

Abstract
Germline mutations in the PTEN tumor-suppressor gene cause autosomal-dominant conditions such as Cowden and Bannayan-Riley-Ruvalcaba syndromes with variable presentations, including hamartomatous gastrointestinal tumors, dermatologic abnormalities, neurologic symptoms, and elevated cancer risk. We describe a father and son with extensive hamartomatous gastrointestinal polyposis who both developed early-onset esophageal cancer. Exome sequencing identified a novel germline PTEN frameshift mutation (c.568_569insC, p.V191S_fs*11). In addition, a missense mutation of SMAD7 (c.115G>A, p.G39R) with an allele frequency of 0.3% in the Exome Variant Server was detected in both affected individuals. Fluorescence in-situ hybridization for PTEN in the resected esophageal cancer specimen demonstrated no PTEN copy loss in malignant cells, however, immunohistochemistry demonstrated loss of PTEN protein expression. While the risks of many cancers are elevated in the PTEN hamartoma tumor syndromes, esophageal adenocarcinoma has not been previously reported. Esophageal adenocarcinoma and extensive polyposis/ganglioneuromatosis could represent less-common features of these syndromes, potentially correlating with this novel PTEN frameshift and early protein termination genotype. Alternatively, because simultaneous disruption of both the PTEN and TGF-β/SMAD4 pathways is associated with development of esophageal cancer in a mouse model, and SMAD4 mutations cause gastrointestinal hamartomas in Juvenile Polyposis Syndrome, the SMAD7 mutation may represent an additional modifier of these individuals’ PTEN-mutant phenotype.
Keywords: PTEN, SMAD7, esophageal cancer, Cowden Syndrome, PTEN Hamartoma Tumor Syndrome
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) was originally described as a somatically mutated tumor-suppressor gene in brain, breast, and prostate cancers.[1] Germline loss-of-function PTEN mutations are responsible for Cowden, Bannayan-Riley-Ruvalcaba, and other syndromes, known collectively as the PTEN hamartoma tumor syndrome (PHTS).[2–5] The autosomal-dominant and highly-penetrant PHTS conditions are characterized by a broad range of manifestations including macrocephaly, skin abnormalities, neurologic problems, and hamartomatous or ganglioneuromatous gastrointestinal polyposis.[6,7] Harmartomatous polyps of the stomach and colorectum define the related but distinct autosomal-dominant Juvenile Polyposis Syndrome (JPS), which results from germline mutations of SMAD4 or BMPR1A disrupting signaling through the bone morphogenetic protein (BMP)/SMAD4 pathway.[8,9]
PHTS confers vastly increased lifetime risk of many cancers, including breast (85%), thyroid (35%), colon (9%), kidney (34%), and endometrial (28%) malignancies.[10,11] PTEN terminates growth factor receptor signaling in the phosphatidylinositol-3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway by dephosphorylating phosphatidylinositol-3,4,5-trisphosphate (PIP3).[12] Loss of PTEN function leads to increased cellular growth, proliferation, angiogenesis, and survival signaling.[6,12] In this report we describe a novel PTEN frameshift mutation and a SMAD7 missense mutation occurring in a father and son who had a syndrome of gastrointestinal hamartomatous and ganglioneuromatous polyposis, and who both developed esophageal adenocarcinoma, which has not previously been reported as a feature of PHTS.
Materials and Methods
Patients were enrolled under an Institutional Review Board-approved protocol and provided informed consent. Tissues available included blood from both affected patients, a thyroid resection specimen from the proband, and an esophageal resection specimen from the proband’s son. DNA was recovered from peripheral leukocytes. SMAD4 and BMPR1A were screened for mutations and deletion/duplications as described.[13,14] Exome sequencing of the proband was performed by Centrillion Biosciences (Palo Alto, CA) using the SureSelect Human All Exon v.4 51Mb kit (Agilent Technologies, Santa Clara, CA) and HiSeq 2000 Sequencer (Illumina, San Diego, CA). Sequence alignment employed the Burroughs-Wheeler Aligner (BWA-MEM),[15] with processing and variant calling by the Genome Analysis Toolkit pipeline.[16] Variant frequencies were from the Exome Sequencing Project Exome Variant Server (EVS).[17] After filtering, candidate mutations included those that were heterozygous (due to presumed autosomal dominant inheritance), were rare in the EVS population, and were predicted to be damaging (Supplemental Table). Top candidate mutations were confirmed by PCR with Sanger sequencing. Fluorescence in-situ hybridization (FISH) was performed using probes for PTEN and the chromosome 10 centromere (CEP10) according to manufacturer specifications (Abbott Laboratories, Abbott Park, IL). Slides were counterstained with DAPI and 200 interphase nuclei were analyzed. Immunohistochemistry (IHC) for PTEN expression was performed as described with mouse monoclonal antibody 6H2.1 at 1:100 dilution (Dako, Carpinteria, CA),[18] while SMAD7 IHC employed rabbit monoclonal antibody SC-11932 at 1:20 dilution (Santa Cruz Biotechnology, Dallas, TX).
Results
Clinical Features
The proband, a European-American male, presented at age 41 with dysphagia, weight loss, and abdominal pain and was found to have adenocarcinoma of the distal esophagus and multiple gastric, duodenal, and colonic juvenile polyps (Figure 1A, Patient II-2). He underwent esophagectomy, which revealed node-positive disease, followed by adjuvant chemoradiation. Four years later he underwent total thyroidectomy for papillary thyroid cancer. At age 47, colonoscopy revealed persistent colonic polyposis, including a large polyp in the transverse colon, and he underwent subtotal colectomy. Pathology showed generalized juvenile polyposis of the colon. He continued to have regular surveillance and removal of gastric polyps, however, at age 54 he experienced progressive dysphagia and was diagnosed with squamous cell carcinoma at the esophagogastric anastomosis. He underwent palliative chemoradiotherapy and died at age 57.
Figure 1.
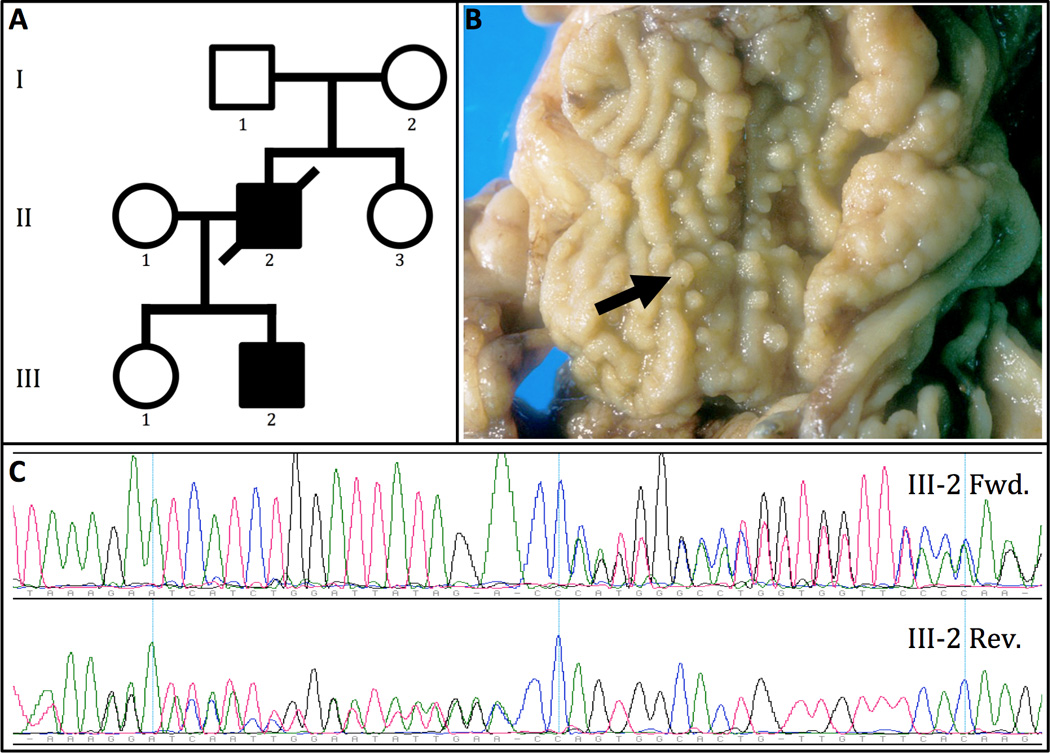
(A) PTEN Hamartoma Tumor Syndrome and esophageal cancer family. Solid shading indicates affected individuals who both had colonic polyposis and esophageal adenocarcinoma. Individuals I-1, I-2, II-3, and III-1 had no apparent symptoms. The proband, (Patient II-2), had esophageal cancer, thyroid cancer, and multiple juvenile polyps of the colon. Patient III-2 also had esophageal cancer, as well as ganglioneuromatous polyps of the stomach, duodenum and colon, and thyroid nodules. (B) Colectomy specimen from Patient III-2, showing diffuse polypoid ganglioneuromas with the arrow marking a representative ganglioneuromatous polyp. (C) Forward (Fwd.) and reverse (Rev.) Sanger sequencing confirmed a germline PTEN frameshift mutation in Patients II-2 and III-2.
Due to the proband’s presumed JPS diagnosis and development of esophageal cancer at a young age, his son (Patient III-2) had regular upper and lower endoscopic screening, which identified extensive gastroduodenal and colonic polyps and polypoid ganglioneuromas. Of note, Patient III-2 was treated for an intracranial arteriovenous malformation (AVM) at age 21 and had a facial trichilemmoma. With colonic lesions too numerous for endoscopic removal, he underwent subtotal colectomy at age 30. Pathology showed inflammatory polyps, tubular adenoma, and diffuse polypoid ganglioneuromas (Figure 1B). He continued upper endoscopic surveillance and was well until age 33, when a distal esophageal lesion was confirmed as node-positive adenocarcinoma. He likewise underwent esophagectomy and had neoadjuvant chemoradiotherapy. Both patients were lifelong non-smokers who did not abuse alcohol.
Sequencing
The proband’s numerous juvenile polyps and lack of PHTS features such as macrocephaly, trichilemmoma, or intellectual disability led to a JPS diagnosis, yet sequencing and multiplex ligation-dependent probe amplification revealed no mutations or deletion/duplications in coding or promoter regions of SMAD4 or BMPR1A. Exome sequencing was therefore performed to search for germline mutations in other potential disease-associated genes. This identified a novel heterozygous single-base insertion in the PTEN gene (c.568_569insC, p.V191S_fs*11), predicted to cause a frameshift with premature termination in the 5th coding exon. Sanger sequencing confirmed the presence of the mutation in both the proband and his son (Figure 1C). This mutation has not previously been reported in large series of PHTS patients or the Online Mendelian Inheritance in Man and Human Gene Mutation databases.[10,19,20]
While this PTEN mutation was considered likely to be responsible for the proband’s symptoms and multiple cancers, the list of candidate mutations was examined for variants occurring in known GI cancer pathways, which might influence the proband’s clinical phenotype. Out of 72 rare variant candidates (Supplementary Table), a germline missense mutation in SMAD7 (c.115G>A, p.G39R) stood out due to its role in the TGF-β/SMAD4 pathway, mutations in which are known to cause Juvenile Polyposis Syndrome. This mutation was confirmed by Sanger sequencing in Patients II-2 and III-2. The SMAD7 G39R mutation is reported in dbSNP (rs144204026), but is rare with an allele frequency of 0.3% (36/10904 chromosomes) in EVS.[17] The G39R alteration occurs in a conserved region and in silico mutation analysis tools predict it to be damaging.[21]
PTEN and SMAD7 in Tumor Specimens
Immunohistochemistry was performed to determine whether malignant tissues expressed PTEN protein (Figure 2A–C). In Patient II-2’s thyroid cancer and Patient III-2’s esophageal cancer specimens, PTEN protein was weakly expressed in normal tissue, but completely absent in malignant cells. In contrast to this, IHC revealed expression of SMAD7 protein in tumor that was similar to background expression levels (Figure 2D). To investigate whether PTEN allelic loss had occurred, FISH was performed on malignant tissue from Patient III-2’s esophagectomy specimen (Figure 2E). Hybridization revealed two PTEN copies in all 200 malignant nuclei examined, suggesting that deletion of the wild-type PTEN allele is not responsible for disrupting PTEN protein expression in this tumor.
Figure 2.
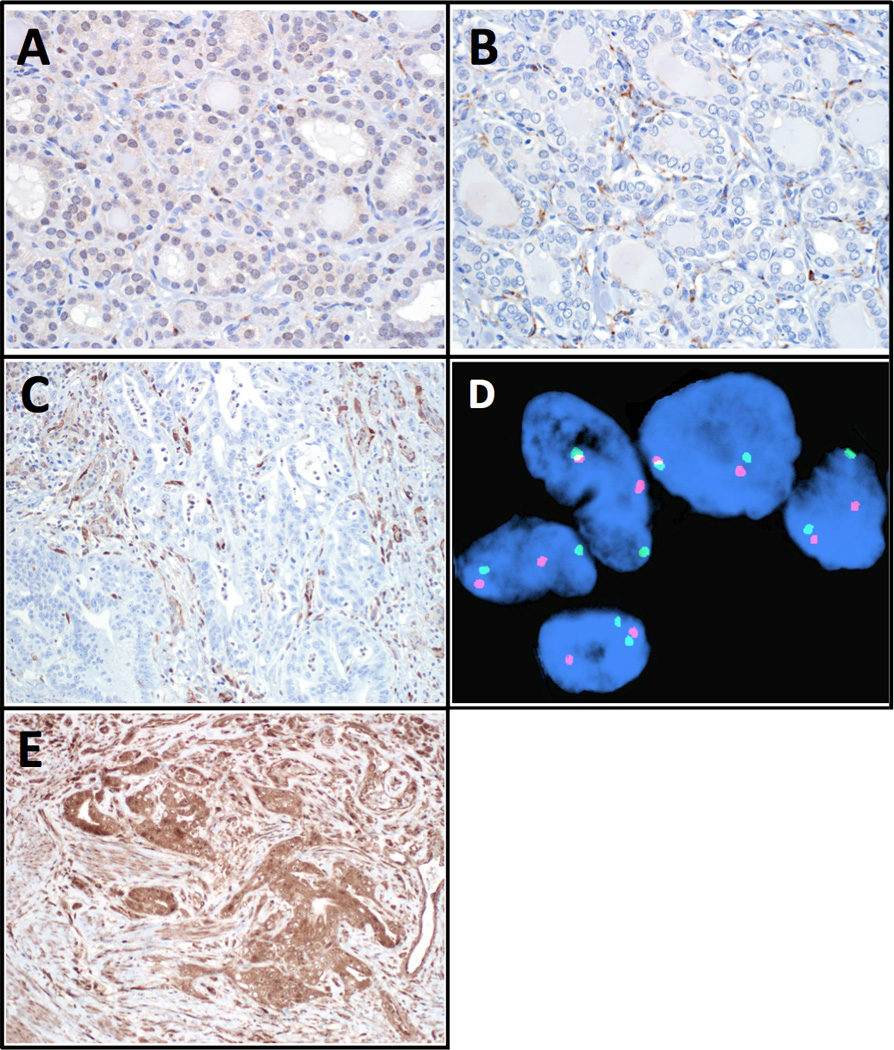
Immunohistochemical staining reveals weak PTEN protein expression throughout Patient II-2’s normal thyroid tissue (A), which is absent in his papillary thyroid cancer (B). Both images magnified 400×. Similar loss of PTEN expression is observed in esophageal adenocarcinoma cells from Patient III-2 (C, 200× magnification). In (B) and (C), non-cancerous endothelial cells retain PTEN expression. Esophageal adenocarcinoma from Patient III-2 demonstrates preserved expression of SMAD7 protein, with staining similar to that seen in surrounding cells (D, 200× magnification). Fluorescent in-situ hybridization demonstrates two PTEN copies in malignant esophageal cancer cells from Patient III-2 (E). In these representative cells, red fluorescence marks PTEN and green fluorescence marks the chromosome 10 centromere.
Discussion
The PTEN mutation in this family (c.568_569insC) has not been previously observed, but occurs in a frequently-mutated region of PTEN exon 5, and is similar to several reported mutations.[10] Of 290 probands with PTEN mutations causing PHTS reported in a large registry, 13 had termination or frameshift mutations within 20 residues of this V191S_fs*11 mutation.[6] The same registry reported a higher incidence of colorectal cancer among patients with PTEN frameshift mutations compared to missense or promoter mutations, while another registry reported lower rates of thyroid cancer in missense mutation patients.[7,10] The present family demonstrates clinical features consistent with these genotype-phenotype correlations, with thyroid cancer occurring in Patient II-2, and prominent colonic involvement in both affected patients. Although DNA from Parents I-1 and I-2 was not available for analysis, as they are alive and reportedly without symptoms, we speculate that the PTEN mutation arose de novo in Patient II-2. Notably, the proband survived more than 16 years after diagnosis of Stage III esophageal adenocarcinoma (EAC), which compares favorably with the 2-year median survival expected in Stage III patients.[22] Whether EAC developing in the setting of PHTS is associated with better prognosis is difficult to infer from a single family.
Loss of PTEN protein expression in these patients’ malignant tissue matches reports from PHTS-associated tumors of the breast, ovary, cerebellum, and thyroid.[18,23–26] PTEN gene dosage affects tumor susceptibility, with the reduced protein expression in patients with germline mutations predisposing them to develop hamartomas, which may retain PTEN expression.[23,27] In cancer cells, however, a second-hit eliminates expression from the wild-type allele. Recognized PHTS second-hit mechanisms include promoter methylation, chromosomal loss of heterozygosity (LOH), and new somatic mutations.[23–25] In Patient III-2, normal PTEN copy number by FISH argues against chromosomal loss, but copy-neutral LOH and other genetic or epigenetic changes remain possible.
Whereas PHTS shows high penetrance, expressivity of its diverse features is variable and the proband’s phenotype of prominent juvenile polyps led to PHTS initially being less-strongly suspected. In retrospect, Patient II-2’s clinical features at presentation for colectomy give a significant 29% risk of having a PTEN mutation by the Cleveland Clinic Calculator.[6] Additional PHTS features present in Patient III-2 (AVM, trichilemmoma, ganglioneuromas) raise his risk to >99%. Although whole-exome sequencing, rather than PTEN mutational screening, may therefore have been unnecessary to make a PHTS diagnosis, it contributed intriguing additional information in light of both patients’ unusual presentation of esophageal cancer at a young age.
Esophageal adenocarcinoma has not been reported in PHTS registries with long-term follow-up.[7,10] In some respects, this is surprising. Although somatic PTEN mutations are uncommon in esophageal cancer, alterations in PTEN expression commonly occur in EAC and esophageal squamous cell carcinoma (ESCC).[12,28] In a study of 117 resected EACs, 38% showed absent or markedly reduced PTEN staining by IHC, and PTEN deficiency independently correlated with worse disease-free and overall survival.[29] Similarly, in 97 ESCCs, 50.5% showed loss of nuclear PTEN IHC staining, which also correlated with worse outcome.[30] Patients with germline haploinsufficiency for PTEN, who develop other GI cancers at rates many times higher than unaffected individuals, might therefore be expected to show increased susceptibility to esophageal cancer. While EAC could simply be a less-common manifestation of PHTS, its rarity in long-term follow-up of large numbers of PHTS patients makes its presence in these cases suggestive of additional modifying genetic characteristics.
The SMAD7 G39R mutation could represent such a modifier. SMAD7 negatively regulates the transforming growth factor beta (TGF-β) superfamily pathway in a finely-tuned feedback loop, where it targets TGF-β receptors for ubiquitination and proteasomal degradation and blocks receptor/effector protein association.[31,32] Although the TGF-β and BMP pathways both converge on SMAD4 to exert their effects, they have distinct functions, and whereas mutations in BMPR1A and SMAD4 cause JPS, mutations in TGF-β receptor-associated SMADs (SMAD2-3 and SMAD7), have not been found in hamartomatous tumor syndromes.[33]
The TGF-β pathway has a complex relationship to cancer development, serving as both a pro- and anti-proliferative and apoptotic signal in different cell types and contexts,[32,34] and recent research suggests an important role for SMAD7 in cancer susceptibility, progression, and evasion of immune surveillance.[32,34–38] Loss of SMAD7 protein causes decreased colorectal cancer cell growth in vitro, but in vivo also decreases the ability of tumor infiltrating lymphocytes to induce cancer cell apoptosis, thereby promoting metastasis.[32,34] Smad7 knockout increases rates of hepatocellular carcinoma (HCC) formation after diethylnitrosamine injection in mice,[38] and negative SMAD7 staining by IHC correlates with worse survival in human esophageal squamous and pancreatic cancers.[39,40] IHC results in Patient III-2 esophageal tumor specimen demonstrate intact levels of SMAD7 protein expression, however, whether the function of this protein is affected by the G39R mutation remains unknown. Strikingly, in a mouse model of conditional dual Smad4/Pten keratinocyte-specific knockout, 100% of Smad4/Pten-deficient mice developed esophagogastric squamous cell carcinomas by 2 months, while in mice with only Pten deficiency, no cancers developed by 8 months.[41] SMAD4 activation increases PTEN protein levels, while Akt directly sequesters SMAD3, down-regulating the TGF-β pathway.[41] Disruption of two closely-regulated and interacting pathways could explain enhanced tumor development in the dual-knockout mouse.[41]
If loss of SMAD4 promotes cancer, dysfunction of a SMAD4 signaling inhibitor might be expected to have the opposite effect, however, data showing tumorigenic effects of SMAD7 loss, and association of single-nucleotide polymorphisms (SNPs) causing decreased SMAD7 function with colorectal cancer in multiple populations,[35,37,42–44] suggest that perturbation of the TGF-β pathway at the level of either SMAD7 or SMAD4 promotes tumorigenesis. Perhaps most germane to this family, the microRNA 216a/217, which is upregulated in recurrent HCC specimens, was found to promote HCC recurrence and sorafenib resistance by direct inhibition of both PTEN and SMAD7.[45] Finally, sequencing of SMAD7 in patients with SNP haplotypes deemed at high-risk for colorectal cancer revealed the G39R allele in in 2 of 35 individuals,[37] a frequency significantly higher than in the general population (36 of 10904 chromosomes by EVS, p=0.02 by Fisher exact test). This suggests the SMAD7 G39R mutation could represent an attenuated allele, which while only weakly tumorigenic by itself, promotes EAC development in this family’s PTEN-deficient background, similar to the role proposed for succinate dehydrogenase-family (SDHx) variants in modifying breast cancer risk in PHTS patients.[11]
In summary, we report novel PTEN and rare SMAD7 mutations in a family with gastrointestinal polyposis and esophageal adenocarcinoma. Emergence of EAC in these patients is possibly influenced by their coexisting SMAD7 mutation, or may represent a less-common manifestation of PHTS. Exome sequencing of additional affected families could further define the contribution of rare mutations in genes other than PTEN to PHTS phenotypic variation. Beyond colonoscopic and other surveillance recommended for PHTS patients,[10] these results support including baseline upper endoscopy, with repeat screening and biopsy of suspicious lesions in patients with upper GI polyps.
Supplementary Material
The supplementary table supplies the list of the 72 high-priority candidate variants identified from exome sequencing of peripheral leukocyte DNA from the proband, Patient II-2. Columns (left to right) list the gene name, genetic coding change and chromosomal coordinates, the predicted amino-acid change, the full gene name, information on the function of the gene based on the Online Mendelian Inheritance in Man (OMIM) database, the coding sequence change, and what type of mutation is predicted. These candidates were selected based on the following filtering strategy: Raw data were first analyzed according to the Genome Analysis Toolkit Pipeline. Variant calls were then compared to a local University of Iowa exome database to filter high-frequency, likely artifactual, variants. This raw list of variants was then inspected for mutations in known polyposis-associated genes, which revealed the PTEN mutation, which was confirmed by Sanger sequencing in the other affected patient. As esophageal cancer is not a standard PTEN hamartoma tumor syndrome cancer, it was hypothesized that additional deleterious variants in GI-cancer related pathways might act as modifiers of this syndrome. A strategy designed to identify only high-quality, likely deleterious variants with autosomal-dominant inheritance was then applied to the raw data using the following filters: quality per read depth >5; not on the X chromosome; minimum 10× coverage; non-intronic, frameshift or non-synonymous variant, not present or with minor allele frequency of <0.01 in dbSNP, BLOSUM score <0; and heterozygous. Of the resulting 72 variants, 2 besides PTEN were frameshifts. One of these was involved in autosomal recessive deafness (TPRN) and was judged unlikely to contribute to esophageal cancer. The other, CRIPAK has been reported to negatively regulate a putative tumor suppressor, but inspection of the supposedly mutated region revealed extremely repetitive sequence, which suggests that this variant call is an artifact of sequencing or sequence fragment mapping. Four additional variants resulted in nonsense mutations. These have roles in retinol metabolism (DHRS9), extracellular matrix (FBN3), major histocompatibility complex transcription (ZXDC), and unknown function (SSPO). None of these are known to have major roles in cancer or polyposis. The remaining 65 variants were manually annotated using OMIM, and assessed for potential involvement in cancer or polyposis-related pathways. From these variants, SMAD7 immediately stood out due to its role in the TGF-β/SMAD4 pathway, dysfunction of which is responsible for Juvenile Polyposis Syndrome. The SMAD7 mutation was confirmed in the proband and his affected son by Sanger sequencing.
Acknowledgements
We are grateful for our patients’ generosity in participating in this research.
Supported by NIH 5T32#CA148062-04 (SKS and JEM).
Footnotes
Financial Disclosures: None
Contributor Information
Scott K. Sherman, Email: scott-sherman@uiowa.edu.
Jessica E. Maxwell, Email: jessica-maxwell@uiowa.edu.
Qining Qian, Email: qining-qian@uiowa.edu.
Andrew M. Bellizzi, Email: andrew-bellizzi@uiowa.edu.
Terry A. Braun, Email: terry-braun@uiowa.edu.
Mark D. Iannettoni, Email: mark-iannettoni@uiowa.edu.
Benjamin W. Darbro, Email: benjamin-darbro@uiowa.edu.
References
- 1.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 2.Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B, Frants RR, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13:114–116. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 3.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 4.Longy M, Coulon V, Duboue B, David A, Larregue M, Eng C, et al. Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J Med Genet. 1998;35:886–889. doi: 10.1136/jmg.35.11.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163:114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- 6.Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88:42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieuwenhuis MH, Kets CM, Murphy-Ryan M, Yntema HG, Evans DG, Colas C, et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer. 2014;13:57–63. doi: 10.1007/s10689-013-9674-3. [DOI] [PubMed] [Google Scholar]
- 8.Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 9.Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 10.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ngeow J, Stanuch K, Mester JL, Barnholtz-Sloan JS, Eng C. Second Malignant Neoplasms in Patients With Cowden Syndrome With Underlying Germline PTEN Mutations. J Clin Oncol. 2014 Apr 28; doi: 10.1200/JCO.2013.53.6656. [Epub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hildebrandt MA, Yang H, Hung MC, Izzo JG, Huang M, Lin J, et al. Genetic variations in the PI3K/PTEN/AKT/mTOR pathway are associated with clinical outcomes in esophageal cancer patients treated with chemoradiotherapy. J Clin Oncol. 2009;27:857–871. doi: 10.1200/JCO.2008.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howe JR, Chinnathambi S, Calva D, Bair J, Pechman B, Salamon A, et al. A family with two consecutive nonsense mutations in BMPR1A causing juvenile polyposis. Cancer Genet Cytogenet. 2008;181:52–54. doi: 10.1016/j.cancergencyto.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, Howe JR. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet. 2009;75:79–85. doi: 10.1111/j.1399-0004.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Heart Lung and Blood Institute Exome Sequencing Project Exome Variant Server. [Accessed April 16, 2014]; Available at: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 18.Barletta JA, Bellizzi AM, Hornick JL. Immunohistochemical staining of thyroidectomy specimens for PTEN can aid in the identification of patients with Cowden syndrome. Am J Surg Pathol. 2011;35:1505–1511. doi: 10.1097/PAS.0b013e31822fbc7d. [DOI] [PubMed] [Google Scholar]
- 19.Online Mendelian Inheritance in Man Database. [Accessed April 11, 2014]; Available at: http://www.omim.org. [Google Scholar]
- 20.Human Gene Mutation Database. [Accessed April 11, 2014]; Available at: http://www.hgmd.org. [Google Scholar]
- 21.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rice TW, Rusch VW, Ishwaran H, Blackstone EH. Cancer of the esophagus and esophagogastric junction: data-driven staging for the seventh edition of the American Joint Committee on Cancer/International Union Against Cancer Cancer Staging Manuals. Cancer. 2010;116:3763–3773. doi: 10.1002/cncr.25146. [DOI] [PubMed] [Google Scholar]
- 23.Reifenberger J, Rauch L, Beckmann MW, Megahed M, Ruzicka T, Reifenberger G. Cowden's disease: clinical and molecular genetic findings in a patient with a novel PTEN germline mutation. Br J Dermatol. 2003;148:1040–1046. doi: 10.1046/j.1365-2133.2003.05322.x. [DOI] [PubMed] [Google Scholar]
- 24.Cho MY, Kim HS, Eng C, Kim DS, Kang SJ, Eom M, et al. First report of ovarian dysgerminoma in Cowden syndrome with germline PTEN mutation and PTEN-related 10q loss of tumor heterozygosity. Am J Surg Pathol. 2008;32:1258–1264. doi: 10.1097/PAS.0b013e31816be8b7. [DOI] [PubMed] [Google Scholar]
- 25.Zhou XP, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet. 2003;73:1191–1198. doi: 10.1086/379382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peiro G, Adrover E, Guijarro J, Ballester I, Jimenez MJ, Planelles M, et al. Synchronous bilateral breast carcinoma in a patient with cowden syndrome: a case report with morphologic, immunohistochemical and genetic analysis. Breast J. 2010;16:77–81. doi: 10.1111/j.1524-4741.2009.00846.x. [DOI] [PubMed] [Google Scholar]
- 27.Ngeow J, He X, Mester JL, Lei J, Romigh T, Orloff MS, et al. Utility of PTEN protein dosage in predicting for underlying germline PTEN mutations among patients presenting with thyroid cancer and Cowden-like phenotypes. J Clin Endocrinol Metab. 2012;97:E2320–E2327. doi: 10.1210/jc.2012-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu YC, Lam KY, Tang JC, Srivastava G. Mutational analysis of the PTEN/MMAC1 gene in primary oesophageal squamous cell carcinomas. Mol Pathol. 1999;52:353–356. doi: 10.1136/mp.52.6.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bettstetter M, Berezowska S, Keller G, Walch A, Feuchtinger A, Slotta-Huspenina J, et al. Epidermal growth factor receptor, phosphatidylinositol-3-kinase catalytic subunit/PTEN, and KRAS/NRAS/BRAF in primary resected esophageal adenocarcinomas: loss of PTEN is associated with worse clinical outcome. Hum Pathol. 2013;44:829–836. doi: 10.1016/j.humpath.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Tachibana M, Shibakita M, Ohno S, Kinugasa S, Yoshimura H, Ueda S, et al. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94:1955–1960. doi: 10.1002/cncr.0678. [DOI] [PubMed] [Google Scholar]
- 31.Li R, Rosendahl A, Brodin G, Cheng AM, Ahgren A, Sundquist C, et al. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J Immunol. 2006;176:6777–6784. doi: 10.4049/jimmunol.176.11.6777. [DOI] [PubMed] [Google Scholar]
- 32.Rizzo A, De Mare V, Rocchi C, Stolfi C, Colantoni A, Neurath MF, et al. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis. 2014 Feb. doi: 10.1093/carcin/bgu027. [Epub] [DOI] [PubMed] [Google Scholar]
- 33.Dahdaleh FS, Carr JC, Calva D, Howe JR. Juvenile polyposis and other intestinal polyposis syndromes with microdeletions of chromosome 10q22–23. Clin Genet. 2012;81:110–116. doi: 10.1111/j.1399-0004.2011.01763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stolfi C, De Simone V, Colantoni A, Franze E, Ribichini E, Fantini MC, et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis. 2014;5:e1073. doi: 10.1038/cddis.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slattery ML, Herrick J, Curtin K, Samowitz W, Wolff RK, Caan BJ, et al. Increased risk of colon cancer associated with a genetic polymorphism of SMAD7. Cancer Res. 2010;70:1479–1485. doi: 10.1158/0008-5472.CAN-08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salot S, Gude R. MTA1-mediated transcriptional repression of SMAD7 in breast cancer cell lines. Eur J Cancer. 2013;49:492–499. doi: 10.1016/j.ejca.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 37.Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Zhao J, Chu ES, Mok MT, Go MY, Man K, et al. Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J Pathol. 2013;230:441–452. doi: 10.1002/path.4206. [DOI] [PubMed] [Google Scholar]
- 39.Osawa H, Nakajima M, Kato H, Fukuchi M, Kuwano H. Prognostic value of the expression of Smad6 and Smad7, as inhibitory Smads of the TGF-beta superfamily, in esophageal squamous cell carcinoma. Anticancer Res. 2004;24:3703–3709. [PubMed] [Google Scholar]
- 40.Wang P, Fan J, Chen Z, Meng ZQ, Luo JM, Lin JH, et al. Low-level expression of Smad7 correlates with lymph node metastasis and poor prognosis in patients with pancreatic cancer. Ann Surg Oncol. 2009;16:826–835. doi: 10.1245/s10434-008-0284-5. [DOI] [PubMed] [Google Scholar]
- 41.Teng Y, Sun AN, Pan XC, Yang G, Yang LL, Wang MR, et al. Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res. 2006;66:6972–6981. doi: 10.1158/0008-5472.CAN-06-0507. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y, Sun Z, Zhang A, Zhang J. SMAD7 rs12953717 polymorphism contributes to increased risk of colorectal cancer. Tumour Biol. 2014;35:695–699. doi: 10.1007/s13277-013-1095-2. [DOI] [PubMed] [Google Scholar]
- 43.Kirac I, Matosevic P, Augustin G, Simunovic I, Hostic V, Zupancic S, et al. SMAD7 variant rs4939827 is associated with colorectal cancer risk in Croatian population. PLoS One. 2013;8:e74042. doi: 10.1371/journal.pone.0074042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang B, Jia WH, Matsuo K, Shin A, Xiang YB, Matsuda K, et al. Genome-wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in East Asians. Int J Cancer. 2014 doi: 10.1002/ijc.28733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. doi: 10.1002/hep.26369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The supplementary table supplies the list of the 72 high-priority candidate variants identified from exome sequencing of peripheral leukocyte DNA from the proband, Patient II-2. Columns (left to right) list the gene name, genetic coding change and chromosomal coordinates, the predicted amino-acid change, the full gene name, information on the function of the gene based on the Online Mendelian Inheritance in Man (OMIM) database, the coding sequence change, and what type of mutation is predicted. These candidates were selected based on the following filtering strategy: Raw data were first analyzed according to the Genome Analysis Toolkit Pipeline. Variant calls were then compared to a local University of Iowa exome database to filter high-frequency, likely artifactual, variants. This raw list of variants was then inspected for mutations in known polyposis-associated genes, which revealed the PTEN mutation, which was confirmed by Sanger sequencing in the other affected patient. As esophageal cancer is not a standard PTEN hamartoma tumor syndrome cancer, it was hypothesized that additional deleterious variants in GI-cancer related pathways might act as modifiers of this syndrome. A strategy designed to identify only high-quality, likely deleterious variants with autosomal-dominant inheritance was then applied to the raw data using the following filters: quality per read depth >5; not on the X chromosome; minimum 10× coverage; non-intronic, frameshift or non-synonymous variant, not present or with minor allele frequency of <0.01 in dbSNP, BLOSUM score <0; and heterozygous. Of the resulting 72 variants, 2 besides PTEN were frameshifts. One of these was involved in autosomal recessive deafness (TPRN) and was judged unlikely to contribute to esophageal cancer. The other, CRIPAK has been reported to negatively regulate a putative tumor suppressor, but inspection of the supposedly mutated region revealed extremely repetitive sequence, which suggests that this variant call is an artifact of sequencing or sequence fragment mapping. Four additional variants resulted in nonsense mutations. These have roles in retinol metabolism (DHRS9), extracellular matrix (FBN3), major histocompatibility complex transcription (ZXDC), and unknown function (SSPO). None of these are known to have major roles in cancer or polyposis. The remaining 65 variants were manually annotated using OMIM, and assessed for potential involvement in cancer or polyposis-related pathways. From these variants, SMAD7 immediately stood out due to its role in the TGF-β/SMAD4 pathway, dysfunction of which is responsible for Juvenile Polyposis Syndrome. The SMAD7 mutation was confirmed in the proband and his affected son by Sanger sequencing.