hyperoxidation, but with PrxII being about 10-fold more sensitive than PrxIII [61], and the S. typhimurium AhpC is naturally rather robust against hyperoxidation. For all of these enzymes mutations that alter the stability of the decamer and/or the FF active site region and/or the FF C-terminal region influence catalysis and the proclivity toward hyperoxidation. In one detailed study, a set of four C-terminal residues were identified that differed between human PrxII and PrxIII, and by mutating these, PrxII was made less sensitive to hyperoxidation, and PrxIII was made more sensitive [62]. In a study of S. typhimurium AhpC, the thermodynamic influence of decamer formation on the stability of the FF active site was shown in that a point mutation far from the active site but that disrupted decamer formation both decreased the kcat/KM for peroxidation by a factor of 100 and also decreased the enzyme’s sensitivity to hyperoxidation [63]. Furthermore, using S. typhimurium AhpC we have recently shown that even very subtle mutations, such as the CP-> Ser, CR-> Ser or CR-> Ala mutations that are often used to study Prx properties actually can measurably shift the FF/LU equilibrium and change the enzyme properties [60]. This latter result provides a sobering reminder that in some cases the approach used to study Prx catalysis may alter the very properties one is interested in characterizing.
Also, closely related to this, post-translational modifications can modulate activity and sensitivity to hyperoxidation and may have important physiological roles. It has been recently shown that acetylation of a single C-terminal lysine [64] or nitration of a single C-terminal tyrosine [65] that occur in vivo destabilize the FF state of human PrxII and make the enzyme more robust, and, in the case of acetylation, enhance its chaperone activity [66]. Recently, we have suggested that targeting the stabilization of either the FF or the LU conformation of Prxs provides a novel strategy for Prx inhibitor design [60]. Stabilizing the FF form would lead to Prx inhibition through promoting hyperoxidation, and stabilizing the LU form would lead to Prx inhibition through slowing peroxidation. Because such inhibitors need not target the conserved active site there is a greater opportunity for developing selective inhibitors.
Pointers to further information
In this primer on the basics of the structural, catalytic, and conformational properties of Prxs, I have sought to emphasize how the modulation of the stabilities of their FF and LU conformations is a crucially important factor that influences the activities of Prxs and is also a factor that during evolution will have been subjected to optimization for each Prx so as to make it well suited for its purpose. Given the apparent importance of the Prxs both for redox defense and for modulating redox signaling, these principles should aid researchers in designing and interpreting studies that sort out the roles of Prxs as well as in successfully targeting Prxs for therapeutic interventions. For those interested in learning more about Prxs, recent reviews and articles provide more in-depth information about structure-function relations and enzymology (e.g. [29]), about the potential roles of Prxs as triage agents [67], and chaperones (e.g. [26, 68]) and in redox signaling, aging, and disease (e.g. [69–76]), about the use of Prxs in nanotechnology [27], and about Srx [51, 77].
Highlights.
Peroxiredoxins are crucial for redox homeostasis of most organisms
Some peroxiredoxins have a build in sensitivity to oxidative inactivation
The five chemical steps and two conformations relating to function are described
Peroxiredoxin function is exquisitely governed by a conformational equilibrium
Acknowledgments
This study was supported in part by National Institutes of Health grant RO1 GM050389.
I thank members of my research group and collaborators for making many of the contributions reported here, and former graduate student Andrea Hall for making the image that forms the basis of figure 3A. This work was supported in part by National Institutes of Health grant R01 GM50389 to Leslie B. Poole and P.A.K..
ABBREVIATIONS
- Prx
peroxiredoxin
- Gpx
glutathione peroxidase
- TSA
thiol-specific antioxidant
- AhpC
alkyl hydroperoxide reductase C
- CP
peroxidatic Cys
- CR
resolving Cys
- FF
fully folded
- LU
locally unfolded
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loew O. A New Enzyme of General Occurrence in Organisms. Science. 1900;11:701–702. doi: 10.1126/science.11.279.701. [DOI] [PubMed] [Google Scholar]
- 2.Mills GC. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem. 1957;229:189–197. [PubMed] [Google Scholar]
- 3.Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem. 1994;269:27670–27678. [PubMed] [Google Scholar]
- 5.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 6.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann B, Hecht HJ, Flohe L. Peroxiredoxins. Biol Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 8.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci USA. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 10.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 11.Leyens G, Donnay I, Knoops B. Cloning of bovine peroxiredoxins-gene expression in bovine tissues and amino acid sequence comparison with rat, mouse and primate peroxiredoxins. Comp Biochem Physiol B Biochem Mol Biol. 2003;136:943–955. doi: 10.1016/s1096-4959(03)00290-2. [DOI] [PubMed] [Google Scholar]
- 12.Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J. 2010;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 13.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Rad Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Gretes MC, Poole LB, Karplus PA. Peroxiredoxins in parasites. Antioxid Redox Signal. 2012;17:608–633. doi: 10.1089/ars.2011.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 16.Copley SD, Novak WR, Babbitt PC. Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry. 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T, Kado Y, Yamaguchi T, Matsumura H, Ishikawa K, Inoue T. Crystal structure of peroxiredoxin from Aeropyrum pernix K1 complexed with its substrate, hydrogen peroxide. J Biochem. 2010;147:109–115. doi: 10.1093/jb/mvp154. [DOI] [PubMed] [Google Scholar]
- 18.Hall A, Parsonage D, Poole LB, Karplus PA. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarma GN, Nickel C, Rahlfs S, Fischer M, Becker K, Karplus PA. Crystal structure of a novel Plasmodium falciparum 1-Cys peroxiredoxin. J Mol Biol. 2005;346:1021–1034. doi: 10.1016/j.jmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 20.Qiu W, Dong A, Pizarro JC, Botchkarsev A, Min J, Wernimont AK, Hills T, Hui R, Artz JD. Crystal structures from the Plasmodium peroxiredoxins: new insights into oligomerization and product binding. BMC Struct Biol. 2012;12:2. doi: 10.1186/1472-6807-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S, Peterson NA, Kim MY, Kim CY, Hung LW, Yu M, Lekin T, Segelke BW, Lott JS, Baker EN. Crystal Structure of AhpE from Mycobacterium tuberculosis, a 1-Cys peroxiredoxin. J Mol Biol. 2005;346:1035–1046. doi: 10.1016/j.jmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 22.Gretes MC, Karplus PA. Observed octameric assembly of a Plasmodium yoelii peroxiredoxin can be explained by the replacement of native “ball-and-socket” interacting residues by an affinity tag. Protein Sci. 2013;22:1445–1452. doi: 10.1002/pro.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karplus PA, Hall A. Structural survey of the peroxiredoxins. Subcell Biochem. 2007;44:41–60. doi: 10.1007/978-1-4020-6051-9_3. [DOI] [PubMed] [Google Scholar]
- 24.Saccoccia F, Di Micco P, Boumis G, Brunori M, Koutris I, Miele AE, Morea V, Sriratana P, Williams DL, Bellelli A, Angelucci F. Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure. 2012;20:429–439. doi: 10.1016/j.str.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Angelucci F, Saccoccia F, Ardini M, Boumis G, Brunori M, Di Leandro L, Ippoliti R, Miele AE, Natoli G, Scotti S, Bellelli A. Switching between the alternative structures and functions of a 2-Cys peroxiredoxin, by site-directed mutagenesis. J Mol Biol. 2013;425:4556–4568. doi: 10.1016/j.jmb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Phillips AJ, Littlejohn J, Yewdall NA, Zhu T, Valery C, Pearce FG, Mitra AK, Radjainia M, Gerrard JA. Peroxiredoxin is a versatile self-assembling tecton for protein nanotechnology. Biomacromolecules. 2014;15:1871–1881. doi: 10.1021/bm500261u. [DOI] [PubMed] [Google Scholar]
- 28.Nelson KJ, Knutson ST, Soito L, Klomsiri C, Poole LB, Fetrow JS. Analysis of the peroxiredoxin family: using active-site structure and sequence information for global classification and residue analysis. Proteins. 2011;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soito L, Williamson C, Knutson ST, Fetrow JS, Poole LB, Nelson KJ. PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acid Res. 2011;39:D332–337. doi: 10.1093/nar/gkq1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aden J, Wallgren M, Storm P, Weise CF, Christiansen A, Schroder WP, Funk C, Wolf-Watz M. Extraordinary mus-ms backbone dynamics in Arabidopsis thaliana peroxiredoxin Q. Biochim Biophys Acta. 2011;1814:1880–1890. doi: 10.1016/j.bbapap.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 32.Barranco-Medina S, Kakorin S, Lazaro JJ, Dietz KJ. Thermodynamics of the dimer-decamer transition of reduced human and plant 2-cys peroxiredoxin. Biochemistry. 2008;47:7196–7204. doi: 10.1021/bi8002956. [DOI] [PubMed] [Google Scholar]
- 33.Matsumura T, Okamoto K, Iwahara S, Hori H, Takahashi Y, Nishino T, Abe Y. Dimer-oligomer interconversion of wild-type and mutant rat 2-Cys peroxiredoxin: disulfide formation at dimer-dimer interfaces is not essential for decamerization. J Biol Chem. 2008;283:284–293. doi: 10.1074/jbc.M705753200. [DOI] [PubMed] [Google Scholar]
- 34.Park JW, Piszczek G, Rhee SG, Chock PB. Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry. 2011;50:3204–3210. doi: 10.1021/bi101373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 36.Hall A, Sankaran B, Poole LB, Karplus PA. Structural changes common to catalysis in the Tpx peroxiredoxin subfamily. J Mol Biol. 2009;393:867–881. doi: 10.1016/j.jmb.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao Z, Tavender TJ, Roszak AW, Cogdell RJ, Bulleid NJ. Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a non-disulfide-bonded intermediate in the catalytic cycle. J Biol Chem. 2011;286:42257–42266. doi: 10.1074/jbc.M111.298810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lian FM, Yu J, Ma XX, Yu XJ, Chen Y, Zhou CZ. Structural snapshots of yeast alkyl hydroperoxide reductase Ahp1 peroxiredoxin reveal a novel two-cysteine mechanism of electron transfer to eliminate reactive oxygen species. J Biol Chem. 2012;287:17077–17087. doi: 10.1074/jbc.M112.357368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poole LB, Reynolds CM, Wood ZA, Karplus PA, Ellis HR, Li Calzi M. AhpF and other NADH:peroxiredoxin oxidoreductases, homologues of low Mr thioredoxin reductase. Eur J Biochem. 2000;267:6126–6133. doi: 10.1046/j.1432-1327.2000.01704.x. [DOI] [PubMed] [Google Scholar]
- 40.Budde H, Flohe L, Hofmann B, Nimtz M. Verification of the interaction of a tryparedoxin peroxidase with tryparedoxin by ESI-MS/MS. Biol Chem. 2003;384:1305–1309. doi: 10.1515/BC.2003.146. [DOI] [PubMed] [Google Scholar]
- 41.Jonsson TJ, Ellis HR, Poole LB. Cysteine reactivity and thiol-disulfide interchange pathways in AhpF and AhpC of the bacterial alkyl hydroperoxide reductase system. Biochemistry. 2007;46:5709–5721. doi: 10.1021/bi7001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poole LB. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, Winterbourn CC. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 45.Nelson KJ, Parsonage D, Karplus PA, Poole LB. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Meth Enzymol. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheah FC, Peskin AV, Wong FL, Ithnin A, Othman A, Winterbourn CC. Increased basal oxidation of peroxiredoxin 2 and limited peroxiredoxin recycling in glucose-6-phosphate dehydrogenase-deficient erythrocytes from newborn infants. FASEB J. 2014;28:3205–3210. doi: 10.1096/fj.14-250050. [DOI] [PubMed] [Google Scholar]
- 47.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 48.Cao Z, Subramaniam S, Bulleid NJ. Lack of an efficient endoplasmic reticulum-localized recycling system protects peroxiredoxin IV from hyperoxidation. J Biol Chem. 2014;289:5490–5498. doi: 10.1074/jbc.M113.529305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 50.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 51.Lowther WT, Haynes AC. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid Redox Signal. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jonsson TJ, Johnson LC, Lowther WT. Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature. 2008;451:98–101. doi: 10.1038/nature06415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277:38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 54.Karplus PA, Poole LB. Peroxiredoxins as molecular triage agents, sacrificing themselves to enhance cell survival during a peroxide attack. Mol Cell. 2012;45:275–278. doi: 10.1016/j.molcel.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Day AM, Brown JD, Taylor SR, Rand JD, Morgan BA, Veal EA. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol Cell. 2012;45:398–408. doi: 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 56.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, Jonsson TJ, Poole LB, Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kil IS, Lee SK, Ryu KW, Woo HA, Hu MC, Bae SH, Rhee SG. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell. 2012;46:584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 58.Koo KH, Lee S, Jeong SY, Kim ET, Kim HJ, Kim K, Song K, Chae HZ. Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch Biochem Biophys. 2002;397:312–318. doi: 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- 59.Sayed AA, Williams DL. Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J Biol Chem. 2004;279:26159–26166. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 60.Perkins A, Nelson KJ, Williams JR, Parsonage D, Poole LB, Karplus PA. The sensitive balance between the fully folded and locally unfolded conformations of a model peroxiredoxin. Biochemistry. 2013;52:8708–8721. doi: 10.1021/bi4011573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cox AG, Pearson AG, Pullar JM, Jonsson TJ, Lowther WT, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem J. 2009;421:51–58. doi: 10.1042/BJ20090242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haynes AC, Qian J, Reisz JA, Furdui CM, Lowther WT. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem. 2013;288:29714–29723. doi: 10.1074/jbc.M113.473470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci USA. 2008;105:9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Randall LM, Manta B, Hugo M, Gil M, Batthyany C, Trujillo M, Poole LB, Denicola A. Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem. 2014;289:15536–15543. doi: 10.1074/jbc.M113.539213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan Y, Jin JH, Yu Y, Wang J. Significant Enhancement of hPrx1 Chaperone Activity through Lysine Acetylation. Chembiochem. 2014;15:1773–1776. doi: 10.1002/cbic.201402164. [DOI] [PubMed] [Google Scholar]
- 67.Veal EA, Tomalin LE, Morgan BA, Day AM. The fission yeast Schizosaccharomyces pombe as a model to understand how peroxiredoxins influence cell responses to hydrogen peroxide. Biochem Soc Trans. 2014;42:909–916. doi: 10.1042/BST20140059. [DOI] [PubMed] [Google Scholar]
- 68.Konig J, Galliardt H, Jutte P, Schaper S, Dittmann L, Dietz KJ. The conformational bases for the two functionalities of 2-cysteine peroxiredoxins as peroxidase and chaperone. J Exp Bot. 2013;64:3483–3497. doi: 10.1093/jxb/ert184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nystrom T, Yang J, Molin M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Gene Dev. 2012;26:2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park J, Lee S, Lee S, Kang SW. 2-cys peroxiredoxins: emerging hubs determining redox dependency of Mammalian signaling networks. Int J Cell Biol. 2014;2014:715867. doi: 10.1155/2014/715867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 74.Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014;289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang T, Diaz AJ, Yen Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer (Dove Med Press) 2014;6:73–80. doi: 10.2147/BCTT.S61281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]