

Abstract
Oculopharyngeal muscular dystrophy (OPMD) is a rare myopathy for which validated outcome measures are lacking, posing a barrier to clinical trials. Our goal was to identify factors associated with impaired mobility in OPMD in order to guide development of surrogate endpoints in future clinical trials. 144 individuals with OPMD were included in this retrospective, single-center study. We made novel use of parametric time-to-event analysis to model age at initial use of assistive device for ambulation. We hypothesized that limb weakness and other markers of disease severity are associated with earlier use of assistive devices. 23.6% of individuals (34/144) progressed to use of assistive devices (mean age 66.0±9.6 y). Earlier age at assistive device was associated with hip flexion Medical Research Council grade ≤ 3 (p<0.0001), earlier disease onset (p<0.0001), and lack of blepharoptosis surgery (p=0.011). Markers of dysphagia severity were not associated with earlier progression to assistive devices. Our study is the first to show a statistical association between hip flexion weakness and impaired mobility in OPMD, indicating that hip flexion strength could be explored as a surrogate endpoint for use in clinical trials. Since severity of disease features may be discordant within individuals, composite outcome measures are warranted.
Keywords: Oculopharyngeal muscular dystrophy, Outcome measures, Mobility impairment, Time-to-event analysis, Natural history
1. Introduction
Oculopharyngeal muscular dystrophy (OPMD) is a rare, late-onset myopathy with worldwide incidence [1, 2]. Autosomal-dominant OPMD is caused by heterozygous mutations in the PABPN1 gene consisting of triplet-repeat (GCN) expansions coding for alanine [3, 4]. Large disease clusters occur in New Mexico, Quebec and Israel due to founder effects [5–7], though prevalence in the U.S is unknown. While OPMD was first described in 1915 [8] and its causative mutation discovered in 1998 [3], clinical trials have been few [4]. A critical barrier to designing clinical trials is the lack of validated outcome measures that can track disease progression and treatment effects [9].
While the most conspicuous features of OPMD are ptosis and dysphagia, a major limitation to relying on ptosis or dysphagia measures as markers of OPMD progression is that surgical interventions are often performed for these symptoms, thus altering their natural history [10, 11]. Because OPMD also causes limb weakness, experts have proposed using limb strength as an outcome measure [4]. However, before surrogate endpoints such as muscle strength can be used in clinical trials that support marketing approval of a drug, researchers must demonstrate that the surrogate measure is associated with impaired function [9]. Yet few studies have investigated the functional consequences of limb myopathy in OPMD. We found only 7 studies of OPMD with ≥ 10 participants that reported mobility impairment, with frequencies ranging from 9–81% [5, 7, 12–16]. No study demonstrated a correlation between muscle strength and impaired mobility in OPMD.
Our aim in this study was to identify factors statistically associated with impaired mobility in OPMD, using data from the largest cohort of OPMD patients in the U.S. Our primary outcome variable was age at first use of assistive device for ambulation. We hypothesized that limb weakness and other markers of disease severity are associated with earlier use of assistive devices. By identifying clinical variables associated with impaired mobility, we sought to identify potential surrogate endpoints for use in future clinical trials. Secondarily, we report estimates of minimum disease prevalence in New Mexico.
2. Patients and methods
2.1 Sample
This study was a retrospective chart review. Since our report of the New Mexico OPMD cluster [5], we established a dedicated OPMD clinic that has served as the tertiary referral center for the state. Using administrative records, we identified all patients with suspected OPMD referred to us between January 1, 2001, and December 31, 2011. This study was approved by the University of New Mexico’s Human Research Protections Office. Requirement for written informed consent was waived. We followed the STROBE statement for reporting of observational studies [17].
2.2 Inclusion and exclusion criteria
We included cases meeting established criteria for diagnosis of OPMD: 1) late-onset ptosis (or previous corrective surgery for ptosis) and dysphagia, and positive family history affecting ≥2 generations, OR 2) positive genetic test for OPMD [2]. We excluded cases with a negative OPMD gene test and cases with clinical data supporting a diagnosis other than OPMD (onset of ptosis or dysphagia before age 30 y, severe external ophthalmoplegia before age 60 y, or clinical or electromyographic myotonia). We excluded cases if use of assistive device (see Section 2.4.1) occurred before disease onset.
2.3 Prevalence
For prevalence calculations, we included all individuals with confirmed OPMD who resided in New Mexico and were alive at the end of 2011. To ascertain which patients were living at the end of 2011, we obtained records from the National Death Index. We used population sizes from the 2010 U.S. Census [18].
2.4 Analysis
2.4.1 Dependent variable
The primary outcome was age at first use of assistive device for ambulation (hereafter, “assistive device”; defined as cane, walker, scooter, or wheelchair). Age at first use of assistive device was ascertained based on historical information and physician observation at neuromuscular clinic visits. Individuals who temporarily used an assistive device (for example, due to recent trauma) were not considered to have reached the primary outcome.
2.4.2 Independent variables
Independent variables included gender, ethnicity (Hispanic/not Hispanic), age at earliest symptom (ptosis or dysphagia), hip flexion strength, blepharoptosis surgery (yes/no), dysphagia treatment (yes/no), gastrostomy (yes/no), body mass index (BMI), and Functional Comorbidity Index (FCI).
Blepharoptosis surgery included frontalis sling suspension, levator resection, and upper lid blepharoplasty [10]. Dysphagia procedures included esophageal dilatation, cricopharyngeal botulinum toxin injection, or cricopharyngeal myotomy. Interventions for ptosis and dysphagia are typically performed when symptoms are severe. When dysphagia is very severe, weight loss may occur and individuals may undergo gastrostomy [2, 19]. We therefore included BMI and gastrostomy as covariates related to dysphagia severity. To calculate BMI for individuals with missing heights, we imputed missing observations using means for males and females. For individuals with multiple weight measurements during the period of follow-up, we used weight recorded closest to time of first use of assistive device; for individuals who did not progress to use of assistive device, we used weight at last visit.
We abstracted strength in 15 muscle groups. During the study period, four attending physicians performed neuromuscular examinations; strength was documented using a 10-point Medical Research Council (MRC) scale. Since inter-rater reliability of MRC scores was not established, we used a conservative approach and converted the 10-point scale to a simplified 6-point scale (0, 1, 2, 3, 4, 5). If right and left muscle groups differed in score, an average was assigned before converting to the simplified scale. Strength at the visit closest to time of first use of assistive device was abstracted; if an individual had not progressed to use of assistive device by the last visit, then MRC score at the last visit was used. For time-to-event statistical analysis, we used hip flexion strength as an indicator of lower extremity myopathy since this muscle group was most affected in previous reports [16, 20]. In time-to-event models, we categorized hip flexion strength using a binary variable (MRC ≥4 vs. MRC ≤3).
Because comorbidity was a potential confounder of impaired mobility, we used the FCI as a covariate in our model. The FCI is a validated comorbidity index predictive of physical function; it is scored by assigning 1 point to each of 18 medical conditions (including arthritis, stroke, degenerative disk disease, and obesity) that are known to affect physical function [21]. In addition, we reported prevalence of peripheral neuropathy, cognitive impairment, cardiac disease, pneumonia and cancer. We counted medical conditions only if they were documented as diagnoses in clinic notes.
2.4.3 Statistics
We performed univariate comparisons of use of assistive device and other variables using t-tests and Wilcoxon tests for continuous measures and χ2 and Fisher’s exact tests for categorical measures (STATA 11.2, StataCorp LP, College Station, TX). For univariate comparisons, use of assistive device was a categorical variable (yes/no); this variable is distinct from the dependent variable used in our time-to-event analysis (age at first use of assistive device).
We used parametric time-to-event analysis (i.e., parametric survival analysis) to model age at first use of assistive device (PROC LIFEREG in SAS 9.3, SAS Institute, Cary, NC) [22]. The two methods that are most often employed to analyze time-to-event data are Kaplan-Meier estimation along with log-rank/Wilcoxon tests, which produce non-parametric estimates of the time-to-event distribution and comparisons between groups, and Cox (proportional hazards) regression, a semi-parametric procedure which models the effect of multiple predictors without assuming an underlying distribution but does assume proportional hazards between levels of predictors.
For both Kaplan-Meier and Cox methods, the standard form of data is either exact time-to-event or right-censored data (where the time recorded is only a lower bound on actual time). Our data were more complicated than this, however, because they included not only right-censored but also interval-censored and left-censored observations. We defined censoring as follows: When age at first use of assistive device (“age at first use”) was specified in the medical record, it was classified as uncensored. If age at first use occurred during a time interval (e.g., between two clinic visits), then it was classified as interval-censored. If age at first use occurred prior to the first visit but no further information was available to determine specific age, then it was classified as left-censored. If no assistive device was used by the last visit, then the dependent variable was classified as right-censored.
Standard software packages such as SAS do not allow data with such complex censoring for full estimation of survival distributions and inference on regression parameters in the Kaplan-Meier or Cox framework. Parametric survival analysis (LIFEREG in SAS) does provide a full solution to the problem for such data, with the main difference being that an explicit form for the survival (time-to-event) distribution must be specified (similar to assuming normal distributions in conventional regression). We assumed a Weibull form for the distribution, which is the most common form used, and that appeared reasonable in the regression diagnostics [22, 23].
The parametric time-to-event method allowed us to quantify the effect of various independent variables on the dependent time-to-event variable using multivariable regression models. In our study, the dependent time-to-event variable was age at initial use of assistive device, and the independent variables were gender, ethnicity, age at earliest symptom, history of blepharoptosis surgery, history of dysphagia treatment, gastrostomy, hip flexion strength, BMI, and FCI.
We summarized the results of the parametric time-to-event analysis in a table showing regression coefficients for each independent variable, 95% confidence intervals for the coefficients, and p-values. Each regression coefficient was interpreted as the change in age (in years) at initial use of assistive device for each unit change in the independent variable. The p-values associated with each regression coefficient indicate the statistical significance of each independent variable in the model. We used backward elimination to yield a final, parsimonious model that included only independent variables whose coefficients had p-values < 0.05. The final model was used to generate predicted time-to-event curves, assuming various values of the independent variables (R 3.0.3, R Foundation for Statistical Computing, Vienna, Austria) [24]. To verify the validity of using a binary variable for hip flexion strength, we constructed paired Wald tests of the coefficients for each MRC grade in the time-to-event model.
In accordance with the STROBE statement [17], we performed sensitivity analyses to evaluate the robustness of our results if study assumptions were slightly modified. In the first sensitivity analysis, we included individuals who had been excluded due to possible but unconfirmed OPMD (i.e., cases in which some but not all inclusion criteria for OPMD were met; such cases could represent early, less severe cases of OPMD). In the second sensitivity analysis, we included in our model a term corresponding to confirmed (GCN)13(GCN)10 genotype. This was to account for the possibility that model predictions differed for the group with confirmed (GCN)13(GCN)10 genotype.
3. Results
3.1 Patient characteristics
Of 170 individuals referred to our OPMD clinic with suspected OPMD, 144 met inclusion/exclusion criteria (Figure 1). Table 1 shows the demographic, clinical and genetic characteristics of the sample, for which there was 342 patient-years of follow-up. Mean age at earliest symptom was 52.5±6.9 y. All Hispanic patients with genetic test results had the (GCN)13(GCN)10 genotype. No individual in our OPMD clinic who met the clinical criteria for OPMD diagnosis (late-onset ptosis and dysphagia, and family history) and who underwent genetic testing had a negative genetic test result. Thirteen non-Hispanic individuals (9.0%) were of the following ancestries: French-Canadian, French, German, and non-Hispanic American.
Figure 1.
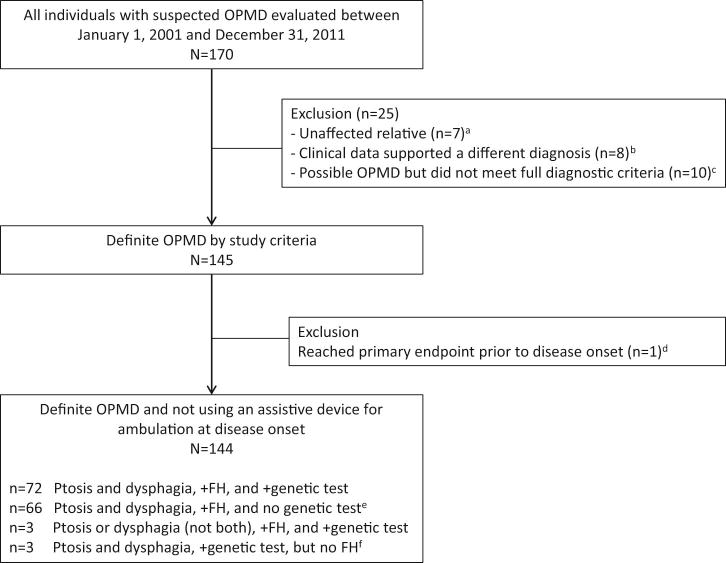
Flow chart showing derivation of study sample.
Abbreviations: FH = family history
aThese individuals had a positive family history of OPMD, normal neuromuscular examination, and negative genetic test for OPMD
bNone of these individuals had a positive family history of OPMD, and 7 of the 8 cases had a negative genetic test for OPMD. Three individuals were diagnosed with seronegative myasthenia gravis, 2 had severe external ophthalmoplegia before age 60 y, and 3 individuals had onset of ptosis and/or dysphagia before age 30 y.
cSix of the 10 individuals had a positive family history of OPMD, but did not meet full diagnostic criteria because they had only one cardinal symptom (ptosis or dysphagia, but not both).
dThis individual lost independent ambulation in youth secondary to trauma.
eThirty of these individuals were related to someone with a positive genetic test
fThese individuals had either a biological parent whose medical history was unknown or a parent who died before the age of 50 y.
Table 1.
Demographic, clinical, and genetic characteristics of the cohort (N=144).
| n (%) or mean (SD); median (IQR) | |
|---|---|
| Demographic characteristics | |
| Male sex | 60 (41.7) |
| Resident of New Mexico | 137 (95.1) |
| Hispanic ethnicity | 131 (91.0) |
| Clinical characteristics and follow-up | |
| Age at earliest symptom (ptosis or dysphagia), y | 52.5 (6.9); 53 (48–56)a |
| Ptosis | 141 (97.9) |
| Dysphagia | 144 (100) |
| Positive family history in ≥2 generations | 141 (97.9) |
| Age at first neuromuscular clinic visit, y | 63.1 (9.0); 62.5 (57.2–68.3) |
| Number of clinic visits | 2.8 (2.4); 2 (1–4) |
| Duration of follow-up, y | 2.4 (2.9); 1.1 (0–4.2) |
| Age at last neuromuscular clinic visit, y | 65.5 (9.0); 64.8 (59.2–71.1) |
| Genetic characteristics | |
| PABPN1 genotypeb | |
| GCN13, GCN10 | 56 (38.9) |
| GCN14, GCN10 | 2 (1.4)c |
| GCN15, GCN10 | 1 (0.7)c |
| Positive genetic test – genotype unknown | 19 (13.2)d |
| Genetic test not performed | 66 (45.8)e |
Abbreviations: SD = standard deviation; IQR = interquartile range
There were 6 missing observations (4.2%) for age at earliest symptom. First symptom was ptosis in 52, dysphagia in 37, and both in 49.
Wild-type alleles: (GCN)10; dominant alleles: (GCN)12–17. Genetic testing was performed through Athena Diagnostics, Inc (Worcester, MA).
These individuals were not Hispanic.
7 of these individuals belonged to a family in which the GCN13, GCN10 genotype was confirmed.
30 of these individuals belonged to a family in which at least one member had genetic testing that confirmed OPMD; 24 were from families with the GCN13, GCN10 genotype.
3.2 Prevalence
One hundred and thirty-eight individuals resided in New Mexico (including 10 non-Hispanic cases). Fifteen individuals (10.4%) died by the close of 2011 (mean age at death 74.6±7.6 y; range 64.7–87.2 y) and were excluded from prevalence calculations. Of these, 13 had resided in New Mexico (all Hispanic). Incorporating U.S. census data [18], the minimum prevalence of OPMD in New Mexico was 6.1 per 100,000. Among Hispanics in New Mexico, the minimum prevalence was 12.1 per 100,000. Among non-Hispanics, the minimum prevalence was 0.9 per 100,000.
3.3 Clinical course and comorbidities
Table 2 shows clinical procedures and comorbidities of the cohort. 77.1% (111/144) underwent blepharoptosis surgery (mean age 60.0±6.4 y; mean disease duration at first procedure 6.8±5.6 y), 39.6% (57/144) underwent procedures for dysphagia (mean age 64.4±8.7 y; mean disease duration at first procedure 11.1±8.0 y). The most commonly performed procedure for dysphagia was esophageal dilatation (32.6%; 47/144). Only 2 individuals underwent cricopharyngeal myotomy. Of those who underwent blepharoptosis surgery, 43% (48/111) had surgeries more than once (median 2, range 2 to 5). Of those who underwent procedures for dysphagia, 67% (38/57) underwent procedures more than once (median 3.5, range 2 to 13). Gastrostomy for nutritional support was performed in 6.9% of individuals (10/144) late in the disease (mean age 74.4±9.6 y; mean disease duration 18.9±8.7 y). The mean BMI of those who underwent gastrostomy was significantly lower than those who did not undergo gastrostomy (19.7±3.4 vs 25.6±4.5, p=0.0001, t-test).
Table 2.
Clinical course and comorbidities of the cohort (N=144).
| n (%) or mean (SD); median (IQR) | |
|---|---|
| Treatment of ptosis and dysphagia | |
| At least one blepharoptosis surgery | 111 (77.1) |
| Age at first blepharoptosis surgery, y | 60.0 (6.4); 59 (55–64) |
| Disease duration at first blepharoptosis surgery, y | 6.8 (5.6); 5 (2–10)a |
| At least one procedure for dysphagia | 57 (39.6) |
| Esophageal dilatation | 47 (32.6)b |
| Cricopharyngeal botulinum toxin injection | 25 (17.4)b |
| Cricopharyngeal myotomy | 2 (1.4) |
| Age at first procedure for dysphagia, y | 64.4 (8.7); 65 (58–70) |
| Disease duration at first procedure for dysphagia, y | 11.1 (8.0); 9 (5–17)c |
| Markers of nutritional status | |
| Gastrostomy | 10 (6.9) |
| Age at gastrostomy, y | 74.4 (9.6); 71 (67–84) |
| Disease duration at gastrostomy, y | 18.9 (8.7); 21 (12.5–24.5)d |
| Body mass index, kg/m2 | 25.3 (4.6); 24.8 (22.4–28.2)e |
| Comorbidities | |
| Functional Comorbidity Index | 2.4 (1.9); 2 (1–3) |
| Peripheral neuropathy | 8 (5.6) |
| Cognitive impairment | 6 (4.2)f |
| Cardiac electrical disturbance | 3 (2.1) |
| Cardiomyopathy | 2 (1.4) |
| History of pneumonia | 23 (16.0) |
| History of any cancer | 21 (14.6)g |
Abbreviations: SD = standard deviation; IQR = interquartile range
Calculated using n=107 due to 4 individuals who had blepharoptosis surgery but were missing age at earliest symptom.
1 individual underwent esophageal dilatation and cricopharyngeal botulinum toxin injection as a combined procedure.
Calculated using n=55 due to 2 individuals who underwent dysphagia procedures but were missing age at earliest symptom.
Calculated using n=8 due to 2 individuals who underwent gastrostomy but were missing age at earliest symptom.
There were 27 missing observations (18.8%) for height.
5 individuals had adult-onset cognitive impairment not otherwise specified, and 1 had intellectual impairment.
Cancer types included: breast (n=5), colon (n=4), prostate (n=4), lung (n=2), basal cell (n=2), Hodgkin’s lymphoma (n=1), acute promyelocytic leukemia (n=1), bladder (n=1), papillary thyroid (n=1), and endometrial (n=1).
The study cohort had a relatively low prevalence of comorbidities that affect physical function, as assessed by the FCI. The FCI ranges from 0–18, with higher scores indicating greater comorbidities. The median score on the FCI for our sample was 2 (interquartile range 1–3). Peripheral neuropathy, cognitive impairment, and cardiac disease were uncommon. A history of pneumonia was found in 16.0% (23/144); while this may reflect susceptibility to aspiration in OPMD, medical records did not allow us to distinguish aspiration pneumonia from other types. No pattern of unusual cancers was observed.
3.4 Pattern of limb weakness
Table e-1 (online) shows the pattern of weakness in 15 muscle groups. The pelvic girdle muscles (hip flexors, hip extensors, hip abductors, and hip adductors) were most commonly weak compared with other body regions. 76.4% (110/144) had some degree of hip flexion weakness. There was no case in which an individual had weakness in shoulder abductors, knee extensors, or ankle dorsiflexors without weakness of the hip flexors. Symmetric weakness was observed in nearly all cases.
Table 3 shows the degree of weakness by age group for the two muscle groups with the fewest missing observations: hip flexors (n=141) and shoulder abductors (n=130). As expected, in older age groups there was a higher proportion of individuals with weaker MRC grades.
Table 3.
Degree of proximal limb weakness by age group. Counts are shown, with percentages in parentheses.
| Muscle group | MRC 0 | MRC 1 | MRC 2 | MRC 3 | MRC 4 | MRC 5 | Total number |
|---|---|---|---|---|---|---|---|
| Shoulder abductors (n=130) | |||||||
| Age 30–39 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (100) | 0 (0) | 1 |
| Age 40–49 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 3 |
| Age 50–59 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 5 (13.5) | 32 (86.5) | 37 |
| Age 60–69 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 23 (44.2) | 29 (55.8) | 52 |
| Age 70–79 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 11 (35.5) | 20 (64.5) | 31 |
| Age 80–89 y | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 4 (66.7) | 1 (16.7) | 6 |
|
| |||||||
| Total number | 0 (0) | 0 (0) | 1 (0.8) | 0 (0) | 44 (33.8) | 85 (65.4) | 130 |
|
| |||||||
| Hip flexors (n=141) | |||||||
| Age 30–39 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (100) | 0 (0) | 1 |
| Age 40–49 y | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 0 (0) | 3 |
| Age 50–59 y | 0 (0) | 0 (0) | 0 (0) | 3 (7.7) | 19 (48.7) | 17 (43.6) | 39 |
| Age 60–69 y | 0 (0) | 0 (0) | 3 (5.3) | 11 (19.3) | 31 (54.4) | 12 (21.1) | 57 |
| Age 70–79 y | 0 (0) | 0 (0) | 4 (11.4) | 2 (5.7) | 27 (77.1) | 2 (5.7) | 35 |
| Age 80–89 y | 0 (0) | 0 (0) | 2 (33.3) | 0 (0) | 4 (66.7) | 0 (0) | 6 |
|
| |||||||
| Total number | 0 (0) | 0 (0) | 9 (6.4) | 16 (11.3) | 85 (60.3) | 31 (22.0) | 141 |
3.5 Impaired mobility and associated variables
Thirty-four individuals (23.6%) progressed to use of an assistive device (mean age 66.0±9.6 y; mean disease duration 14.0±7.3 y). Of these, 19 individuals used a cane, 9 used a walker, 3 used a scooter, and 3 used a wheelchair. Of 28 individuals who initially used a hand-held assistive device, 3 later used a scooter or wheelchair. Hip flexion strength was associated with use of assistive device (76.0% of individuals with MRC ≤3 vs. 12.9% of individuals with MRC ≥4, χ2-test, p<0.001). In contrast, markers of ptosis and dysphagia severity (blepharoptosis surgery, dysphagia treatment, gastrostomy, and BMI) were not associated with use of assistive device. Mean age at earliest symptom did not differ statistically between the groups that did and did not use assistive devices (52.0±8.6 y vs. 52.7±6.3 y, p=0.605, t-test).
Table 4 shows results of time-to-event analyses for the primary outcome of age at initial use of assistive device. In the final model, 3 independent variables were significantly associated with age at first use of assistive device for ambulation: hip flexion weakness, age at symptom onset, and blepharoptosis surgery. Hip flexion MRC ≤3 was associated with a 12.3 y earlier progression to use of assistive device (95% CI 6.8 y, 17.7 y; p<0.0001) compared with MRC ≥4. A 10-year delay in symptom onset was associated with a 5.5 y delay in progression to use of assistive device (95% CI 2.8 y, 8.2 y; p<0.0001). Blepharoptosis surgery was associated with a 7.3 y delay in progression to use of assistive device compared with no blepharoptosis surgery (95% CI 1.7 y, 12.9 y; p=0.011).
Table 4.
Analysis of effects in parametric time-to-event models (full model and final model), in which the dependent variable is age at first use of assistive device for ambulation.
| Independent variable | Regression coefficient | 95% confidence interval | p-value |
|---|---|---|---|
| Full model | |||
| Hip flexion strengtha | |||
| MRC ≥4 | 12.60 | 7.41, 17.78 | 0.008 |
| MRC ≤3 | Ref | Ref | |
| Gender | |||
| Female | −0.71 | −5.50, 4.09 | 0.773 |
| Male | Ref | Ref | |
| Ethnicity | |||
| Not Hispanic | −1.63 | −8.56, 5.29 | 0.644 |
| Hispanic | Ref | Ref | |
| Age at earliest symptom, yb | 0.36 | 0.04, 0.68 | 0.027 |
| BMI, kg/m2 | −0.34 | −0.96, 0.29 | 0.290 |
| Blepharoptosis surgery | |||
| Yes | 8.28 | 3.35, 13.20 | 0.001 |
| No | Ref | Ref | |
| Dysphagia treatment | |||
| Yes | −0.15 | −4.87, 4.58 | 0.952 |
| No | Ref | Ref | |
| Gastrostomy | |||
| Yes | 7.77 | −2.51, 18.04 | 0.139 |
| No | Ref | Ref | |
| FCIc | −3.35 | −7.22, 0.51 | 0.089 |
|
| |||
| Final model | |||
| Hip flexion strength | |||
| MRC ≥4 | 12.27 | 6.81, 17.73 | <0.0001 |
| MRC ≤3 | Ref | Ref | |
| Age at earliest symptom, y | 0.55 | 0.28, 0.82 | <0.0001 |
| Blepharoptosis surgery | |||
| Yes | 7.28 | 1.70, 12.85 | 0.011 |
| No | Ref | Ref | |
Abbreviations: Ref = reference; BMI = body mass index; FCI = Functional Comorbidity Index
For categorical independent variables (hip flexion strength, gender, ethnicity, blepharoptosis surgery, dysphagia treatment, and gastrostomy), the regression coefficient for one value of the variable is used as the reference. The regression coefficient for the other value of the variable is interpreted as the number of years that first use of assistive device is delayed (for positive regression coefficients) or earlier (for negative regression coefficients) relative to the reference.
For continuous independent variables (age at earliest symptom, BMI, and FCI), the regression coefficient is interpreted as the number of years that first use of assistive device is delayed (for positive regression coefficients) or earlier (for negative regression coefficients), for each unit increase in the independent variable.
FCI scores were positively skewed; therefore, a square root transformation was used in the regression model.
We examined whether grouping of MRC scores into 2 categories (MRC ≤3 and MRC >4) was valid for our time-to-event analysis. Paired tests of coefficients demonstrated no significant difference between the coefficients of MRC 4 and 5 (p=0.41, Wald test) and no significant difference between the coefficients of MRC 2 and 3 (p=0.74, Wald test; there were no individuals in our dataset with hip flexion MRC <2).
Figure 2 depicts predicted probability curves for ambulating without an assistive device by age. For illustrative purposes, we show curves for model scenarios with the following assumptions: age of disease onset of 45 y vs. 65 y, and hip flexion MRC of ≤3 vs. >4. As Figure 2 illustrates, earlier disease onset and marked hip flexor weakness (MRC ≤3) are independently associated with earlier progression to use of an assistive device. Our analysis demonstrates that the group of patients with marked hip flexor weakness (MRC ≤3) has poorer mobility outcomes, even after controlling for age of disease onset and comorbidities.
Figure 2.
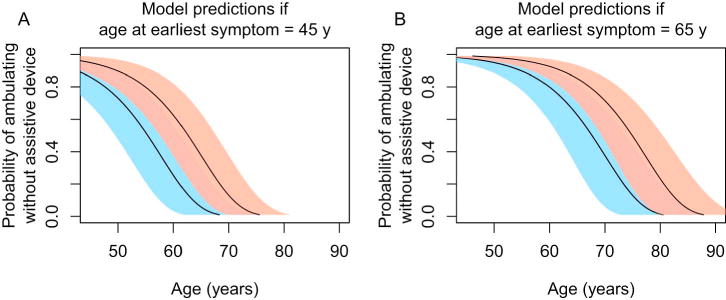
Predicted probabilities of ambulating without assistive device by age.
The final time-to-event statistical model was used to generate predicted probability curves for ambulating without assistive device by age. The solid line and pink shaded regions show the predicted probabilities and 95% pointwise confidence intervals if hip flexion MRC grade is ≥ 4. The solid line and blue shaded regions show the predicted probabilities and 95% pointwise confidence intervals if hip flexion strength MRC grade is ≤ 3. Panel A demonstrates the predicted probabilities if age at earliest symptom is assumed to be 45 y. Panel B demonstrates the predicted probabilities if age at earliest symptom is assumed to be 65 y. All illustrated curves assume a history of blepharoptosis surgery. The plots show that marked hip flexion weakness and younger age at symptom onset are each associated with greater probability of using an assistive device at a younger age. MRC=Medical Research Council.
Sensitivity analyses showed that inclusion of 10 cases with possible but unconfirmed OPMD in the time-to-event model did not significantly alter our findings (results not shown). Inclusion of a term corresponding to confirmed (GCN)13(GCN)10 genotype was not significant in the model (p=0.157).
4. Discussion
This was a retrospective study of the largest group of OPMD patients in the U.S., spanning 342 patient-years of follow-up. We achieved the aim of identifying clinical variables associated with impaired mobility. Specifically, we show that greater hip flexion weakness is strongly associated with earlier use of assistive devices for ambulation, even after controlling for age at symptom onset and for comorbidities that affect physical function. Our findings confirm that OPMD is a physically disabling disease: 23.6% (34/144) of individuals progressed to use of assistive devices after an average disease duration of 14 y, indicating that impaired mobility contributes to disease burden.
Contrary to our hypothesis, we found that markers of ptosis and dysphagia severity were not associated with earlier use of assistive devices for ambulation. This raises the possibility that the severity of cardinal symptoms may be discordant within individuals. Previous authors observed that ptosis and dysphagia severity are not always concordant [7]. Other studies found limb-girdle weakness to be the presenting complaint in some cases, occurring before ptosis or dysphagia [13, 16, 25]. Overall severity may also relate to age of onset, since it has previously been observed in French-Canadian cases that patients with earlier disease onset tend to have more severe progression [20]. We confirm that even among individuals with the same PABPN1 genotype, disease severity is highly variable [13, 15, 20].
There are currently no validated outcome measures for OPMD, which is a barrier to clinical trial design. Our study has implications for outcome measure development. First, we show that limb strength is a potential surrogate endpoint. Establishing efficacy of a new therapy requires proof that it influences a clinically meaningful endpoint (defined as a direct measure of “how a patient feels, functions, or survives”) [26]. Yet clinically meaningful endpoints, such as survival or loss of independent ambulation, may take years to occur in slowly progressive diseases. The U.S. Food and Drug Administration permits use of surrogate endpoints that substitute for clinically meaningful endpoints, predict effects of therapeutic interventions, and change over shorter time periods [9]. Muscle strength is not a clinically meaningful endpoint (since it is not a direct measure of function), but can serve as a surrogate endpoint if it predicts loss of function (e.g., impaired ambulation). By demonstrating a statistical association between hip flexion strength and impaired mobility in OPMD, we have provided justification for consideration of muscle strength as a surrogate endpoint. Validation of muscle strength as a surrogate endpoint in OPMD will require a prospective study to confirm that it predicts loss of ambulatory function and to demonstrate that strength measurements are responsive to change over time. Second, our results have implications for development of a clinical rating scale for OPMD. While previous studies have used a single measure (such as swallow or ptosis assessment) to judge the severity of OPMD [3, 10], our study suggests that adequate disease assessment will require a composite measure that incorporates multiple symptom domains. This approach has been used to develop rating scales for other neuromuscular diseases, such as amyotrophic lateral sclerosis and inclusion body myositis [27, 28]. Development and validation of an OPMD clinical rating scale that is responsive to change over time will facilitate the design of future clinical trials.
A secondary aim of our study was to provide the first prevalence estimates of OPMD for New Mexico. We found that the minimum prevalence of OPMD is 6.1 per 100,000, and much greater among Hispanics in the state (12.1 per 100,000). Since it is doubtful that our study included all patients with OPMD in New Mexico, the actual prevalence is presumably higher. It is likely that OPMD is the most common adult muscular dystrophy in New Mexico. Our estimate of minimum prevalence among Hispanic New Mexicans is lower than that reported among Bukharian Jews in Israel (1:600), although the Israeli estimate derives from a smaller absolute number of cases (n=117) [29]. Our estimate for the minimum prevalence of OPMD among non-Hispanic New Mexicans reflects the prevalence of OPMD independent of the founder effect in New Mexico. Thus, 0.9 per 100,000 may approximate the minimum prevalence of OPMD in the general U.S. population. This is comparable to the estimated minimum prevalence of 0.5 per 100,000 in the general population in France [30].
Our study provides information on the contemporary natural history and management of OPMD. We show that OPMD continues to be a highly morbid disease, with 77.1% (111/144) developing sufficiently severe ptosis to warrant blepharoptosis surgery at an average of 6.8 y from disease onset, and 39.6% (57/144) developing sufficiently severe dysphagia to warrant invasive procedures at an average of 11.1 y from disease onset. Many patients underwent these procedures repeatedly. Gastrostomy for nutritional support was an end-stage procedure in our cohort, occurring in 6.9% (10/144) of individuals at an average of 18.9 y from disease onset; patients who underwent gastrostomy had significantly lower BMI compared with those who did not. As currently described in the literature, cricopharyngeal myotomy is the standard intervention for dysphagia in OPMD [2]. Yet in our cohort, esophageal dilatation was performed in 32.6% (47/144) and myotomy in just 1.4% (2/144). Regional variations may exist in management of OPMD, or the typical management of patients may have changed in recent years. Comparative studies are needed to determine the relative utility of cricopharyngeal myotomy, esophageal dilatation, and cricopharyngeal botulinum toxin injection for treatment of dysphagia in OPMD.
There have been conflicting reports regarding the association of peripheral neuropathy [31, 32] and cognitive impairment with autosomal-dominant OPMD [33, 34]. Our data do not support such associations, since prevalences of these comorbidities in our cohort were similar to those in the general, older population [35, 36]. PABPN1 has been shown to play a role in alternative cleavage and polyadenylation of messenger RNA, with triplet-repeat expanded PABPN1 leading to enhanced use of proximal cleavage sites [37]. A shift to proximal cleavage sites occurs in cancer [38], raising the question of whether cancer risk is increased in OPMD. The 14.6% (21/144) cancer prevalence in our sample does not suggest an increased cancer risk compared with cancer prevalence in the general, older population [39], nor was a pattern of unusual cancers observed.
Our novel use of parametric time-to-event statistical methods allowed us to include left- and interval-censored observations, which are typically excluded from analyses using the Kaplan-Meier and Cox proportional hazards models [23]. Exclusion of observations in rare disease research is highly detrimental as it further reduces already-limited sample sizes. Through use of parametric time-to-event models, we were able to evaluate associations between various clinical variables and our primary outcome with greater statistical power, yielding estimates of time-to-event with relatively tight confidence intervals (Fig. 2). This method may have applicability to other studies of rare diseases.
We included cases in which the diagnosis of OPMD was made purely clinically (i.e., based on symptoms of late-onset ptosis and dysphagia, and family history), without molecular confirmation. Nevertheless, the majority of subjects (75%; 108/144) had molecular confirmation of OPMD or were related to someone with molecular confirmation. All individuals in our clinic who met clinical criteria for a diagnosis of OPMD and underwent genetic testing had a positive test (n=72). Thus, in our clinic, the likelihood of fulfilling clinical criteria for OPMD (late-onset ptosis and dysphagia, and family history) and having a negative genetic test is less than 1in 72 (<2%). It is therefore unlikely that our sample was strongly biased by incorrect case ascertainment.
Our study had several limitations. Individuals with mild or advanced disease may have been less likely to present for evaluation in our clinic. We assumed that characteristics that were not recorded in the medical record were absent. This source of bias would lead to underestimation of clinical characteristics and outcomes. Age at disease onset was reported by patients and thus subject to inaccuracies of recall; recall accuracy may have been worse in older patients. Since our study was single-center, generalizability is a concern; however, age of onset and frequency of limb weakness in our sample is comparable to that reported in cohorts from many other geographic regions [12, 13, 15, 20, 40]. Furthermore, 9% of our cohort were not of Hispanic New Mexican ancestry, and thus represented the disease in the general population. Finally, the majority of individuals with genotype information bore the (GCN)13(GCN)10 genotype, the most commonly observed expansion size [3]. Therefore, our results are likely generalizable.
Recent work on basic mechanisms of OPMD raises hopes for new treatments [41, 42]; however, a major obstacle to conducting clinical trials is lack of validated outcome measures to assess disease progression and treatment effects. Our study demonstrates that hip flexion weakness is statistically associated with impaired ambulation, and supports consideration of muscle strength as a surrogate endpoint in future trials. Moreover, our results imply that composite outcome measures are needed since cardinal symptoms of OPMD may differ in severity within individuals. We emphasize that OPMD should not be viewed as a myopathy mainly limited to eyelid and pharyngeal muscles, and that the generalized myopathy in OPMD has important functional consequences.
Supplementary Material
Highlights.
Hip flexion weakness is associated with increased risk of impaired mobility in oculopharyngeal muscular dystrophy
A subset of patients has marked hip flexion weakness and poorer outcome
Limb weakness does not strongly correlate with severity of other disease symptoms
Muscle strength may be considered as a surrogate endpoint in future clinical trials
Composite outcome measures are warranted in future clinical trials
Acknowledgments
This work was partly supported by a Muscular Dystrophy Association Clinical Research Training Grant (S.Y.) and the NIH/NCRR/NCATS (University of New Mexico Clinical and Translational Science Center, 8UL1TR000041).
Abbreviations
- OPMD
oculopharyngeal muscular dystrophy
- BMI
body mass index
- FCI
Functional Comorbidity Index
- MRC
Medical Research Council
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Victor M, Hayes R, Adams RD. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the evelids. New Engl J Med. 1962;267:1267–72. doi: 10.1056/NEJM196212202672501. [DOI] [PubMed] [Google Scholar]
- 2.Brais B, Tome F. Oculopharyngeal muscular dystrophy. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. pp. 1147–1162. [Google Scholar]
- 3.Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–7. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 4.Raz V, Butler-Browne G, van Engelen B, Brais B. 191st ENMC international workshop: recent advances in oculopharyngeal muscular dystrophy research: from bench to bedside 8–10 June 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:516–23. doi: 10.1016/j.nmd.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Becher MW, Morrison L, Davis LE, et al. Oculopharyngeal muscular dystrophy in Hispanic New Mexicans. JAMA. 2001;286:2437–40. doi: 10.1001/jama.286.19.2437. [DOI] [PubMed] [Google Scholar]
- 6.Barbeau A. The syndrome of hereditary late onset ptosis and dysphagia in French-Canada. In: Kuhn E, editor. Symposium uber Progressive Muskeldystrophie. Berlin: Springer-Verlag; 1966. pp. 102–9. [Google Scholar]
- 7.Blumen SC, Nisipeanu P, Sadeh M, Asherov A, Tome FM, Korczyn AD. Clinical features of oculopharyngeal muscular dystrophy among Bukhara Jews. Neuromuscul Disord. 1993;3:575–7. doi: 10.1016/0960-8966(93)90119-5. [DOI] [PubMed] [Google Scholar]
- 8.Taylor EW. Progressive vagus-glossopharyngeal paralysis with ptosis: a contribution to the group of family diseases. J Nerve Ment Dis. 1915;42:129–139. [Google Scholar]
- 9.Mendell JR, Csimma C, McDonald CM, et al. Challenges in drug development for muscle disease: a stakeholders’ meeting. Muscle Nerve. 2007;35:8–16. doi: 10.1002/mus.20686. [DOI] [PubMed] [Google Scholar]
- 10.Allen RC, Jaramillo J, Black R, et al. Clinical characterization and blepharoptosis surgery outcomes in Hispanic New Mexicans with oculopharyngeal muscular dystrophy. Ophthal Plast Reconstr Surg. 2009;25:103–8. doi: 10.1097/IOP.0b013e3181994e21. [DOI] [PubMed] [Google Scholar]
- 11.Coiffier L, Perie S, Laforet P, Eymard B, St Guily JL. Long-term results of cricopharyngeal myotomy in oculopharyngeal muscular dystrophy. Otolaryngol Head Neck Surg. 2006;135:218–22. doi: 10.1016/j.otohns.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Muller T, Schroder R, Zierz S. GCG repeats and phenotype in oculopharyngeal muscular dystrophy. Muscle Nerve. 2001;24:120–2. doi: 10.1002/1097-4598(200101)24:1<120::aid-mus17>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Van Der Sluijs BM, Hoefsloot LH, Padberg GW, Van Der Maarel SM, Van Engelen BG. Oculopharyngeal muscular dystrophy with limb girdle weakness as major complaint. J Neurol. 2003;250:1307–12. doi: 10.1007/s00415-003-0201-6. [DOI] [PubMed] [Google Scholar]
- 14.Rivera D, Mejia-Lopez H, Pompa-Mera EN, et al. Two different PABPN1 expanded alleles in a Mexican population with oculopharyngeal muscular dystrophy arising from independent founder effects. Br J Ophthalmol. 2008;92:998–1002. doi: 10.1136/bjo.2007.131482. [DOI] [PubMed] [Google Scholar]
- 15.Tondo M, Gamez J, Gutierrez-Rivas E, Medel-Jimenez R, Martorell L. Genotype and phenotype study of 34 Spanish patients diagnosed with oculopharyngeal muscular dystrophy. J Neurol. 2012;259(8):1546–52. doi: 10.1007/s00415-011-6374-5. [DOI] [PubMed] [Google Scholar]
- 16.Witting N, Mensah A, Kober L, et al. Ocular, bulbar, limb, and cardiopulmonary involvement in oculopharyngeal muscular dystrophy. Acta Neurol Scand. 2014;130(2):125–30. doi: 10.1111/ane.12244. [DOI] [PubMed] [Google Scholar]
- 17.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370:1453–7. doi: 10.1016/S0140-6736(07)61602-X. [DOI] [PubMed] [Google Scholar]
- 18.U.S. Census Bureau. Census Interactive Population Search. 2010 Available at http://www.census.gov/2010census/popmap/ipmtext.php?fl=35. Accessed 2014 Mar 13.
- 19.Medici M, Pizzarossa C, Skuk D, Yorio D, Emmanuelli G, Mesa R. Oculopharyngeal muscular dystrophy in Uruguay. Neuromuscul Disord. 1997;7(Suppl 1):S50–2. doi: 10.1016/s0960-8966(97)00082-5. [DOI] [PubMed] [Google Scholar]
- 20.Bouchard JP, Brais B, Brunet D, Gould PV, Rouleau GA. Recent studies on oculopharyngeal muscular dystrophy in Quebec. Neuromuscul Disord. 1997;7(Suppl 1):S22–9. doi: 10.1016/s0960-8966(97)00077-1. [DOI] [PubMed] [Google Scholar]
- 21.Groll DL, To T, Bombardier C, Wright JG. The development of a comorbidity index with physical function as the outcome. J Clin Epidemiol. 2005;58:595–602. doi: 10.1016/j.jclinepi.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 22.SAS Institute Inc. SAS/STAT® 9.3 User’s Guide. SAS Institute Inc; Cary, NC: 2011. pp. 3765–3874. [Google Scholar]
- 23.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. 2. New Jersey: John Wiley & Sons; 2002. pp. 1–56. [Google Scholar]
- 24.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2014. Available at http://www.R-project.org/. Accessed 2014 May 23. [Google Scholar]
- 25.Fardeau M, Tome FM. Oculopharyngeal muscular dystrophy in France. Neuromuscul Disord. 1997;7(Suppl 1):S30–3. doi: 10.1016/s0960-8966(97)00078-3. [DOI] [PubMed] [Google Scholar]
- 26.Lesko LJ, Atkinson AJ., Jr Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol. 2001;41:347–66. doi: 10.1146/annurev.pharmtox.41.1.347. [DOI] [PubMed] [Google Scholar]
- 27.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 28.Jackson CE, Barohn RJ, Gronseth G, Pandya S, Herbelin L. Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity. Muscle Nerve. 2008;37:473–6. doi: 10.1002/mus.20958. [DOI] [PubMed] [Google Scholar]
- 29.Blumen SC, Nisipeanu P, Sadeh M, et al. Epidemiology and inheritance of oculopharyngeal muscular dystrophy in Israel. Neuromuscul Disord. 1997;7(Suppl 1):S38–40. doi: 10.1016/s0960-8966(97)00080-1. [DOI] [PubMed] [Google Scholar]
- 30.Brunet G, Tome FM, Samson F, Robert JM, Fardeau M. Oculopharyngeal muscular dystrophy. A census of French families and genealogic study. Rev Neurol (Paris) 1990;146:425–9. (in French) [PubMed] [Google Scholar]
- 31.Hardiman O, Halperin JJ, Farrell MA, Shapiro BE, Wray SH, Brown RH., Jr Neuropathic findings in oculopharyngeal muscular dystrophy. A report of seven cases and a review of the literature. Arch Neurol. 1993;50:481–8. doi: 10.1001/archneur.1993.00540050033011. [DOI] [PubMed] [Google Scholar]
- 32.Finsterer J. Involvement of the peripheral nerves in oculopharyngeal muscular dystrophy. Clin Neurophysiol. 2010;121:803–4. doi: 10.1016/j.clinph.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 33.Blumen SC, Bouchard JP, Brais B, et al. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology. 2009;73:596–601. doi: 10.1212/WNL.0b013e3181b388a3. [DOI] [PubMed] [Google Scholar]
- 34.Dubbioso R, Moretta P, Manganelli F, et al. Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophy. J Neurol. 2012;259(5):833–7. doi: 10.1007/s00415-011-6255-y. [DOI] [PubMed] [Google Scholar]
- 35.Martyn CN, Hughes RA. Epidemiology of peripheral neuropathy. J Neurol Neurosurg Psychiatry. 1997;62:310–8. doi: 10.1136/jnnp.62.4.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen RC, Roberts RO, Knopman DS, et al. Prevalence of mild cognitive impairment is higher in men. The Mayo Clinic Study of Aging. Neurology. 2010;75:889–97. doi: 10.1212/WNL.0b013e3181f11d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenal M, Elkon R, Loayza-Puch F, et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–53. doi: 10.1016/j.cell.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. Summary Health Statistics for U.S. Adults: National Health Interview Survey. 2012 Available at http://www.cdc.gov/nchs/data/series/sr_10/sr10_260.pdf. p. 26. Accessed 2014 May 15.
- 40.Mirabella M, Silvestri G, de Rosa G, et al. GCG genetic expansions in Italian patients with oculopharyngeal muscular dystrophy. Neurology. 2000;54:608–14. doi: 10.1212/wnl.54.3.608. [DOI] [PubMed] [Google Scholar]
- 41.Barbezier N, Chartier A, Bidet Y, et al. Antiprion drugs 6-aminophenanthridine and guanabenz reduce PABPN1 toxicity and aggregation in oculopharyngeal muscular dystrophy. EMBO Mol Med. 2011;3:35–49. doi: 10.1002/emmm.201000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abu-Baker A, Laganiere J, Gaudet R, et al. Lithium chloride attenuates cell death in oculopharyngeal muscular dystrophy by perturbing Wnt/beta-catenin pathway. Cell Death Dis. 2013;4:e821. doi: 10.1038/cddis.2013.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.