

Abstract
Spermidine N-acetyltransferase, encoded by the gene speG, catalyzes the initial step in the degradation of polyamines and is a critical enzyme for determining the polyamine concentrations in bacteria. In Escherichia coli, studies have shown that SpeG is the enzyme responsible for acetylating spermidine under stress conditions and for preventing spermidine toxicity. Not all bacteria contain speG, and many bacterial pathogens have developed strategies to either acquire or silence it for pathogenesis. Here, we present thorough kinetic analyses combined with structural characterization of the VCA0947 SpeG enzyme from the important human pathogen Vibrio cholerae. Our studies revealed the unexpected presence of a previously unknown allosteric site and an unusual dodecameric structure for a member of the Gcn5-related N-acetyltransferase (GNAT) superfamily. We show that SpeG forms dodecamers in solution and in crystals and describe its three-dimensional structure in several ligand-free and liganded structures. Importantly, these structural data define the first view of a polyamine bound in an allosteric site of an N-acetyltransferase. Kinetic characterization of SpeG from V. cholerae showed that it acetylates spermidine and spermine. The behavior of this enzyme is complex and exhibits sigmoidal curves and substrate inhibition. We performed a detailed non-linear regression kinetic analysis to simultaneously fit families of substrate saturation curves to uncover a simple kinetic mechanism that explains the apparent complexity of this enzyme. Our results provide a fundamental understanding of the bacterial SpeG enzyme, which will be key towards understanding the regulation of polyamine levels in bacteria during pathogenesis.
Keywords: spermidine/spermine, dodecamer, allosteric enzyme, GNAT, acetyltransferase
Introduction
Bacteria have adopted many strategies for survival in diverse environments, some of which can increase their virulence and pathogenicity. One such strategy is the utilization, acquisition, or silencing of the speG gene [1–5]. This gene encodes the spermidine N-acetyltransferase protein, which catalyzes the transfer of an acetyl group from acetyl coenzyme A (AcCoA) to the primary amino group(s) of the cationic polyamine. Acetylation neutralizes the charge of the polyamine, which is then typically excreted from the cell. In the absence of speG in E. coli, there is decreased cell viability due to inhibition of protein synthesis by spermidine [4]. It has also been shown that SpeG becomes more active under stressful conditions including cold temperatures [6–7] and poor nutrient availability [3]. Heat shock, alkaline shift, and ethanol treatment produce increased concentrations of monoacetylated spermidine in E. coli, though it has not been demonstrated whether SpeG becomes stimulated under these conditions [8]. SpeG is a member of the Gcn5-related N-acetyltransferase (GNAT) superfamily, which is characterized by a fold composed of a series of α-helices that encompass a mixed parallel/antiparallel β-sheet and conserved AcCoA-binding site [9, 10]. Typically, GNAT proteins have been found as monomers or homodimers in solution and use either a direct transfer or ping-pong kinetic mechanism [9]. For some time it was believed that polyamine production was ubiquitous amongst organisms; however, recent evidence indicates that some bacteria like Staphylococcus aureus do not produce polyamines and are hypersensitive to them [1, 5]. To overcome this hypersensitivity during infection, S. aureus USA-300 has acquired speG on an arginine catabolic mobile element (ACME) through horizontal gene transfer, which results in increased biofilm formation and virulence [1]. In contrast, Shigella has silenced or deleted speG to increase its pathogenicity and survival in macrophages [2]. Studies have also shown that polyamines such as putrescine, spermidine, and spermine are found in very high concentrations in the human gut and may influence colonization of V. cholerae [11]. Biofilms in V. cholerae are important for its survival, and it has been shown that high concentrations of spermidine can disrupt biofilm formation. Although deleting the spermidine import gene potD1 in V. cholerae causes an increase in biofilm formation [12], the role of SpeG may also contribute to reducing spermidine concentrations in the cell. Overall, utilization of SpeG to prevent polyamine toxicity appears to be common for both pathogenic and non-pathogenic bacteria [1, 4, 6, 11].
Due to the importance of speG in bacterial pathogenicity, we sought to gain a more in-depth understanding of the three-dimensional structure of SpeG from bacteria and its mechanism for polyamine acetylation. We chose to study SpeG from the pathogen V. cholerae, which causes the deadly disease cholera. Homologs of SpeG from V. cholerae include SpeG from other known human pathogens such as Escherichia coli, Shigella, Yersinia pestis, Coxiella burnetii and S. aureus (Fig. 1a). Similar to the previously characterized SpeG enzyme from E. coli, we found that SpeG from V. cholerae acetylates spermidine/spermine, but not putrescine or cadaverine. A thorough kinetic characterization showed that SpeG uses a bireactant random steady-state mechanism for catalysis. Additionally, the V. cholerae SpeG enzyme has a dodecameric structure, which is unusual for members of the GNAT superfamily. Another unexpected feature of the SpeG structure was the revelation of a previously unknown allosteric site that binds spermidine/spermine. The residues that comprise the allosteric site are conserved amongst SpeG homologs but not other polyamine acetyltransferases (Fig. 1a); therefore, we propose that the presence of this allosteric site places the enzyme in a separate class of polyamine N-acetyltransferases. The structure and kinetic mechanism presented in this work provide fundamental details that are necessary and important for understanding SpeG function in bacteria.
Fig. 1.
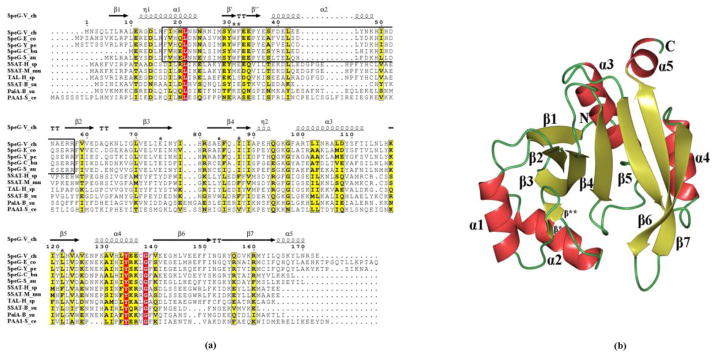
Sequence alignment of SpeG and overall structural fold. (a) Sequence alignment of SpeG from V. cholerae (SpeG-V_ch) with other SpeG homologs and known polyamine N-acetyltransferase family members, including: SpeG from E. coli (SpeG-E_co), SpeG from Y. pestis (SpeG-Y_pe), SpeG from C. burnetii (SpeG-C_bu), SpeG from S. aureus (SpeG-S_au), spermidine/spermine N1-acetyltransferase (SSAT) from Homo sapiens (SSAT-H_sa), SSAT from Mus musculus (SSAT-M_mu), diamine N-acetyltransferase from Homo sapiens (TAL-H_sa), SSAT from Bacillus subtilis (SSAT-B_su), polyamine N-acetyltransferase from B. subtilis (PaiA-B_su), polyamine N-acetyltransferase from Saccharomyces cerevisiae (PAA1-S_ce). Secondary structure elements of SpeG from V. cholerae are indicated above the sequence. The higher sequence identity is highlighted in red and low in yellow. The helices α1, α2 and the interconnected loop between them involved in polyamine binding in the allosteric site of the SpeG proteins are shown in the box with black borders. Conserved residues at the substrate-binding site between SpeG homologs are indicated above the sequence by black stars. The Sequence alignment of SpeG from V. cholerae with other SpeG proteins and known polyamine N-acetyltransferase family members were performed using CLUSTALW [56] and formatted in ESPript [57]. (b) Ribbon diagram of the SpeG monomer. The secondary structure elements are labeled.
Results
Identification of substrates
To identify potential substrates for the previously uncharacterized SpeG enzyme from V. cholerae, we screened a library composed of ninety-five compounds from multiple substrate classes including polyamines, amino acids, antibiotics and different metabolites [13]. The screen yielded four positive hits for the polyamine substrates: spermine, spermidine, N1-acetylspermine, and putrescine (1400, 1400, 497, and 3.3 nmol/min/mg, respectively; data from [13]). N1-acetylspermine and putrescine were only acetylated under reaction conditions with high concentrations of enzyme (10 μM; [13]), whereas spermine and spermidine were acetylated even at low concentrations of enzyme (0.06 μM, this manuscript). Acetylation of cadaverine or other polyamines [13] was not detected even at high concentrations of enzyme. From the results of the broad-substrate screen, it appeared that spermine and spermidine were the preferential substrates for SpeG. Therefore, we used these compounds to further characterize the enzyme.
Dodecameric structure of SpeG
We determined six structures of SpeG from V. cholerae, including the ligand-free, binary complexes with either polyamine (spermidine/spermine) in the allosteric sites or AcCoA in the substrate-binding sites, and a ternary complex with spermine in the allosteric sites and CoA in the substrate-binding sites (Table 1). Five ligand-free SpeG structures were determined in different crystal forms by screening a variety of crystallization conditions and were deposited in the protein data bank (PDB). The SpeG ligand-free structures presented in this work were crystallized in high space group symmetry (I222 and P42212) and contain three monomers per asymmetric unit (Table 1). Three other ligand-free SpeG structures were obtained in the monoclinic space group C2 with six monomers per asymmetric unit and will be characterized in a subsequent manuscript. The structure of SpeG in the binary complex with polyamine (spermidine/spermine) was determined in the I222 space group containing three monomers in the asymmetric unit, while the binary complex with AcCoA and the ternary complex with spermine and CoA were determined in the P3 space group with 12 monomers per asymmetric unit (Table 1).
Table 1.
Data collection, structure determination, and refinement statistics&.
| SpeG ligand-free form | SpeG ligand-free form | SpeG-spermidine | SpeG-spermine | SpeG-AcCoA | SpeG-CoA-spermine | |
|---|---|---|---|---|---|---|
|
| ||||||
| Crystal parameters | ||||||
| Resolution (Å) | 32.0–1.89 (1.94–1.89) | 30.0–2.83 (2.87–2.83) | 30.0–2.3 (2.34–2.3) | 30.0–1.85 (1.88–1.85) | 30.0–2.08 (2.12–2.08) | 30.0–2.6 (2.64–2.6) |
| Space group | I222 | P42212 | I222 | I222 | P3 | P3 |
| Unit cell parameters | ||||||
| a, b, c (Å) | 71.7, 134.5, 137.1 | 131.5, 131.5, 72.7 | 73.4, 136.0, 139.8 | 71.6, 135.9, 140.5 | 176.7, 176.7, 67.0 | 176.9, 176.9, 67.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 |
| Matthews coefficient (Å3/Da) | 2.6 | 2.5 | 2.6 | 2.6 | 2.4 | 2.4 |
| Solvent content (%) | 52.9 | 50.7 | 53.2 | 52.2 | 48.8 | 48.9 |
|
| ||||||
| Data Collection | ||||||
| Completeness (%) | 98.3 (94.7) | 98.8 (99.7) | 98.8 (100) | 99.6 (99.8) | 99.4 (95.2) | 96.2 (100) |
| No of unique reflections | 52181 | 15565 | 30239 | 59130 | 140107 | 68437 |
| I/σ(I) | 13.6 (2.8) | 19.4 (2.0) | 43.8 (7.9) | 38.8 (6.6) | 38.1 (2.8) | 15.0 (2.9) |
| Rmerge (%) | 0.07 (0.6) | 0.12 (0.7) | 0.06 (0.2) | 0.08 (0.3) | 0.08 (0.6) | 0.17 (0.6) |
| Redundancy | 7.6 (6.4) | 3.5 (3.4) | 7.2 (7.4) | 5.2 (5.1) | 5.4 (5.0) | 4.7 (5.3) |
| Wilson B-factor (Å2) | 43.6 | 68.2 | 57.3 | 35.3 | 33.8 | 36.5 |
|
| ||||||
| Refinement | ||||||
| R (%)/Rfree (%) | 19.1/23.8 | 19.7/26.5 | 19.6/25.4 | 14.9/18.4 | 17.1/23.9 | 18.5/23.8 |
| RMSD bond length (Å) | 0.016 | 0.01 | 0.016 | 0.019 | 0.013 | 0.014 |
| RMSD bond angle (°) | 1.77 | 1.39 | 1.92 | 1.93 | 1.75 | 1.96 |
| Average B value (Å2) | ||||||
| All atoms | 43.2 | 67.3 | 54.8 | 30.9 | 37.1 | 37 |
| Biological Ligand | - | - | 51 | 33 | 58 | 68 (CoA)/39 (SPM) |
| No of atoms | ||||||
| Protein | 4308 | 4288 | 4373 | 4398 | 17191 | 18174 |
| Biological Ligand/Other | -/64 | - | 30 | 42 | 612/31 | 611/21 |
| Ligands | ||||||
| Water molecules | 276 | 26 | 131 | 559 | 278 | 325 |
|
| ||||||
| Ramachandran analysis# | ||||||
| Favoured (%)/n | 98.1/507 | 91.7/461 | 98.1/510 | 99.4/529 | 96.3/1935 | 89.8/1811 |
| Allowed (%)/n | 1.9/10 | 8.3/42 | 1.9/10 | 0.6/3 | 3.7/74 | 10.2/205 |
| Outlier (%)/n | - | - | - | - | - | - |
Data for the highest resolution shell are given in parentheses. The abbreviations RMSD and ASU stand for root-mean-square deviation and asymmetric unit, respectively.
Defined by validation program MOLPROBITY
Similar to other GNATs, the overall fold of a SpeG monomer has a mixed α/β architecture (Fig. 1b) [9]. Two interconnected β-sheets, with four strands in one and three strands in the other side, form a V-like shape. Five α-helices surround these β-sheets with α1 and α2 on one face and α3, α4, and α5 on the opposite face of the sheets (Fig. 1b). The β4-strand and the α3-helix have previously been shown to be important for substrate and AcCoA binding and contain critical residues involved in catalysis [9, 10]. The β-bulge formed by the β4 and β5 parallel strands is where the pantotheine moiety of AcCoA is located in the active site and is characteristic of members of the GNAT superfamily [9, 10].
The crystal asymmetric units vary in the number of SpeG monomers, but in each case SpeG appears to be dodecameric with overall dimensions of ~103 ×103 × 72 Å (Fig. 2a). Gel filtration chromatography and dynamic light scattering (DLS) both show that the solution state is a 270 kDa species consistent with the dodecameric state of the protein observed in the crystal (Fig. S1).
Fig. 2.

Dodecameric structure of SpeG. (a) Ribbon diagram of front and vertical side view of SpeG dodecamer. Two alternating subunits of the hexamer are colored in yellow (top) and green (bottom). (b) Ribbon diagram of SpeG dodecamer in complex with polyamine (red). (c) Alternating subunits of the hexamer. Residues corresponding to labeled loops form the internal region between subunits. (d) Ribbon diagram of the typical GNAT dimer shown in complex with coenzyme (tan). The subunits from adjacent hexamers forming the dimer are colored in green and orange. Strands involved in intersubunit interactions are labeled. (e) Ribbon diagram of SpeG dodecamer in complex with AcCoA (tan).
This is an unusual oligomeric state for a GNAT, as proteins in this family are typically monomeric or dimeric [9, 10]. Although we obtained structures of the SpeG protein with a variety of space groups and different numbers of monomers in the asymmetric unit cell, all of them show that the dodecamer contains monomers with similar intersubunit interactions (RMSD values between SpeG monomers are given in Table S1). The SpeG dodecamer can be described as a dimer of hexamers, where the two hexamers stack one on top of the other as shown in Fig 2a. Each hexamer is composed of six adjacent monomers, and the residues on the following loops form the interface between the monomers of the hexamer: β1-α1, α1-α2, α2-β2, β4-α3, β6-β7 (Fig. 2c). Each monomer from one hexamer interacts with a monomer from the second hexamer to form a GNAT dimer that is similar to the dimeric structure of an aminoglycoside 6′-N-acetyltransferase [9]. The intermolecular surface in each of the six GNAT dimers consists of a β-sheet that is created from three C-terminal β-strands β5-β6-β7 and loops between strands β3-β4 and β6-β7 from each monomer (Fig. 2d). The recently deposited homologous structure of SpeG from C. burnetii with 55 % identity to the V. cholerae enzyme represents a similar dodecameric architecture (PDB ID: 3TTH).
Acetyl-coenzyme A binding site
To define the cofactor-binding site in SpeG, we determined the structure of the binary complex of SpeG with AcCoA (Fig. 2e, 3). Co-crystallization of SpeG with very high concentrations of AcCoA (20 mM) was necessary to obtain a structure with AcCoA bound in all twelve monomers of the dodecamer in the canonical cofactor-binding site [9]. We found that residues involved in binding AcCoA to SpeG are conserved amongst GNATs (Fig. 1a), and a schematic diagram of the AcCoA binding mode is presented in Fig. 3.
Fig. 3.
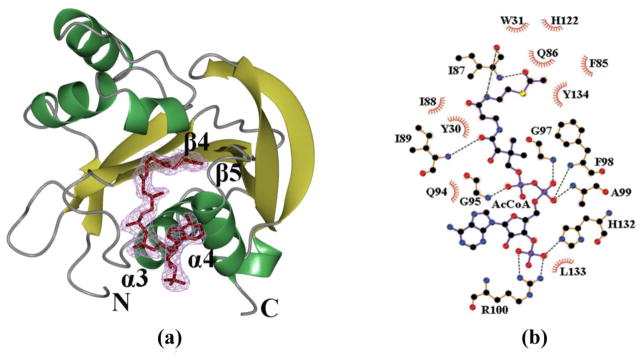
AcCoA-binding site. (a) Ribbon diagram of the SpeG monomer with bound AcCoA (tan) at the substrate-binding site. Secondary structure elements forming the cofactor-binding site are labeled. The electron density map (2Fo-Fc) for AcCoA is shown at the 1σ contour level in magenta. (b) LigPlot+ diagrams of AcCoA binding sites. Hydrogen bonds for cofactor on the LigPlot+ are shown as black broken lines. In the LigPlot+ diagram, water molecules are colored in brown, oxygen atoms in red, carbon atoms in black, nitrogen atoms in blue, sulfur atom in yellow and phosphorus atoms in purple. The hydrophobic contacts are presented as a red circular arc with radial spikes.
In each SpeG monomer, AcCoA binds in the cleft created by strands β4, β5 and helices α3, α4 (Fig. 3a). The pyrophosphate moiety of AcCoA interacts with the main chain nitrogen atoms of residues Gly95, Gly97, Phe98 and Ala99, which form the so-called P-loop (Fig. 3b). This set of residues, which is also known as “motif A,” is the most conserved feature of GNATs [9]. The AcCoA in the active site of each monomer of the SpeG dodecamer generally has the 3′-phosphate of AcCoA interacting with the amino groups of the side chains of Arg100 and His132 (Fig. 3b); however, there is variability in the position of the adenine moiety. Two distinct conformations were identified. In one conformation, the adenine ring is located close to the P-loop and forms hydrogen bonds with the main chain oxygen of Gly95, whereas in the second conformation it is located close to Lys129. The position of the adenine ring of AcCoA is likely to be affected by contacts with symmetry equivalent monomers that stabilize one conformation compared to another. The pantothenate moiety of AcCoA is located close to the loop between β4 and α3. In this position, the pantothenate moiety forms hydrogen bonds with two conserved residues Ile87 and Ile89 (Fig. 3b). The sulfhydryl and acetyl groups of AcCoA are situated in the splay between strands β4 and β5, which is consistent with other GNAT structures [9]. In all monomers, the acetyl group is hydrogen-bonded to the main-chain nitrogen atom of Ile87 and the sulfur atom is located in close proximity to Tyr134, which is also a conserved residue among GNATs. This residue is proposed to function as the general acid to protonate the thiolate anion of CoA after transfer of the acetyl group to the substrate [9].
We also observed that the C-terminus of SpeG is affected by binding AcCoA in its active site. In structures that were ligand-free or in complex with polyamine, the C-terminus of a symmetry-related monomer occupies the AcCoA-binding site in one of the three monomers of the asymmetric unit. These structures show the carbonyl oxygen of the C-terminal residue Glu173 hydrogen bonds with the conserved residues Gly95, Gly92, and Ala99 of “motifA.” In the AcCoA binary complex structure, the C-terminus is disordered and the last residue observed in the electron density is 171. The presence of the C-terminus in the AcCoA binding pocket could, in part, explain why numerous attempts to obtain a complex with AcCoA or CoA at low concentrations were unsuccessful. In different SpeG homologs there is variability of the length of the C-terminus, but its role is not presently understood.
Polyamine allosteric site
Since it appeared that spermidine/spermine were the preferred substrates from our broad-substrate screening assay, we used these ligands for co-crystallization trials with SpeG. The binary spermidine/spermine liganded structures revealed that both polyamines bind in an unexpected pocket outside of the predicted SpeG active site. In each hexamer the polyamine binds in an extensive pocket spanning two neighboring monomers, creating a total of twelve sites (Fig. 2b, 2c). These unusual ligand-binding locations have not been previously described for any published GNAT structure. Since they are located outside of the active site, we refer to them as allosteric polyamine-binding sites.
The allosteric spermidine/spermine binding pocket involves a combination of hydrophobic and polar interactions. All hydrophobic interactions between the polyamine and SpeG include only side-chains of the bottom (green) monomer: Phe17, Ile18, Leu21, Met28, Trp31, Leu42, Leu45, Leu70, and Ile88 (Fig. 4). Spermidine and spermine have similar hydrogen bonding contacts in the allosteric sites of all monomers of the SpeG dodecamer (Fig. 4). The N10/N14 amine of the bound spermidine/spermine forms hydrogen bonds with the main chain carbonyl oxygens of His49, Ile50, and Asp52, and with the side-chain OE2 carboxyl of Glu55 that is mediated by a water molecule (Fig. 4a). The N6/N10 amine of spermidine/spermine forms hydrogen-bonds with the carboxyl group of Glu41 of the bottom monomer (Fig. 4a, 4b). Because of their difference in length, spermidine and spermine establish distinct hydrogen-bonds with the bottom (green) monomer (Fig. 4c). Specifically, the N1 amine of the spermidine makes hydrogen-bonds with the Glu33 carboxyl, while the N5 amine of spermine binds through two water molecules to the Asn22 and Glu34 carboxyl groups on one side of the polyamine and Glu37 and Glu41 carboxyl groups on the other side, respectively (Fig. 4a). The end of the spermine with N1 interacts with the OE1 oxygen of Glu33 and through a water molecule with OE1 of Glu72 (Fig. 4a). Additionally, we have observed a conformational change of the position of Tyr36 when spermidine is bound in the allosteric site compared to spermine (Fig. 4c). All residues involved in hydrogen-bonding with the ligand in this binding site are highly conserved among SpeG homologs (Fig. 1a).
Fig. 4.
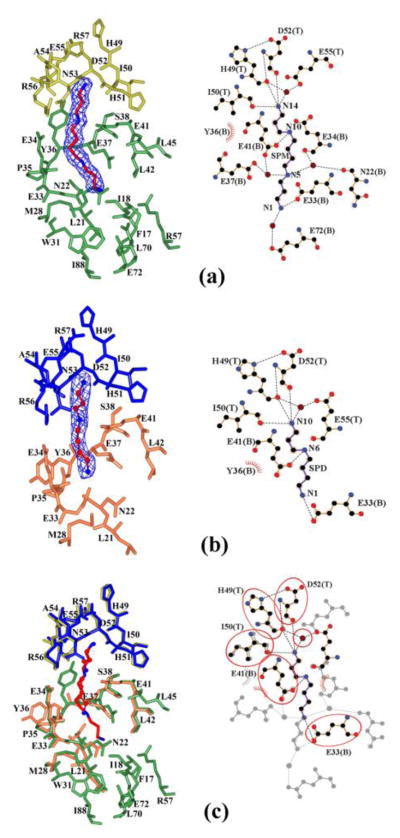
Allosteric polyamine binding-site. The spermine/spermidine allosteric site is shown: top (T) and bottom (B) monomers for the spermine binding pocket (a) are colored in yellow and green, respectively; top (T) and bottom (B) subunits for spermidine (b) are in blue and orange, respectively. (c) Superposition of spermine and spermidine binding pockets. Spermine and spermidine are shown as a stick and a ball-and-stick model, respectively, where carbon atoms are in red and nitrogen atoms in blue. The electron density map (2Fo-Fc) for polyamines is shown at the 1σ contour level in blue. Additionally, (a)/(b), and (c) represent LigPlot+ diagrams of spermine/spermidine binding sites and their superposition, respectively. Hydrogen bonds for polyamine on the LigPlot+ are shown as black broken lines. Similar interactions are highlighted by ovals. In the LigPlot+ diagram, water molecules are colored in brown, oxygen atoms in red, carbon atoms in black, and nitrogen atoms in blue. The hydrophobic contacts are presented as a red circular arc with radial spikes.
Conformational differences between SpeG ligand-free and liganded structures
Superposition of individual monomers of the SpeG ligand-free and liganded SpeG structures with polyamine or coenzyme revealed significant differences in the active site of the enzyme. The structural comparison for this analysis was performed using LSQKAB from the CCP4 program suit [14]. The root-mean square deviation (RMSD) values between the Cα atoms of SpeG in the ligand-free and liganded structures are presented in Table S1. All SpeG structures show similar RMSD values of ~0.3 or 0.4 Å when measured between Cα atoms of the monomers within the asymmetric unit or between monomers of SpeG in the liganded state, respectively. The high RMSD values > 1.2 Å were calculated between SpeG in ligand-free and liganded structures. A major conformational change was observed in the position of the loop connecting helices α1 and α2 (Fig. 5). In all structures of liganded SpeG, this loop moves by about 15 Å (measured between Cα atoms of Phe32 in the SpeG ligand-free and liganded structures) towards the loop located between β6 and β7, close to the center of the β-bulge. The secondary structural composition of this loop changes depending upon the presence of ligand in the structure: from two short β strands ((β* and β** in ligand-free) to one short α-helix (α* in liganded) (Fig. 5). Movement of this loop in all liganded structures also brings the conserved Tyr30 in the active site near the position of the acetyl group of AcCoA, which is required for proper acetyl transfer in the substrate-binding site in known GNATs. In the ligand-free SpeG structure this loop has a different conformation and faces towards strands β2 and β3, creating a more open active site. These data suggest that the conformational change of the loop is induced by either polyamine binding in the allosteric site or coenzyme binding in the active site. Another conformational change observed between ligand-free and cofactor-liganded structures was the difference in the position of helix α4 where the ribose and adenine moiety of AcCoA binds in the active site (Fig. 5).
Fig. 5.
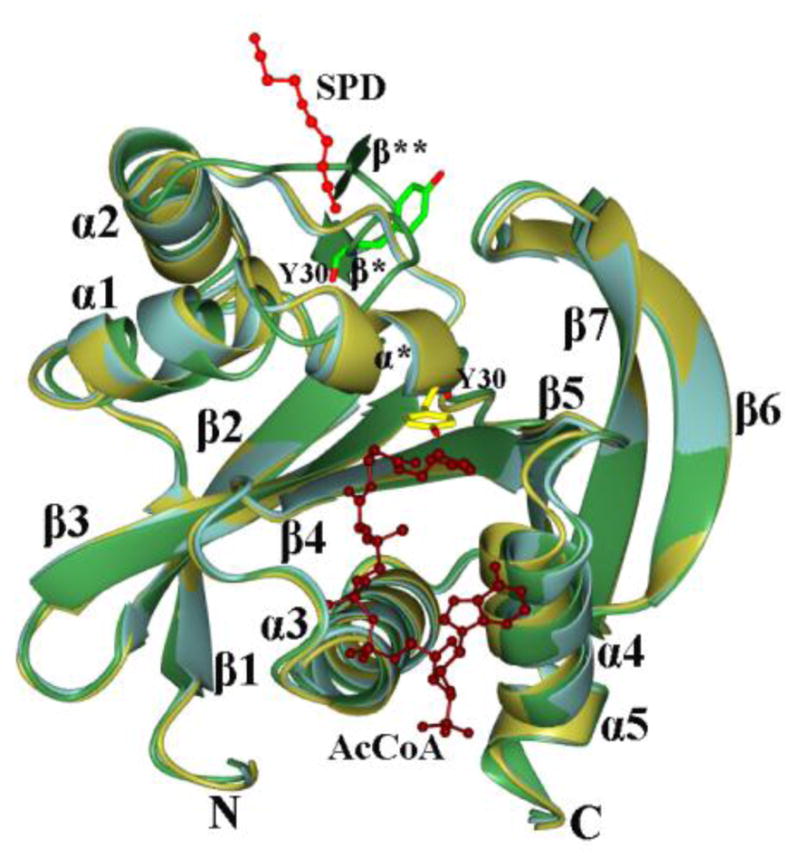
Superposition of SpeG in ligand-free and liganded structures. The SpeG subunit in the ligand-free form (green), in complex with polyamine (light blue) in allosteric binding site and in complex with AcCoA (gold) are shown. The secondary structural elements are labeled. Spermine (red) and AcCoA (tan) are drawn as ball-and-stick models. The Tyr30 residue located on the loop between helices α1 and α2 are shown as cylinder model in light green in the ligand-free form structure and yellow in the liganded structure.
A comparison of the ligand-free structures determined in the P42212 space group and I222 space group show that the ligand-free form may or may not include salts or buffer molecules. This is consistent with other ligand-free structures from a different space group that will be discussed separately. The structure determined in P42212 does not have any ligands bound but the structure with I222 symmetry shows a CHES buffer molecule or a sulfate ion bound close to the loop between helices α1 and α2. The position of this loop is identical regardless of the presence of these molecules. It appears that binding specific ligands to SpeG, but not binding of non-specific additives such as CHES or sulfate, induces the change in the position of the allosteric loop.
Predicted polyamine substrate binding site
To investigate the placement of polyamine in the active site, we quickly soaked crystals of SpeG that already had AcCoA bound in the active sites with spermine. This produced a structure of SpeG containing CoA in twelve active sites and spermine in six of the allosteric sites with a similar binding mode as seen in prior complexes; however, no spermine was bound in the active sites (Fig. 6a). The position of the allosteric loop between helices α1 and α2 remained the same as that observed in structures of SpeG in complex with AcCoA in the substrate-binding sites or spermidine/spermine in the allosteric sites. Unfortunately, longer soaks with spermine caused the crystals to dissolve, and co-crystallization of SpeG with high concentrations of spermidine/spermine did not yield a structure of the enzyme with polyamine in the active site. Since the allosteric sites are occupied by polyamine and the active sites contain CoA, we can hypothesize where the substrate polyamine may bind. The predicted substrate-binding pocket is located between two loops created by α1-α2 and β6-β7 (Fig. 6b, 6c), which form the canonical GNAT substrate-binding site [9]. This pocket is lined by amino acids Trp31, Phe32, Ile74, Glu75, Glu84, Phe85, Gln86, Ile87, Leu121, His122, Val123, and Phe149. The inner surface of the pocket is negatively charged, suggesting that SpeG prefers to bind to positively charged molecules like polyamines (Fig. 6c). Residues Trp31, Phe32, Glu75, Glu84, Ile87, Leu121 and Val123 are highly conserved among SpeG homologs and thus may be important for binding the substrate polyamine (Fig. 1a). Based on the size of the cavity, position of acetyl group of AcCoA and superposition with a related structure of spermine/spermidine N1-acetyltransferase from mouse in complex with spermine (the only known polyamine N-acetyltransferase structure in complex with polyamine (PDBID 3BJ8)) we manually docked spermine into the predicted substrate-binding pocket of a SpeG monomer in complex with cofactor and spermine in the allosteric binding site (Fig. 6b). In the model, the N1 atom of spermine (or spermidine) is located in close proximity to the acetyl group of AcCoA. The N5 atom of spermine (or N6 of spermidine) is likely to bind to the OE1 atom of Gln86. The N10 and N14 atoms of spermine (or N10 spermidine) would hydrogen bond to Glu84 and Glu75. Therefore, the model suggests that the conserved Glu84 and Glu75 could be important for binding and selectivity towards long polyamines like spermidine or spermine at the enzyme active site.
Fig. 6.
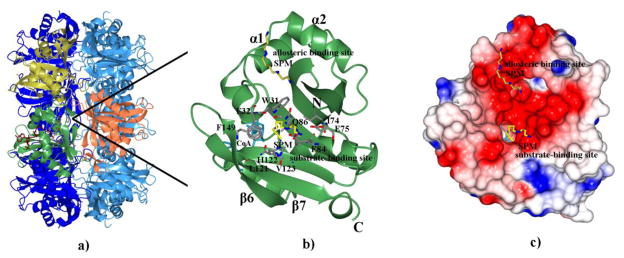
Predicted substrate-binding site. (a) Ribbon diagram of SpeG dodecamer in complex with CoA (tan) and spermine (yellow) in the allosteric binding site. Subunits forming the GNAT dimer are colored in green and orange. The alternating subunit of the hexamer are colored in yellow (top). (b) Modeled spermine (yellow) docked into the predicted substrate-binding site of the SpeG structure in complex with CoA (cyan) and spermine (yellow) in allosteric site is shown in one subunit. Secondary structural elements and residues (grey) that make up the substrate-binding site are labeled. The atoms of the residues making up the substrate-binding pocket are colored: carbon in grey, oxygen in red, and nitrogen in blue. The atoms of the spermine are colored: carbon in yellow and nitrogen in blue. The N- and C-termini are indicated. (c) Electrostatic surface of SpeG monomer in presence of cofactor (cyan) and polyamine in the allosteric binding site with spermine docked into the substrate-binding site.
We noticed that PEG can compete with polyamine binding in the allosteric site when we soaked crystals in 20% PEG6000 for cryo-protection. In the binary complex with AcCoA, a PEG molecule is bound in the allosteric site in two of the 12 monomers. The competition between spermine and the crystallizing agent PEG may explain why the ternary complex with CoA and spermine does not have all 12 allosteric polyamine-binding sites filled by spermine.
Isothermal Titration Calorimetry
We performed isothermal titration calorimetry (ITC) experiments to establish the binding affinities of spermidine/spermine and AcCoA to SpeG. The heats of dilution for spermine were too high to accurately determine its binding affinity to SpeG; therefore, we used spermidine for further experiments and fitted these data to a model with two binding sites (Fig. S2). Consistent with the structural data, this analysis suggests that two types of binding interactions occur between the protein and spermidine. The first site (1 per subunit) has a high affinity (Kd1 = 1.6 μM) and the second site has a low affinity (Kd2 = 833 μM) for spermidine, potentially corresponding to the allosteric and substrate-binding sites, respectively. The integrated data for AcCoA were fitted using a model with one site per subunit. The analysis showed that AcCoA binds to SpeG with a low affinity (Kd = 610 μM) in the absence of spermidine. These data confirm that spermidine binds to SpeG with two different affinities and AcCoA binds poorly to SpeG in the absence of polyamine.
Kinetic characterization
To kinetically characterize SpeG, we produced saturation curves for each substrate (AcCoA, spermine, spermidine) and estimated the kinetic parameters using a modified Hill equation, as described in Experimental Procedures and previously [15]. AcCoA curves, obtained by holding spermine at a constant concentration while varying the concentration of AcCoA, showed that the enzyme’s apparent affinity for AcCoA decreased significantly as the spermine concentration was increased (Fig. 7, Table 2). Additionally, SpeG exhibits substrate inhibition by AcCoA at lower spermine concentrations (Fig. 7). Polyamine curves show strong sigmoidicity and indicate that the enzyme has a higher apparent affinity for spermine than spermidine (Table 2). In all polyamine curves, the apparent affinity for spermine or spermidine decreases and the maximal velocity increases as the concentration of AcCoA increases (Fig. 7, Table 2).
Fig. 7.
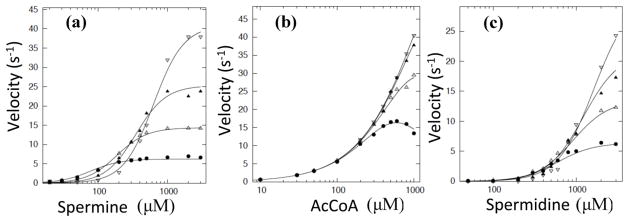
Series of substrate curves for spermine, spermidine, and AcCoA. (a) Spermine curves at varying concentrations of AcCoA (filled circle is 0.1 mM, open triangle is 0.25 mM, filled triangle is 0.5 mM, open upside down triangle is 1 mM). (b) AcCoA curves at varying concentrations of spermine (filled circle is 0.5 mM, open triangle is 1 mM, filled triangle is 2 mM, open upside down triangle is 3 mM). (c) Spermidine curves at varying concentrations of AcCoA (filled circle is 0.1 mM, open triangle is 0.25 mM, filled triangle is 0.5 mM, open upside down triangle is 1 mM). Curves were fit using the bireactant random steady-state 3 model (Model X) as described in Materials and Methods.
Table 2.
Estimated Kinetic Parameters for SpeG from V. cholerae. All data were generated based on the procedures described in Materials and Methods.
| Spermine curves | [AcCoA] mM | S0.5 μM | n | Vmax U/mg | kcat s−1 |
|---|---|---|---|---|---|
| 0.1 | 99 ± 3 | 1.79 ± 0.07 | 19.4 ± 0.2 | 6.79 | |
| 0.25 | 180 ± 4 | 2.02 ± 0.07 | 41.3 ± 0.4 | 14.5 | |
| 0.5 | 326 ± 15 | 2.24 ± 0.24 | 68.7 ± 1.8 | 24.0 | |
| 1 | 579 ± 13 | 2.62 ± 0.11 | 111 ± 1.4 | 38.9 |
| Spermidine curves | [AcCoA] mM | S0.5 μM | n | Vmax U/mg | kcat s−1 |
|---|---|---|---|---|---|
| 0.1 | 554 ± 20 | 2.50 ± 0.20 | 18.8 ± 0.49 | 6.58 | |
| 0.25 | 813 ± 23 | 2.17 ± 0.10 | 37.6 ± 0.73 | 13.2 | |
| 0.5 | 1010 ± 60 | 2.38 ± 0.18 | 53.1 ± 2.09 | 18.6 | |
| 1 | 1320 ± 96 | 2.57 ± 0.24 | 76.2 ± 4.4 | 26.7 |
| AcCoA curves | [Spermine] mM | S0.5 μM | n | Vmax U/mg | kcat s−1 |
|---|---|---|---|---|---|
| 0.5 | 132 ± 20 | 1.83 ± 0.40 | 48.1 ± 3.1 | 16.8 | |
| 1 | 369 ± 51 | 1.27 ± 0.10 | 110 ± 8.1 | 38.5 | |
| 2 | 897 ± 185 | 1.12 ± 0.07 | 209 ± 25 | 73.2 | |
| 3 | 1210 ± 123 | 1.07 ± 0.03 | 263 ±16 | 92.1 |
A comparison of the kinetic parameters for the recombinant V. cholerae SpeG enzyme (VcSpeG) characterized in this work to the previously characterized recombinant SpeG enzyme from E. coli (EcSpeG) [3] shows that these two enzymes exhibit relatively similar kinetic characteristics. Both EcSpeG and VcSpeG enzymes have comparable specific activities: 255 μmol/min/mg with 3 mM spermidine for EcSpeG [3] and 263 μmol/min/mg in the presence of 3 mM spermine for VcSpeG. Additionally, both enzymes display a higher apparent affinity for spermine over spermidine: 220 μM and 1.29 mM for spermine and spermidine, respectively for EcSpeG (at 10 μM AcCoA) [3], and 99 μM and 554 μM for spermine and spermidine, respectively for VcSpeG (at 100 μM AcCoA). In spite of the similarity with other kinetic parameters, one clear difference in SpeG kinetic parameters between the two organisms is their apparent affinity for AcCoA: 2 μM for EcSpeG in the presence of 3 mM spermidine [3] and 1.21 mM for VcSpeG with 3 mM spermine. We recently showed that the presence of the poly-histidine tag decreased both the maximal velocity and apparent affinity for AcCoA for VcSpeG [16]. Although both of these parameters increase when the tag is removed from the VcSpeG enzyme, the apparent affinity for AcCoA is still three orders of magnitude lower than EcSpeG. It is not clear if the difference in apparent affinity for AcCoA between EcSpeG and VcSpeG is due to the detection limitations of the assay method (e.g. radiometric vs. colorimetric), presence of additives in the enzyme storage buffers (e.g. 1 mM spermidine, 10% glycerol, 0.1 mM EDTA, 50 mM potassium chloride, 6 mM BME, 10 μM FUT-175, and 0.2 % Brij 35 detergent for EcSpeG, [3]), or possible effect of recombinantly producing the VcSpeG enzyme with an affinity tag and retention of residual residues after tag cleavage.
For VcSpeG, the hill coefficient, which gives a measure of the sigmoidicity of the curves, increases for the spermine curves as the concentration of AcCoA increases and remains relatively constant for the spermidine curves (Table 2). In contrast to the polyamine curves, the sigmoidicity decreases for AcCoA curves as the concentration of spermine increases (Table 2). Since we observed curves with sigmoidicity, we hypothesized that they could be induced by a steady-state mechanism or that there may be homotropic allosteric effects.
Kinetic mechanism
To evaluate the kinetic mechanism of SpeG, we tested the fit of specific kinetic models to the data as described (Models I-X, Materials and Methods). When performing the fitting for the kinetic model, the allosteric site was assumed to be saturated with polyamine since the ITC data showed that the binding affinity of the enzyme for spermidine/spermine in the allosteric site is in the low micromolar range. The data collected to determine the kinetic mechanism of the enzyme was in the sub-millimolar range; therefore, the allosteric site was clearly saturated. We first attempted to fit the data to equations for simple kinetic models and then gradually increased the complexity of the equations until a compatible model was obtained (Fig. 6, S3, S4, S5). The ITC data were used to define the initial parameters for testing each model. In general, rapid-equilibrium models did not fit the data properly; however, steady-state models were significantly better. A bireactant random steady-state model was most compatible with all three sets of curves (Fig. 6, S4, S5, S6) and the third iteration of this model (Model X; Materials and Methods; see Scheme 1) produced the best second order Akaike’s information criterion (AICc) values across all three data sets (Fig. S3, S4, S4).
Scheme 1.

SpeG mechanism of polyamine acetylation in bacteria. (a) A general representation of the bireactant random steady-state model. (b) (1) Polyamine binds to the allosteric site of the ligand-free SpeG enzyme at low concentrations, which causes a conformational change of the loop between helices α1 and α2 (marked by an oval); (2) The SpeG enzyme with polyamine in the allosteric site (red) can bind an additional molecule of polyamine (gold) or AcCoA (tan) in a random order. The preferred kinetic path is shown in green and non-preferred path is shown in grey. (3) Rate of product formation is much faster than binding substrate. The scheme shows the ribbon diagams of liganded structures of SpeG in complex with spermine in the allosteric site, the complex of SpeG with AcCoA in active site or the ternary complex of SpeG with spermine in allosteric site and CoA in active site. A modeled SpeG structure with spermine bound in the active site was also used to show potential binary or ternary complexes. In all diagrams, spermine and AcCoA are shown as a cylinder models. Kinetic parameters include koff_a/Ka (1410 s−1μM−1), koff_a (0.000141 s−1), koff_b/Kb (0.0665 s−1μM−1), koff_b (0.0480 s−1), k3 (0.00806 s−1), k4 (0.0000001 s−1) and kp (106 s−1) based on the fitting of a series of spermine substrate curves at varying concentrations of AcCoA to the bireactant random steady-state model as defined in Materials and Methods.
The series of E→EB→EAB steps was necessary to introduce sufficient complexity into the equation to fit the data to a bireactant steady-state model (Materials and Methods, Scheme 1, S3, S4, S5). Failure to introduce these steps led to poor fitting of the data. This is supported by crystallographic data where we observe AcCoA binding to the substrate site in absence of spermidine. The fact that a steady-state mechanism, rather than a rapid-equilibrium mechanism, is most compatible indicates that the binding and dissociation of the substrates is not significantly faster than product formation. The equations for the rapid-equilibrium models do not take into account both the sigmoidicity of the curves and the substrate inhibition seen for AcCoA. Since the steady-state model explains both, it implies that there is more than one path for binding and dissociation of substrates and both routes are not equally favorable [17]. The sigmoidicity is explained by two different paths for binding and dissociation of substrates that are slower than the product release. This leads to equations that are divisions of polynomials of higher order without the need to introduce more complex homotropic allosteric effects [17]. In other words, this means that the observed allosteric spermidine site does not necessarily affect the kinetic properties of the substrate spermidine site. This would be possible based on the ITC and crystallographic data that show that the allosteric site has a much higher affinity for spermidine and would already be occupied and saturated, while the active site is still undersaturated with spermidine in the range of substrate concentrations that were used to determine the kinetic mechanism. The higher order polynomials that we derived mathematically explain the possibility of both observed types of curves in our experiments: sigmoidal curves and ones that reach a maximum and then decrease (substrate inhibition). Thus, the simplest way to explain the apparent complexity we observe with the SpeG enzyme is through a bireactant random steady-state mechanism.
Although the fitting for all three substrates show the lowest AICc values for the bireactant random steady-state model, we cannot discard the possibility that a more complex kinetic mechanism, such as a hybrid bireactant steady-state ping-pong mechanism, could be used [17]. In particular, we observed that the spermidine data produce low AICc values for the rapid-equilibrium Models IV (cooperative ping-pong; Materials and Methods) and VII (modified Hill equation; Materials and Methods), possibly indicating that an underlying hybrid mechanism may be inherent. If this were indeed the case, a series of steps involving AcCoA acetylation of an enzyme intermediate and then further transfer of the acetyl group to the polyamine may occur. An excess of AcCoA could force the enzyme into this state, producing substrate inhibition. Even if such steps do exist, they would not constitute the preferred catalytic path. The preferred path would be where the polyamine enters first followed by AcCoA to transfer the acetyl group to the polyamine, directly or indirectly.
Discussion
Contrary to all published GNATs, our data show that SpeG displays a dodecameric structure with twelve allosteric sites and twelve active sites. The allosteric sites are located between adjacent monomers that form the hexamers and the active sites are located at the interface between monomers of the stacked hexamers. A sequence alignment of SpeG with other polyamine GNATs shows that residues 90 to 140, which correspond to the cofactor and substrate active site regions, are conserved (Fig. 1a). However, residues 17 to 57 located on helices α1, α2 and the interconnected loop between them correspond to the allosteric site region and seem to differ considerably from other polyamine GNATs. Therefore, we propose that the presence of this allosteric site places SpeG in a distinct class of polyamine N-acetyltransferases (Fig. 1a). The architecture of the SpeG allosteric site appears to be designed to accommodate long polyamines like spermidine/spermine. When the allosteric site is occupied with spermidine/spermine, a number of specific interactions occur between the polyamine and the SpeG protein. These include residues that are conserved amongst SpeG homologs: His49, Ile50, Asp52, Glu41, and Glu33. The interaction of spermidine/spermine with Glu33 seems to be essential for specific polyamine recognition in this binding cavity and explains why shorter polyamines like putrescine and cadavarine would not be able to trigger the allosteric loop movement. In fact, the ligand-free structure in P42212 symmetry was obtained by co-crystallization with putrescine, but no molecules of putrescine are bound to the enzyme and the allosteric loop does not move. The conformational change of the allosteric loop is only seen in structures when spermidine/spermine is bound to the allosteric site or AcCoA is bound in the substrate-binding site. Although AcCoA binding can trigger this loop movement, it may be a consequence of the very high concentration of AcCoA used during co-crystallization trials. It seems that the conformation of this loop is reversible and thermodynamically stable in the presence of the coenzyme or polyamine. Since the SpeG binding affinity for spermidine/spermine is greater than for AcCoA, it appears that the most likely scenario for the conformational change of the allosteric loop is due to binding spermidine/spermine to this site. The allosteric properties of the SpeG enzyme could only be seen structurally, so a combination of kinetic and structural analyses was necessary to understand its kinetic mechanism. All of the kinetic data, including sigmoidicity of the curves and substrate inhibition, can be explained by a bireactant random steady-state kinetic mechanism. The ITC data suggest that the allosteric site has a much higher affinity for spermidine than the substrate-binding site. Thus, at low concentrations of polyamine the allosteric site is filled first and converts the enzyme to an active state ready for catalysis once polyamine concentrations increase. Polyamines are present in high millimolar concentrations in the cell, and significant quantities are bound to polyacids like RNA and DNA [18–20]. Our kinetic data indicate that SpeG may regulate polyamine acetylation and subsequent degradation only when free polyamine concentrations are sufficiently high (millimolar range) so as not to interfere with the cellular concentration requirement for polyacid-polyamine binding.
Our proposed mechanism for SpeG polyamine acetylation in bacteria is depicted in Scheme 1. First, the enzyme binds spermidine/spermine in the allosteric site (Scheme 1–1). The presence of the polyamine in the allosteric site induces a shift of a loop to accommodate both AcCoA and an additional molecule of polyamine in the active site in a random order (Scheme 1–2) using a bireactant random steady-state mechanism. Substrate binding in the active site has the appearance of order, using a preferred path (highlighted in Scheme 1) depending upon the values of the constants and rates of the individual steps. The rate of product formation is much faster than the rate of substrate binding; thus, immediate catalysis occurs when both substrates are present (Scheme 1–3). Due to the high affinity for spermidine/spermine, the first step of binding polyamine in the allosteric site does not seem to be necessary for each turnover. It appears that once the polyamine binds in the allosteric site, it may remain there for successive catalytic steps. Although atypical, the fact that an enzyme can use the same molecule in both its allosteric and active sites is not unprecedented. Examples include phosphofructokinase [21–23], UDP-N-acetylglucosamine 2-epimerase [24] and thiamine diphosphate dependent phenylpyruvate decarboxylase [25].
We cannot exclude the possibility that VcSpeG may acetylate other polyamines like norspermidine, which can be present in higher concentrations than spermidine in specific strains of Vibrionaceae. Although most Vibrionaceae are characterized by the presence of norspermidine, there is a large variability regarding the ratio of norspermidine/spermidine concentrations in particular strains [26–28]. Polyamine uptake from the surrounding environment may also influence the identity and cellular concentrations of various polyamines observed in each bacterium. For instance, E. coli can quickly accumulate spermine even though it is not a native molecule found in this bacterium [19]. Therefore, a physiologically relevant substrate for SpeG may be dictated based on the flux of polyamines it encounters within its environment.
Materials and methods
Cloning, protein production, and crystallization
The gene speG from V. cholerae O1 biovar el tor strain N16961 (NCBI accession AAF96843, GI 9658384) was cloned into the pMCSG7 vector using ligation independent cloning as described previously [29, 30]. E. coli BL21 (DE3) magic cells containing this plasmid were grown in Terrific Broth (TB) for native protein and in High Yield M9 SeMet media (Medicilon Inc.) for selenomethionine-labeled SpeG protein. The protein was expressed and purified by immobilized metal-affinity chromatography (IMAC) and size-exclusion chromatography using standard Center for Structural Genomics of Infectious Diseases protocols [13, 31]. Protein for kinetic assays was purified in the absence of β-mercaptoethanol (BME) and the affinity tag was removed as described in Kuhn et al., 2013.
SpeG was crystallized using the sitting drop vapor-diffusion method at 16 or 19 °C with Classics, Classics II or ComPas Crystal Screens from QIAGEN. One microliter of SpeG (concentration 8.6 mg/ml) in 500 mM sodium chloride, 5 mM BME, 10 mM Tris-HCl buffer at pH 8.3 was mixed with reservoir solution (1μl) and equilibrated against reservoir solution (100μl) using 96-well Compact plates from Corning Life Science. Crystals of ligand-free SpeG in the I222 space group were obtained from the condition containing 0.2 M lithium sulfate, 0.02 M acetylcholine, 1 M potassium/sodium tartrate and 0.1 M CHES (N-cyclohexyl-2-aminoethanesulfonic acid) buffer at pH 9.5. Crystals of ligand-free SpeG in the P42212 space group were obtained by co-crystallization with 5 mM putrescine in a condition containing 45 % (w/v) ethanol. All crystals of SpeG in binary complexes with spermidine/spermine or AcCoA were obtained by co-crystallization using a concentration of 2.5 mM spermidine/spermine or 20 mM AcCoA. Proteins were incubated for 30 minutes on ice with ligand prior to setting crystallization screens. Crystals of SpeG in complex with spermidine grew in a condition containing 0.1 M sodium chloride, 30 % (w/v) ethanol and 0.1 M Tris-HCl at pH 8.5. Crystals of SpeG in complex with spermine grew in a condition containing 20 % (w/v) ethanol and 0.1 M Tris-HCl at pH 8.5. Crystals of SpeG in complex with AcCoA grew in a condition containing 0.2 M sodium citrate and 20 % (w/v) PEG 3350. To obtain the ternary complex structure of SpeG with CoA and spermine, a crystal of SpeG in complex with AcCoA was soaked in solution containing 20 % (w/v) PEG 6000 and 5 mM spermine. Suitable crystals of all ligand-free and liganded complexes of SpeG were soaked in appropriate cryo protectant and flash frozen in liquid nitrogen before data collection: 25 % (w/v) 2-methyl-2, 4-pentanediol (MPD) for ligand-free or polyamine-liganded SpeG crystals and 20 % (w/v) PEG 6000 for crystals of SpeG in complex with AcCoA.
Data collection and structure determination
X-ray data sets were collected at Argonne National Laboratory (Argonne, IL) at 100 K at the Structural Biology Center or Life Sciences Collaborative Access Team beamlines for the SpeG ligand-free and liganded-forms, respectively. Data were processed using HKL-3000 [32] for indexing, integration, and scaling. The selenomethionine-labeled SpeG structure of the ligand-free form was solved by the single-wavelength anomalous dispersion (SAD) method with the HKL-3000 suite [32]. The structures of complexes were solved by molecular replacement with PHASER [33] from the CCP4 suite [14] using the ligand-free form structure as the starting model. Gaps, turns, and side chains for all structures were fitted manually using the program COOT [34]. The structures were refined with REFMAC [35]. The water molecules were built with stringent criteria of electron density above 1.2σ in the 2|F0|-|Fc|map using the program ARP/wARP [36]. The X-ray data collection, structure determination and refinement statistics are summarized in Table 1. The final model for each structure was validated by SFCHECK, PROCHECK, ADIT and MOLPROBITY [37–40]. Figures were produced with PyMOL, CCP4MG and LIGPLOT+ [41–43].
Coordinates and Protein Data Bank Codes
Atomic coordinates and structure factors have been deposited in the Brookhaven Protein Data Bank (PDB) [44] with accession codes 4NCZ, 4JJX, 4MHD, 4MI4, 4R57 (HPUB) and 4R87 (HPUB) for the two ligand-free structures, binary complexes with spermidine, spermine, or AcCoA, and the ternary complex with spermine and CoA, respectively.
Determination of the oligomeric state of SpeG in solution by DLS and Gel Filtration
Dynamic Light Scattering (DLS) experiments were performed on a Zetasizer Nano ZEN1600 (Malvern Instrument). Samples contained 1 mg/ml SpeG protein, 0.01 M Tris-HCl pH 8.3 and 500 mM sodium chloride. Measurements were performed at 25 °C. The molecular weight of the oligomers was estimated using Mark-Houwink-Kuhn-Sakurada (MHKS) relations for globular proteins [45]. The DLS data indicate that a monodisperse solution of SpeG has particles with an exclusion diameter of 13.1 nm, corresponding to a molecular weight of 273 kDa. The molecular weight of the SpeG monomer is 21 kDa.
Size-exclusion chromatography for 28 μg of SpeG was conducted using a Superdex-200 column (Amersham Pharmacia) at 4 °C in 10 mM Tris-HCl pH 8.3, 500 mM sodium chloride, and 5 mM BME. Protein peaks were determined by absorption at 280 nm. For calibration of the gel filtration column we used Gel Filtration Calibration HMW (High Molecular Weight) and LMW (Low Molecular Weight) kits from GE Healthcare Inc. with the following protein standards: aprotinin (6.5 kDa); ribonuclease A (13.7 kDa); carbonic anhydrase (29 kDa); ovalbumin (44 kDa); conalbumin (75 kDa); aldolase (158 kDa) and ferritin (440 kDa). The SpeG molecular weight was calculated based on a standard curve produced using the retention times of the protein standards. The elution volume of SpeG was around 165 ml, corresponding to a molecular weight of 250 kDa.
Isothermal titration calorimetry
ITC was used to determine the binding affinity of ligands to the SpeG enzyme with a MicroCal VP-ITC titration microcalorimeter at 22 °C in a 0.3-ml calorimeter cell. The protein and the ligands were diluted into buffer containing 50 mM HEPES pH 8.3 and 100 mM sodium chloride. Ligands (spermidine, spermine, and AcCoA; 30 mM each) were added sequentially to 0.37 mM SpeG protein in 1 μl aliquots at 2-min intervals for a total of 38 injections. The heats of dilution of the ligands in absence of protein were determined and subtracted from SpeG-ligand binding isotherms before curve fitting. Integration of the binding curves (heat evolved/mol of ligand injected vs. ligand/SpeG molar ratio) that provide the values of ΔH° (heat of binding), Kd, (dissociation constant) and n (binding stoichiometry) were determined with Origin v.7.0 [46, 47] (Fig. S3). Unfortunately, we were not able to measure the binding affinity for spermine due to the high heats of dilution beyond the detection range of the instrument.
Enzyme assays
A broad-substrate screening assay was used to determine possible substrates for the SpeG enzyme as described previously [13]. Based on these results, the enzyme was kinetically characterized and its substrate specificity was determined using a slightly modified procedure. 50 μl reactions were initiated using 10 μl of 0.22 μM enzyme in dilution buffer (50 mM bicine pH 9.0 and 500 mM sodium chloride) and were allowed to proceed for 5 min in 50 mM bicine pH 9.0 at 37 °C. The concentrations of acetyl coenzyme-A (AcCoA), spermine, or spermidine were varied depending upon the type of substrate saturation curve being produced. The reactions were stopped and the absorbance at 415 nm was recorded as described previously [13]. For comparative purposes, data were plotted using Origin v.8.1 and the hill coefficient (n), affinity for substrate at half the maximal velocity (S0.5), and maximal velocity (Vmax) were estimated using a modified Hill equation as described previously [15]. One unit of enzyme activity is the number of μmols of CoA produced per minute under the described conditions.
Enzyme kinetic mechanism
A series of substrate curves were produced to test the fitting of different kinetic models by holding one substrate concentration constant (e.g. AcCoA at 0.1, 0.25, 0.5, or 1 mM; spermine at 0.5, 1, 2, or 3 mM) while varying the other substrate concentrations (e.g. spermine or spermidine at 0–3 mM; AcCoA 0–2 mM). These curves were used to determine which kinetic mechanistic model was compatible with the data. To determine the best kinetic model, the data were fitted to various equations. The equations for these models were produced using the King-Altman method on the BioKin webserver (www.biokin.com/king-altman/index.html) [48–50]. The models for the kinetic mechanism were tested with a program that uses a non-linear least squares regression of the Gauss-Newton algorithm with optional damping using an ad hoc program [51, 52]. The best model was selected based on the corrected Akaike information criterion (AICc) values, where the more negative value indicates the best-fitting statistical model of the ones tested [53–55]. In the equations, V is the velocity, Vm is the maximal velocity, A represents either spermine or spermidine, B represents AcCoA, E is the enzyme, acetylated enzyme is F, and CoA is Q in the ping-pong-like models. P represents the acetylated product in the ping-pong-like models, or both acetylated acceptor substrate and CoA in other models. Additional parameters are defined below each equation. Initial testing indicated that the enzyme may use a bireactant random steady-state model. The model was modified through several iterations until the one that was most compatible with the data was obtained.
Model I: Bireactant Ordered A-First
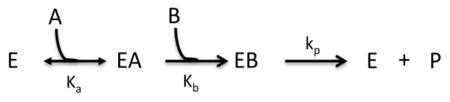
Where Vm = [E]tkp
Model II: Bireactant Ordered B-First
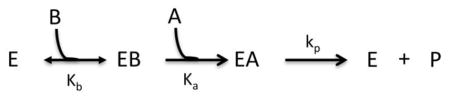
Where Vm = [E]tkp
Model III: Bireactant Ping-Pong
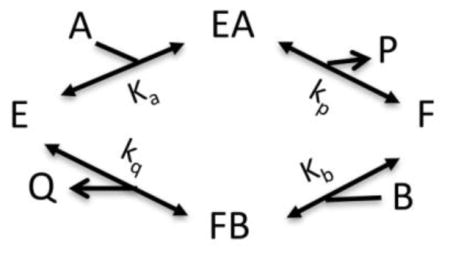
Where Vm = [E]tkcat and
Model IV: Bireactant Cooperative Ping-Pong
Where Vm = [E]tkcat, , n is the coefficient of cooperativity for ligand A, and m is the coefficient of cooperativity for ligand B.
Model V: Bireactant Random Independent
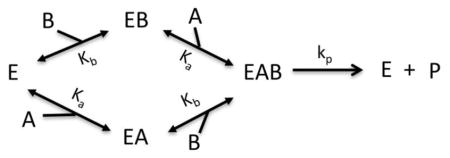
Where Vm = [E]tkp
Model VI: Bireactant Random Cooperative
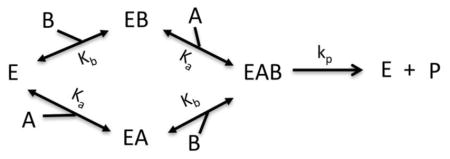
Where Vm = [E]tkp and c is the cooperativity between A and B.
Model VII: Bireactant Modified Hill
Where Vm = [E]tkp, n is the coefficient of cooperativity for ligand A, m is the coefficient of cooperativity for ligand B, and c is the cooperativity between A and B.
Model VIII: Bireactant Random Steady-State
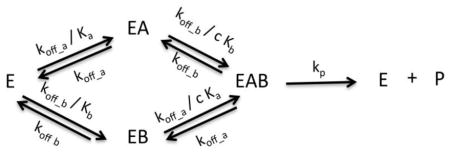
Where n1 = k2k3k4k5, n2 = k1k2k4k5, n3 = k1k2k−3k5 + k−1k3k4k5, d1 = k2k3k4, d2 = k1k2k4, d3 = k2k3k5 + k2k3k−4, d4 = k2k4k5 + k−2k3k4 + k1k2k−3 + k−1k3k4 + k1k2k−4, d5 = k1k4k5 + k1k−2k4, d6 = k2k−3k5 + k2k−3k5 + k2k−3k−4 + k−1k3k5 + k−1k−2k3 + k−1k3k−4, d7 = k−1k4k5 + k−1k−2k4 + k1k−3k5 + k1k−2k−3 + k1k−3k−4, d8 = k−1k−3k5 + k−1k−2k−3 + k−1k−3k−4, , Kd4 = cKd1, Kd2 = cKd3, Kd1 = Ka, Kd3 = Kb, k5 = kp, k−4 = k−1, k−2 = k−3, k−1 = koff_a, k−3 = koff_b, and Vm = [E]tkp
Model IX: Bireactant Random Steady-State 2
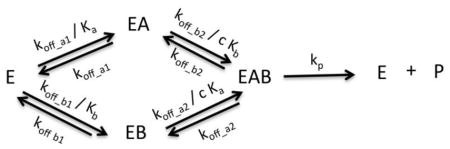
Where n1 = k2k3k4k5, n2 = k1k2k4k5, n3 = k1k2k−3k5 + k−1k3k4k5, d1 = k2k3k4, d2 = k1k2k4, d3 = k2k3k5 + k2k3k−4, d4 = k2k4k5 + k−2k3k4 + k1k2k−3 + k−1k3k4 + k1k2k−4, d5 = k1k4k5 + k1k−2k4, d6 = k2k−3k5 + k2k−3k5 + k2k−3k−4 + k−1k3k5 + k−1k−2k3 + k−1k3k−4, d7 = k−1k4k5 + k−1k−2k4 + k1k−3k5 + k1k−2k−3 + k1k−3k−4, d8 = k−1k−3k5 + k−1k−2k−3 + k−1k−3k−4, , Kd4 = cKd1, Kd2 = cKd3, Kd1 = Ka, Kd3 = Kb, k5 = kp, k−1 = koff_a1, k−3 = koff_b1, k−4 = koff_a2, k−2 = koff_b2, and Vm = [E]tkp
Model X: Bireactant Random Steady-State 3
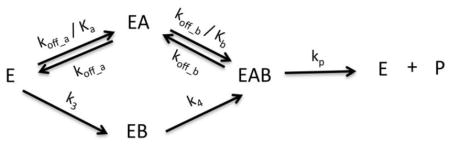
Where n1 = k2k3k4k5, n2 = k1k2k4k5, n3 = k−1k3k4k5, d1 = k2k3k4, d2 = k1k2k4, d3 = k2k3k5, d4 = k2k4k5 + k−2k3k4 + k−1k3k4, d5 = k1k4k5 + k1k−2k4, d6 = k−1k3k5 + k−1k−2k3, d7 = k−1k4k5 + k−1k−2k4, , k5 = kp, k−1 = koff_a, k−2 = koff_b, and Vm = [E]tkp
A general representation of the most compatible model is presented in Scheme 1.
Supplementary Material
Highlights.
Spermidine N-acetyltransferase (SpeG) prevents bacterial polyamine toxicity.
The dodecameric 3D structure of SpeG revealed an unknown polyamine allosteric site.
SpeG acetylates spermidine/spermine using a bireactant random steady-state mechanism.
SpeG has the first described GNAT structure with 12 active and 12 allosteric sites.
Acknowledgments
We would like to extend our sincere appreciation to Samuel Light for his critical review of the manuscript and to Ludmilla Shuvalova and George Minasov for their helpful discussions. Additionally, we would like to thank Lour Volkart and Min Zhou at Argonne National Laboratory for their technical assistance. We would also like to thank the Keck Biophysics Facility at Northwestern University (Evanston Campus) for their assistance with ITC data collection and processing. The data collection for determining crystal structures presented in this manuscript was performed at the LS-CAT and SBC-CAT beamlines at the Advanced Photon Source Science User Facility operated for the U.S. Department of Energy (DOE) and supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contracts No. HHSN272200700058C and HHSN272201200026C (WFA), and the National Science Foundation grant MCB 1024945 (MAB).
Abbreviations
- GNAT
Gcn5-related acetyltransferase superfamily
- AcCoA
acetyl coenzyme A
- CoA
coenzyme A
- ITC
isothermal titration calorimetry
- CHES
N-Cyclohexyl-2-aminoethanesulfonic acid
- PDB
protein data bank
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joshi GS, Spontak JS, Klapper DG, Richardson AR. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol Microbiol. 2011;82(1):9–20. doi: 10.1111/j.1365-2958.2011.07809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbagallo M, Di Martino ML, Marcocci L, Pietrangeli P, De Carolis E, Casalino M, Colonna B, Prosseda G. A new piece of the Shigella Pathogenicity puzzle: spermidine accumulation by silencing of the speG gene [corrected] PLoS One. 2011;6(11):e27226. doi: 10.1371/journal.pone.0027226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukuchi J, Kashiwagi K, Takio K, Igarashi K. Properties and structure of spermidine acetyltransferase in Escherichia coli. J Biol Chem. 1994;269(36):22581–22585. [PubMed] [Google Scholar]
- 4.Fukuchi J, Kashiwagi K, Yamagishi M, Ishihama A, Igarashi K. Decrease in cell viability due to the accumulation of spermidine in spermidine acetyltransferase-dificient mutant of Escherichia coli. J Biol Chem. 1995;270(32):18831–18835. doi: 10.1074/jbc.270.32.18831. [DOI] [PubMed] [Google Scholar]
- 5.Planet PJ, LaRussa SJ, Dana A, Smith H, Xu A, Ryan C, Uhlemann AC, Boundy S, Goldberg J, Narechania A, Kulkarni R, Ratner AJ, Geoghegan JA, Kolokotronis SO, Prince A. Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. M Bio. 2013;4(6):e00889–13. doi: 10.1128/mBio.00889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabor CW. The effects of temperature on the acetylation of spermidine. Biochem Biophys Res Commun. 1968;30(4):339–342. doi: 10.1016/0006-291x(68)90747-x. [DOI] [PubMed] [Google Scholar]
- 7.Limsuwun K, Jones PG. Spermidine acetyltransferase is required to prevent spermidine toxicity at low temperatures in Escherichia coli. J Bacteriol. 2000;182(19):5373–5380. doi: 10.1128/jb.182.19.5373-5380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carper SW, Willis DG, Manning KA, Gerner EW. Spermidine acetylation in response to a variety of stresses in Escherichia coli. J Biol Chem. 1991;266(19):12439–12441. [PubMed] [Google Scholar]
- 9.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vetting MW, de Carvalho LPS, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Goforth JB, Walter NE, Karatan E. Effects of polyamines on Vibrio cholerae virulence properties. PLoS One. 2013;8(4):e60765. doi: 10.1371/journal.pone.0060765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGinnis MW, Parker ZM, Walter NE, Rutkovsky AC, Cartaya-Marin C, Karatan E. Spermidine regulates Vibrio cholerae biofilm formation via transport and signaling pathways. FEMS Microbiol Lett. 2009;299:166–174. doi: 10.1111/j.1574-6968.2009.01744.x. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn ML, Majorek KA, Minor W, Anderson WF. Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N acetyltransferases with unknown substrate specificity. Protein Sci. 2013;22(2):222–230. doi: 10.1002/pro.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystal D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Structural, functional and inhibition studies of a GNAT superfamily protein PA4794: a new C-terminal lysine protein acetyltransferase from Pseudomonas aeruginosa. J Biol Chem. 2013;288(42):30223–30235. doi: 10.1074/jbc.M113.501353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majorek KA, Kuhn ML, Chruszcz M, Anderson WF, Minor W. Double trouble-Buffer selection and His-tag presence may be responsible for nonreproducibility of biomedical experiments. Protein Sci. 2014;23:1359–1368. doi: 10.1002/pro.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segel IH. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: 1975. [Google Scholar]
- 18.Bachrach U, Cohen IJ. Spermidine in the bacterial cell. J Gen Microbiol. 1961;26:1–9. doi: 10.1099/00221287-26-1-1. [DOI] [PubMed] [Google Scholar]
- 19.Tabor CW, Tabor H. Polyamines in microorganisms. Microbiol Rev. 1985;49(1):81–99. doi: 10.1128/mr.49.1.81-99.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igarashi K, Kashiwagi K. Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun. 2000;271:559–564. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- 21.Cabrera R, Ambrosio ALB, Garratt RC, Guixe V, Babul J. Crystallographic structure of phosphofructokinase-2 from Escherichia coli in complex with two ATP molecules. Implications for substrate inhibition. J Mol Biol. 2008;383(3):588–602. doi: 10.1016/j.jmb.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 22.Lowry OH, Passonneau JV. Kinetic evidence for multiple binding sites on phosphofructokinase. J Biol Chem. 1966;241(10):2268–2279. [PubMed] [Google Scholar]
- 23.Passonneau JV, Lowry OH. The role of phosphofructokinase in metabolic regulation. Adv Enzyme Regul. 1964;2:265–274. doi: 10.1016/s0065-2571(64)80018-2. [DOI] [PubMed] [Google Scholar]
- 24.Velloso LM, Bhaskaran SS, Schuch R, Fischetti VA, Stebbins E. A structural basis for the allosteric regulation of non-hydrolysing UDP-GlcNAc 2-epimerases. EMBO Rep. 2008;9(2):199–205. doi: 10.1038/sj.embor.7401154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Versees W, Spaepen S, Wood MDH, Leeper FJ, Vanderleyden J, Steyaert J. Molecular mechanism of allosteric substrate activation in a thiamine diphosphate-dependent decarboxylase. J Biol Chem. 2007;282(48):35269–35278. doi: 10.1074/jbc.M706048200. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto S, Shinoda S, Kawaguchi M, Wakamatsu K, Makita M. Polyamine distribution in Vibrionaceae: norspermidine as a general constituent of Vibrio species. Can J Microbiol. 1983;29:724–728. [Google Scholar]
- 27.Yamamoto S, Chowdhury MAR, Kuroda M, Nakano T, Koumoto Y, Shinoda S. Further study on polyamine compositions in Vibrionaceae. Can J Microbiol. 1991;37:148–153. doi: 10.1139/m91-022. [DOI] [PubMed] [Google Scholar]
- 28.Koei H. Polyamine distribution patterns within the families Aeromonadaceae, Vibrionaceae, Pasteurellaceae, and Halomonadaceae, and related genera of the gamma subclass of the Proteobacteria. J Gen Appl Microbiol. 1997;43:49–59. doi: 10.2323/jgam.43.49. [DOI] [PubMed] [Google Scholar]
- 29.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Express Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 30.Kim Y, Bigelow L, Borovilos M, Dementieva I, Duggan E, Eschenfeldt W, Hatzos C, Joachimiak G, Li H, Malseva N, Mulligan R, Quartey P, Sather A, Stols L, Volkart L, Wu RY, Zhou M, Joachimiak A. High-throughput protein purification for X-ray crystallography and NMR. Adv Protein Chem. 2008;75:85–105. doi: 10.1016/S0065-3233(07)75003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millard CS, Stols L, Quartey P, Kim Y, Dementieva I, Donnelly MI. A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Express Purif. 2003;29:311–320. doi: 10.1016/s1046-5928(03)00063-9. [DOI] [PubMed] [Google Scholar]
- 32.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution from diffraction images to an initial model in minutes. Acta Crystal D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystal D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystal D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystal D. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 38.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystal. 1993;26:283–291. [Google Scholar]
- 39.Yang H, Guranovic V, Dutta S, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystal D. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 40.Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 41.Delano WL. The Pymol Molecular Graphics System. San Carlos, CA: 2002. [Google Scholar]
- 42.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystal D. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 43.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 44.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harding SE. The intrinsic viscosity of biological macromolecules. Progress in measurement, interpretation and application of structure in dilute solution. Prog Biophys Mol Biol. 1997;68:207–262. doi: 10.1016/s0079-6107(97)00027-8. [DOI] [PubMed] [Google Scholar]
- 46.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 47.Zhang T, Johansson JS. An isothermal titration calorimetry study on the binding of four volatile general anesthetics to the hydrophobic core of a four-alpha-helix bundle protein. Biophys J. 2003;85:3279–3285. doi: 10.1016/S0006-3495(03)74746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cornish-Bowden A. An automatic method for deriving steady-state rate equations. Biochem J. 1977;165:55–59. doi: 10.1042/bj1650055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.King EL, Altman C. A Schematic Method of Deriving the Rate Laws for Enzyme-Catalyzed Reactions. J Phys Chem. 1956;60:1375–1378. [Google Scholar]
- 50.Segel IH. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. Wiley; New York: 1975. [Google Scholar]
- 51.Fraser RDB, Suzuki E, editors. Physical principles and techniques of protein chemistry. Part C. Academic Press; New York: 1973. [Google Scholar]
- 52.Ziegler AJ, Florian J, Ballicora MA, Herlinger AW. Alkaline phosphatase inhibition by vanadyl-beta-diketone complexes: electron density effects. J Enzyme Inhib Med Chem. 2009;24:22–28. doi: 10.1080/14756360701841426. [DOI] [PubMed] [Google Scholar]
- 53.Akaike H. A new look at the statistical modelidentification. IEEE Transactions on Automatic Control. 1974;19:716–723. [Google Scholar]
- 54.Burnham KP, Anderson DR. Model selection and inference: a practical information-theoretic approach. Springer; New York: 1998. [Google Scholar]
- 55.Hurvich CM, Tsai CL. Regression and time series model selection in small samples. Biometrika. 1989;76:297–307. [Google Scholar]
- 56.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucl Acids Res. 2014;42(W1):320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.