

Abstract
Mammalian species express nine functional voltage-gated Na+ channels. Three of them, the cardiac-specific isoform Nav1.5 and the neuronal isoforms Nav1.8 and Nav1.9, are relatively resistant to the neurotoxin tetrodotoxin (TTX; IC50 ≥ 1 μM). The other six isoforms are highly sensitive to TTX with IC50 values in the nanomolar range. These isoforms are expressed in the central nervous system (Nav1.1, Nav1.2, Nav1.3, Nav1.6), in the skeletal muscle (Nav1.4), and in the peripheral nervous system (Nav1.6, Nav1.7). The isoform Nav1.5, encoded by the SCN5A gene, is responsible for the upstroke of the action potential in the heart. Mutations in SCN5A are associated with a variety of life-threatening arrhythmias, like long QT syndrome type 3 (LQT3), Brugada syndrome (BrS) or cardiac conduction disease (CCD). Previous immunohistochemical and electrophysiological assays demonstrated the cardiac expression of neuronal and skeletal muscle Na+ channels in the heart of various mammals, which led to far-reaching speculations on their function. However, when comparing the Na+ channel mRNA patterns in the heart of various mammalian species, only minute quantities of transcripts for TTX-sensitive Na+ channels were detectable in whole pig and human hearts, suggesting that these channels are not involved in cardiac excitation phenomena in higher mammals. This conclusion is strongly supported by the fact that mutations in TTX-sensitive Na+ channels were associated with epilepsy or skeletal muscle diseases, rather than with a pathological cardiac phenotype. Moreover, previous data from TTX-intoxicated animals and from cases of human tetrodotoxication showed that low TTX dosages caused at most little alterations of both the cardiac output and the electrocardiogram. Recently, genome-wide association studies identified SCN10A, the gene encoding Nav1.8, as a determinant of cardiac conduction parameters, and mutations in SCN10A have been associated with BrS. These novel findings opened a fascinating new research area in the cardiac ion channel field, and the on-going debate on how SCN10A/Nav1.8 affects cardiac conduction is very exciting.
Structure-function relationships of mammalian voltage-gated Na+ channels
Voltage-gated sodium (Na+) channels are of fundamental importance for excitation processes by triggering the fast upstroke of the action potential. These channels are heteromultimeric proteins consisting of a large pore-forming α subunit (∼260 kDa) and smaller accessory β subunits (Figure 1).1,2 As demonstrated by heterologous expression experiments, the α subunit determines the main electrophysiological and pharmacological properties of a given Na+ channel complex, while the different β subunits modulate the function of the α subunits.3–6
Figure 1.

Structure and function of voltage-gated Na+ channels. A) Proposed membrane topology of voltage-gated Na+ channels. Each of the four domains (DI-IV) is composed of six transmembrane helices. The fourth segment (S4; red) contains regularly arranged positive charges that are important elements of the voltage sensor. The intracellular loop between the third and fourth domain forms the inactivation gate composed of residues isoleucine, phenylalanine, and methionine (IFM motif). The proximal C terminus contains an EF-hand domain involved in the binding of Ca++, and a downstream calmodulin-binding motif (IQ motif). B) Functional states of voltage-gated Na+ channels. C) Whole-cell recordings showing Nav1.5 current family obtained in HEK293 cells. Currents were elicited by test potentials from − 80 mV to various test pulses in 5 or 10 mV increments at a pulsing frequency of 1.0 Hz (holding potential: − 120 mV). D) Single-channel recordings in cell-attached patches using transfected HEK293 cells. Nav1.5 channel activity was measured by stepping from a holding potential of − 120 mV to a test potential of − 20 mV. The arrows indicate the beginning and the end of the test pulse (8 ms). The single-channel amplitude under these conditions was 1.26 pA. Modified from80.
The α subunit is composed of four homologous domains (DI to DIV) that are connected by intracellular linkers (Figure 1A). Each domain contains six transmembrane spanning segments (S1 to S6). The fourth segment (S4) of each domain is an essential element of the voltage sensor. These S4 stretches are characterized by regularly arranged positive charges at every third position. At the resting membrane potential, S4 segments are fixed in a more inward position, and the channel is in its closed state (Figure 1B). A depolarizing voltage pulse leads to a transient outward movement of the S4 in domains I to III, thereby initiating the opening of the pore. The resulting inward current, shown in Figure 1C, depends on the Na+ gradient and the number of activated channels at a certain membrane potential. The pore is formed by the fifth and sixth segments of each domain, and by the connecting extracellular loops, the so-called P loops. These P loops contain key residues for ion selectivity and for binding of natural toxins, like the buffer fish poison tetrodotoxin (TTX). The transition from the open to the inactivated state normally occurs within a few milliseconds after opening (see single-channel traces in Figure 1D). This open-state inactivation is most likely initiated by the S4 in domain IV. This positively charged segment moves when the other three S4 regions are already in a more outward position and when the channel already conducts Na+.7,8 The decelerated response of this S4 in DIV generates the signal for the movement of intracellularly located channel structures that occlude the pore, thereby terminating Na+ influx. Three Na+ channel structures are essentially involved in this fast channel closure: a) the inactivation gate which is formed by the DIII-DIV linker and which contains the hydrophobic residues isoleucine, phenylalanine, and methionine (IFM motif; Figure 1A), b) the inactivation gate receptor formed by amino acid residues in S6 of DIV and in intracellular S4/S5 loops of DIII and DIV, and c) a large portion of the channel C terminus. Inactivation is initiated when the inactivation gate moves towards its receptor. The C terminus finally stabilizes the inactivation gate-occluded channel in a Ca++-dependent manner.9–14 A tight channel closure is crucial for a coordinated action potential termination. In wild-type channels, the fraction of the Na+ current, that persists throughout the action potential, is less than 0.3 % of the transient current. Noteworthy, mutations in channel structures that are essential for fast open-state inactivation often cause an increase of this persistent current fraction, which may result in life-threatening arrhythmias.
The transition from the inactivated state back to the closed state is also called recovery from inactivation. This step is both time- and voltage-dependent. In the closed state, the inactivation gate has finally returned to its initial conformation, and the channel is occluded by its pore. Na+ channels may also enter the inactivated state directly from the closed state. This so-called closed-state inactivation reduces the number of channels available for activation. It occurs when the resting membrane potential becomes more positive, for example when the extracellular potassium concentration is increased. The membrane potential at which half of the cardiac Na+ channels are available for activation is a few mV negative to − 80 mV.15,16
Tissue-specific distribution of mammalian voltage-gated Na+ channels
Nine different functional sodium channel α subunits have been identified by electrophysiological recordings, biochemical purification, and in cloning studies (Figure 2).1,17 The α subunits Nav1.1, Nav1.2, and Nav1.3 are abundantly expressed in various neurons of the central nervous system.1 These isoforms were detected in hybridization experiments using the eel electroplax Na+ channel cDNA as a probe.18,19 This pioneering work was the basis for the discovery of the skeletal muscle channel Nav1.4 and of the major cardiac isoform Nav1.5.20–22 The isoform Nav1.6 is widely expressed in various neurons of the central nervous system. Moreover, Nav1.6 is also present in the peripheral nervous system. It has been detected in dorsal root ganglia (DRG), including motor and sensory neurons, and at the nodes of Ranvier in the sciatic nerve, spinal cord, and the optic nerve.1 The isoforms Nav1.7, Nav1.8, and Nav1.9 are expressed primarily in the peripheral nervous system. Nav1.7 is present in all types of DRG neurons, in Schwann cells, in the sciatic nerve, in sympathetic neurons, and in neuroendocrine cells. Nav1.8 was detected in DRG neurons, in unmyelinated, small-diameter sensory neurons (C-fibres) involved in nociception, and more recently in the heart (see below). Nav1.9 is mainly expressed in sensory neurons of the DRG and trigeminal ganglion.1 It is noteworthy that loss-of-function mutations in the genes encoding Nav1.7 and Nav1.9 (SCN9A and SCN11A, respectively) were associated with the inability to experience pain.23–25
Figure 2.

Phylogenetic tree, tissue distribution, chromosomal localization, and TTX sensitivity of mammalian voltage-gated Na+ channels. The name of an individual channel consists of the chemical symbol of the permeating ion (Na), the principal physiological regulator (voltage), the gene subfamily, and the number of the specific channel isoform, assigned according to the approximate order in which each isoform was identified. Abbreviations: CNS – central nervous system, PNS – peripheral nervous system, TTX – tetrodotoxin. Adapted from17.
In addition to these nine functional sodium channels at least three other Na+ channel sequences have been detected in heart, uterus, smooth muscle, astrocytes, and various neurons of human, rat, and mouse. Because of their low sequence identity to the Nav1 isoforms (approximately 50%), these channels were originally considered as members of a different subfamily (Nav2.x). Predicted protein sequences revealed significant differences in functionally important channel regions, like the voltage sensor, the inactivation gate, and the pore region, which led to their classification as atypical Na+ channels or Nax isoforms. Recent studies suggested that these variants play an important role in body-fluid homeostasis.26
One of the most important compounds in Na+ channel research is the puffer fish poison tetrodotoxin (TTX). The drug acts from the extracellular side by occluding the channel entrance, thereby preventing the flux of monovalent ions through the pore. Radiolabeled toxin has been previously used in channel purification experiments, which were crucial steps for subsequent cloning studies. TTX was also used to map the outer vestibule of the Na+ channel and to identify critical structural elements in the pore. The drug was helpful to separate Na+, K+, and Ca++ currents in native cells and tissues.
TTX can also be used to distinguish between the different Na+ channel isoforms. All neuronal channels, except for Nav1.8 and Nav1.9, as well as the skeletal muscle channel Nav1.4 are highly sensitive towards TTX (IC50 ∼ 1-25 nM).1 The compound forms a complex network of hydrogen-bonds with amino acid residues of the selectivity filter.27,28 One important key residue for high-affinity TTX binding is an aromatic residue in the pore region of domain I (position 374 in rat Nav1.5).29 In cardiac Nav1.5 as well as in neuronal Nav1.8 and Nav1.9 channels the corresponding position is occupied by a non-aromatic residue (cysteine or serine), causing an at least 100-fold lower potency of TTX to occlude the ion conduction pathway. The reported IC50 values are about 1 μM for Nav1.5 and Nav1.9, and 60 to 100 μM for Nav1.8.1
The most important cardiac Na+ channel is the TTX-resistant isoform Nav1.5
Because of its vital importance, the molecular nature of the voltage-activated cardiac Na+ current has been extensively studied during the last decades. There are several lines of evidence demonstrating that the most prominent cardiac Na+ channel is Nav1.5.
TTX sensitivity of cardiac Na+ channels
It has been known since the 1960s that nanomolar concentrations of TTX had no effect on the rate of rise and duration of the mammalian cardiac action potential.30 Biochemical binding assays in the 1980s showed that protein preparations from rat cardiac membranes contained a large portion of low-affinity TTX binding sites.22,31–34 Specific effects were observed with TTX concentrations in the range of about 1 to 10 μM. This is also the approximate concentration to block half of the native cardiac Na+ channels using isolated atrial and ventricular cardiomyocytes.35–37 Very similar data were obtained in Nav1.5-transfected mammalian cells and in cRNA-injected Xenopus oocytes.1,16,38,39 Please note that Nav1.8, the SCN10A gene product, is nearly 100-fold less sensitive towards TTX compared to Nav1.5. To the best of our knowledge, there is no single biochemical study showing a larger quantity of such TTX-resistant receptors in the heart.
Cloning experiments
Hybridization studies in the 1980s using two different Nav1.2 cRNA probes resulted in the isolation of the Nav1.5 cDNA from a rat heart cDNA library.22 Cloning of the more distantly related Nav1.5 channel by this method was undoubtedly due to its abundant expression in the heart. Of little surprise was the detection of positive clones containing brain-type Na+ channel fragments using a brain-type cRNA probe. However, none of the other TTX-resistant Na+ channels, Nav1.8 or Nav1.9, was found.
Electrophysiological properties
Cloned Nav1.5 channels expressed in mammalian cells or Xenopus oocytes showed not only blocking but also gating properties of native channels.16,35,38,40 For example, steady-state activation and inactivation curves are significantly shifted towards more negative voltages in both reconstituted Nav1.5 and native cardiac Na+ channels, when compared to brain-type, skeletal muscle or sensory neuron isoforms, including Nav1.8.41
Widespread expression of Nav1.5 in the heart
Nav1.5 is highly expressed in all types of cardiac myocytes, including the sinus node, the conduction system, atrial and ventricular myocytes.2,42–44 The channel localizes to all important subcellular membrane structures like the intercalated disc regions, the outer plasma membrane and the t-tubular system (Figure 3).37,45
Figure 3.

Subcellular localization of voltage-gated Na+ channels in mouse ventricular myocytes using a specific antibody against Nav1.5. Intense fluorescence signals were obtained at the outer plasma membrane (PM) and at the intercalated disks (*). Staining along the Z lines suggests channel localization in the t-tubular membrane system, as shown at a higher magnification in the right image. Adapted from37.
Knock-out experiments
Complete disruption of SCN5A in mice (Scn5a − / − ) caused severe defects in ventricular morphogenesis, resulting in intrauterine lethality.46 Heterozygous Scn5a +/ − mice showed normal survival, but several cardiac defects including impaired atrioventricular conduction, delayed intramyocardial conduction, increased ventricular refractoriness, and ventricular tachycardia with characteristics of reentrant excitation.46
SCN5A channelopathies and effects of Nav1.5 RNA splicing
Point mutations in SCN5A can lead to life-threatening arrhythmias.47 Interestingly, SCN5A is more often affected than any other cardiac ion channel gene (Figure 4, Table 1). Gain-of-function mutations, such as ΔKPQ, lead to prolonged QTc intervals in the body surface ECG, indicating both a high and widespread expression in ventricles. Furthermore, missplicing of even a smaller fraction of Nav1.5 RNA, resulting in non-functional channels, was related to heart failure.48 More recently, abnormal splicing of SCN5A, resulting in the expression of a Nav1.5/Nav1.5e mixture, was correlated with a BrS-like ECG pattern in myotonic dystrophy type 1.49
Figure 4.

Cardiac currents and number of mutations (red) associated with cardiac arrhythmia. SCN5A is more frequently affected than all the other cardiac ion channel genes. It is likely that considerably more mutations have been recently identified by genetic screenings, and that even novel mutations may not be considered for publication in peer-reviewed journals or online data bases. Furthermore, it must be pointed out that most if not all ion channel subunits interact with other cardiac proteins, whose mutations can affect the cardiac action potential.2 More recently, several SCN10A mutations were identified in BrS patients.74 These mutations were not included, because the physiological significance of SCN10A/Nav1.8 in the heart and its role in shaping the cardiac AP is still a matter of debate. Helpful databases were PubMed/Medline, http://www.fsm.it/cardmoc/ and http://www.qtsyndrome.ch/.
Table 1.
Mutations in cardiac ion channel genes and clinical consequences.
| Current | Channel | Gene | Disease | Number of mutations |
| I Na | Nav1.5 | SCN5A | LQT3 | 87 |
| BrS | 374 | |||
| CCD | 21 | |||
| SSS | 9 | |||
| I to | Kv4.2/4.3 | KCND2/3 | BrS | 2 |
| I Ca | Cav1.2 | CACNA1C | LQT8 | 2 |
| SQTS/BrS | 10 | |||
| ERS | 1 | |||
| CACNB2 | SQTS/BrS | 6 | ||
| IVF/ERS | 3 | |||
| CACNA2D1 | BrS | 2 | ||
| ERV | 1 | |||
| I Ks | KvLQT1 | KCNQ1 | LQT1 | 246 |
| SQTS | 2 | |||
| FAF | 3 | |||
| minK | KCNE1 | LQT5 | 69 | |
| FAF | 16 | |||
| I Kr | hERG | KCNH2 | LQT2 | 297 |
| SQTS | 1 | |||
| BrS | 2 | |||
| MiRPI | KCNE2 | LQT6 | 16 | |
| I K1 | Kir2.1 | KCNJ2 | LQT7 | 29 |
| SQTS | 2 | |||
| FAF | 2 |
Abbreviations: LQT – long QT syndrome, BrS – Brugada syndrome, CCD – cardiac conduction disease, SSS – sick sinus syndrome, SQTS – short QT syndrome, FAF – familiar atrial fibrillation, ERS – early repolarization syndrome, IVF – idiopathic ventricular fibrillation. Helpful databases were PubMed/Medline, http://www.fsm.it/cardmoc/ and http://www.qtsyndrome.ch/.
Expression and function of TTX-sensitive Na+ channels in the mammalian heart
Beside Nav1.5 and its splice variants,50 all TTX-sensitive Na+ channel isoforms have been detected at least in cardiac RNA preparations.16,37,42,51–53 It has been known for a long time, that two families of binding sites for TTX co-exist in the mammalian heart. Coraboeuf and co-workers demonstrated that low concentrations of TTX shortened the action potential of Purkinje fibres and slowed their automatic beating rate.54 A few years later, a smaller fraction with high-affinity TTX binding sites and a larger fraction with low-affinity TTX binding sites were identified in plasma membranes of adult rat hearts, suggesting the presence of neuronal or skeletal muscle Na+ channels in cardiomyocytes.31,34 Successful Na+ channel cloning, the development of RT-PCR, the availability of specific antibodies and advances in electrophysiological recording techniques made it possible to identify the respective transcripts, to show the localization of the channel proteins (Figure 5) and to demonstrate a small TTX-sensitive Na+ current in isolated cardiomyocytes.37,55
Figure 5.
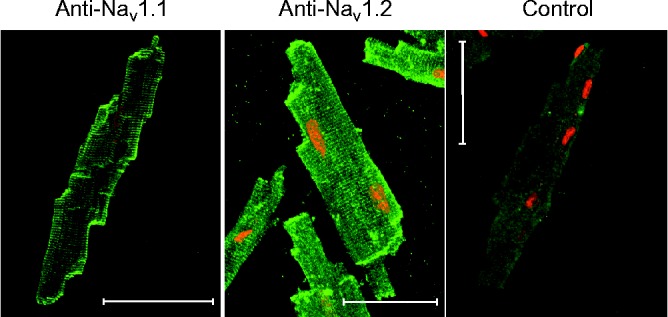
Subcellular localization of Nav1.1 and Nav1.2 in mouse ventricular myocytes. Freshly dissociated cardiac cells were fixed, permeabilized and probed with the primary antibody against Nav1.1 and Nav1.2. Both brain-type Na+ channels were localized at the outer plasma membrane, at the intercalated disk regions and in t-tubular membranes. Scale bars represent 50 μm. Control – antibody against Nav1.2 pre-absorbed against the respective antigen. Adapted from37.
Based on these animal testing data, important functions of the TTX-sensitive Na+ channels in the human heart were suggested (Table 2).51,55–57 However, most of these results were obtained using highly sensitive in vitro assays, and expression levels of TTX-sensitive channels were rarely compared to those of the major isoform Nav1.5. Furthermore, some of the most spectacular data were obtained in small rodents, in particular in mice. It is doubtful whether reliable conclusions for the human heart can be drawn, because important physiological parameters, like stroke volume, heart rate or the action potential shape, are appreciably different between mice and human.
Table 2.
| Suggested function | Species | References | |
| Control of heart rate | Contribution to slow diastolic depolarization | Rabbit | 75,76 |
| Contribution to automaticity and heart rhythm, possible contribution to sick sinus syndrome | Mouse | 51,57 | |
| Contribution to pacemaking and sinus node conduction | Mouse | 44,77 | |
| Cardiac conduction and action potential shape | Higher expression of TTX-sensitive Na+ channels in Purkinje fibres compared to ventricular cells, suggested role for safeguarding conduction | Dog | 42 |
| Expression of Nav1.4 in cardiac Purkinje myocytes, important clinical role in arrhythmia | Dog | 78 | |
| Cardiac contractility | Suggested role for excitation-contraction coupling | Mouse Guinea pig | 55,56,79 |
| Suggested role for action potential propagation in ventricular cardiomyocytes | Mouse Rat | 53,56,80 |
The question whether or not TTX-sensitive Na+ channels are relevant for cardiac excitation could be of interest for a better understanding of cardiac excitation phenomena and for the development and application of antiarrhythmic drugs. If TTX-sensitive Na+ channels exert general effects in the mammalian myocardium, one should expect a) high levels of TTX-sensitive Na+ channel transcripts not only in the mouse, but also in the human heart, b) clinical symptoms and ECG alterations in patients with mutations in genes encoding TTX-sensitive isoforms, c) diminished cardiac conduction and output in animals intoxicated with low, i.e. sub-lethal doses of TTX, and d) impaired cardiac performance in accidentally TTX-intoxicated humans. If this logic is followed, there are four key arguments against any physiological relevance of TTX-sensitive Na+ channels in the non-diseased human myocardium:
Low expression of TTX-sensitive Na+ channels in the heart of higher mammals
Quantitative RT-PCR analysis revealed only minute quantities of transcripts for TTX-sensitive Na+ channels in the whole human heart, when compared to the mouse, rat and dog heart (Figure 6).58 Interestingly, relative transcript levels of TTX-sensitive channels, Nav1.1 to Nav1.4, decreased with increasing heart size. Human and pig hearts were nearly indistinguishable whereas large differences existed between human and mouse. It can be suggested that the mouse heart requires TTX-sensitive Na+ channels in various heart regions to ensure high heart rate and fast conduction.
Figure 6.

Na+ channel transcript patterns in different adult mammalian hearts. Relative transcript levels were obtained by real-time RT-PCR. Relative transcript levels of TTX-sensitive channels decreased with increasing heart size (30% for mouse, 8% for rat, and 4% for pig/human). Furthermore, alternative splicing of Nav1.5 RNA occurred also in a species-dependent manner. For further details see58.
Mutations in neuronal Na+ channels are not associated with cardiac arrhythmia
There are numerous mutations in TTX-sensitive Na+ channel genes related to epilepsy or muscle diseases. None of those channelopathies has been conclusively linked to cardiac dysfunction.59–61 Suspicious ECG alterations are not accompanying clinical features when brain-type or muscle Na+ channels are mutated. Sudden unexpected death in epilepsy (SUDEP) is debated to result from a malfunction of mutated neuronal Na+ channels in the heart. However, the mechanisms underlying SUDEP are still poorly understood. Abnormal cardiac excitation and repolarization phenomena have been observed in epilepsy patients, like QT dispersion, sinus tachycardia, T-wave alternans, bradyarrhythmia or asystole.62 Cardiac dysfunction is most likely triggered by seizures, and causes of SUDEP include several other factors including respiratory failure and dysfunction of the autonomic nervous system.62 To the best of our knowledge, a correlative phenomenon of SUDEP is largely unknown in SCN4A channelopathies, like hypokalemic or hyperkalemic periodic paralysis. Paralytic crisis in patients, however, can be associated with dangerous changes in blood potassium concentrations, resulting in ECG alterations (U wave) and higher susceptibility to ventricular arrhythmia known as torsades de pointes.
No cardiac effects in animals intoxicated with sub-lethal TTX dosages
Studies on the systemic effects of TTX in animals were already published at the end of the 19th century.63 Cardiovascular effects of TTX were intensively studied in the 1960th and 1970th, after the toxin was commercially available as a highly purified powder. The authors provided overwhelming evidence that the heart belongs to the few organs that remain nearly unaffected, even at large sub-lethal or lethal TTX doses that are known to block TTX-sensitive Na+ channels.63 Intoxication often required artificial ventilation, but the heart continued beating regularly. The observed blood pressure reduction in intoxicated animals was sufficiently explained by the reduced vasomotor tone (Figure 7). Heart rate and cardiac output remained unchanged in most studies. In a few cases, bradycardia and reduced cardiac output were noticed, phenomena that were explained by more complex systemic reactions to TTX intoxication, as blockade of nerve fibers resulting in a diminished pressoreflex, a reduced venous return, a block of sympathetic nerve fibers and a slight depressive effect on medullary neurons (Figure 7). Particularly noteworthy is the fact, that compression of the abdominal aorta as well as volume expansion normalized arterial blood pressure and cardiac performance instantaneously. Conduction disturbances were also rarely seen and occurred at high lethal TTX dosages that most likely affected a larger portion of cardiac Nav1.5 channels.63
Figure 7.

Effects of TTX on the cardiovascular system. TTX blocks Na+ channels of the skeletal muscle, the diaphragm, afferent and efferent nerve fibers including the phrenic nerve and the sympathetic nerve system. This leads to muscular incoordination, respiratory dysfunction, a marked decrease in the total peripheral resistance (TPR) and a drop in blood pressure. The toxin also activates the medullary chemoreceptor trigger zone which initiates vomiting. Victims often remain conscious, but cannot speak, move or breathe, suggesting that the brain is largely protected by the blood-brain barrier. Death may occur in as little as 17 minutes after ingesting the toxin (human oral killing dose: 1 to 2 mg). Treatment may involve mechanical ventilation, normal saline infusion, gastric emptying procedures, application of activated charcoal, atropinization, and treatment with dopamine. Prognosis is good if the patient arrives at the emergency department conscious and prior to respiratory arrest. For details see63.
No cardiac effects in cases of tetrodotoxication
Pufferfish poisoning or tetrodotoxication is one of the most common food poisonings along the coast of Asia. A literature screening for reports on outbreaks and cases of tetrodotoxication between 1983 and 2009, including more than 500 patients admitted to an emergency department or an intensive care unit, revealed that cardiac excitation was not significantly impaired in intoxicated victims, as long as sufficient oxygen was provided.63 Patients suffered from severe neurological and neuromuscular symptoms, strongly suggesting that a significant portion of functional TTX-sensitive Na+ channels was blocked (Figure 7). In particular third-degree and fourth-degree intoxicated victims had blood TTX levels above the reported IC50 for TTX-sensitive Na+ channels (Figure 8). At the same time however, cardiovascular manifestations, like hypotension and sinus bradycardia, cannot be considered as general accompanying symptoms, even not in severely intoxicated and mechanically ventilated patients. Except for one victim, who experienced cardiac arrest before treatment by an emergency physician, ECG abnormalities, indicative of cardiac arrhythmias were not reported. Thus, important clinical data are in close agreement with results from animal experiments and suggest that the human heart does not express a physiologically relevant number of TTX-sensitive Na+ channels.63
Figure 8.

Correlation between TTX blood levels and the degree of intoxication. Each data point corresponds to one case of tetrodotoxication. Please note that only in a few cases, blood TTX levels were determined. The figure shows that TTX concentrations fairly correlated with the severity of the patients' symptoms. The clinical grading system for TTX poisoning was introduced by Fukuda and Tani.82 First-degree and second-degree cases are relatively mild cases; blood TTX levels were similar to or lower than the IC50 of TTX-sensitive Na+ channels. The third degree is characterized by more severe disturbances, like ataxia, widespread paralysis, pronounced hyporeflexia, drop in blood pressure, fixed/dilated pupils, cyanosis, and respiratory failure (e.g., dyspnoea, decreased vital capacity or lower forced expiratory volume). Hypothermia also develops, when skeletal muscle contractions and conduction in nerve fibers are gradually blocked. Fourth-degree cases are severely intoxicated victims presenting with cessation of respiration, decreased arterial O2, unconsciousness, bradycardia, and hypotension. TTX concentrations were between 40 and 164 nM. These concentrations would be indeed high enough to block the vast majority of TTX-sensitive Na+ channels in the body. Effects on the heart were rarely seen. Bradycardia was observed in few victims who had a pre-existing chronic disease (diabetes mellitus), and cardiac arrhythmia occurred secondary to hypoxia. Adapted from63.
In conclusion, TTX-sensitive Na+ channels can be detected in the mammalian myocardium by modern electrophysiological and biomolecular techniques. However, there are plausible arguments that these Na+ channel isoforms are not involved in cardiac excitation in higher mammals.
Expression and function of the TTX-resistant isoform Nav1.8 in the heart
Recent genome-wide association studies (GWAS) identified SCN10A, the gene encoding Nav1.8, as a determinant of cardiac conduction parameters, like PR and QRS interval on surface ECG.64–67 These data were very surprising because this channel was thought to be specifically expressed in small- and medium-diameter nociceptive sensory neurons of the dorsal root ganglia (DRG). Previous Northern blot and RT-PCR analysis failed to show even traces of Nav1.8 transcripts in the heart,68,69 and biochemical studies did not provide any evidence pointing to the existence of a fraction of channels with a TTX resistance 100-fold higher than that of Nav1.5. Moreover, Scn10a − / − mice showed only mild cardiac ECG abnormalities.64
During the last four years, numerous studies on the expression and function of SCN10A/Nav1.8 were published, but a uniform picture cannot be reconstructed. In particular, conflicting data were obtained regarding the cell-specificity of expression, and whether the essential function of SCN10A occurs via regulating SCN5A expression or via the electrophysiological properties of the gene product Nav1.8. There are currently some interesting working hypotheses on the function of SCN10A/Nav1.8 in the mammalian heart:
Nav1.8 is specifically expressed in intracardiac neurons
Some authors have shown that Nav1.8 is specifically expressed in murine, canine and human cardiac neurons,70–72 suggesting a function of the SCN10A gene product for cardiac conduction via regulating action potential firing in intracardiac neurons.71 Specific localization in neurons can explain why Nav1.8 expression levels are generally very low in the whole heart, which was consistently observed in various species. Expression of Nav1.8 in cardiac neurons was demonstrated in immunostaining experiments using a specific antibody against Nav1.8.71 Moreover, the Nav1.8 blocker A-803467 reduced Na+ current density and action potential firing frequency in freshly isolated murine intracardiac neurons.71 In human heart atrial appendage tissue, Nav1.8 immunoreactivity was also observed in both cardiac nerve fibres and cardiomyocytes.70 In contrast to this result, however, Nav1.8 could not be detected in mouse atrial or ventricular cardiomyocytes.71,73
SCN10A regulates expression from the SCN5A gene via enhancer/promoter interactions
Recent evidence indicates that SCN10A genetic variants alter cardiac conduction parameters by controlling SCN5A transcript levels.73 The SCN10A gene is located immediately adjacent to the SCN5A gene on chromosome 3p22.2. Genetic regulation of SCN10A has been shown to be mediated by an intronic enhancer located in SCN10A that interacts with the SCN5A promoter. This interaction seems to be crucial for the cellular level of functional Nav1.5 channels, a key determinant for cardiac conduction. Consequently, SCN10A exerts its cardiac effects via controlling transcription from SCN5A, rather than via its gene product Nav1.8. Interestingly, this novel mechanism takes into account that Nav1.5 levels determine upstroke velocity, a major determinant of conduction velocity, and that loss-of-function in Nav1.5 often results in severe conduction disturbances. Notably, these results are also in good agreement with the undetectable Nav1.8 transcript and protein levels in cardiomyocytes,69,71,73 and they do not argue against functional Nav1.8 channels in intracardiac neurons or in the His-Purkinje system.
Nav1.8 is functionally expressed in cardiomyocytes
Data strongly suggesting functional expression of Nav1.8 in cardiomyocytes have also been published. An immunohistochemical study indicated Nav1.8 localization in human cardiomyocytes isolated from the atrial appendage.70 Another study demonstrated that the SCN10A transcript and product is preferentially expressed in the mouse His-Purkinje system.67 In mouse and rabbit ventricular cardiomyocytes, application of the specific Nav1.8 blocker A-803467 reduced the persistent or “late” Na+ current fraction and shortened action potential duration, without affecting the peak current amplitude.41 Interestingly, these effects were only seen in a subset of mouse ventricular cardiomyocytes, suggesting cell-specific expression of Nav1.8 in the heart. More recently, several mutations in SCN10A were identified in BrS patients.74 The authors showed that SCN10A mutations account for 16.7 % of the BrS cases, suggesting that SCN10A is one of the major susceptibility genes for BrS. Consequently, SCN10A affects cardiac excitation not only via regulatory enhancer signals, but also via its gene product Nav1.8. The authors suggested that the clinical phenotype may result from a direct interaction of mutated Nav1.8 channels with wild-type Nav1.5, thereby reducing the number of functional channels in the plasma membrane.74 This hypothesis was established from co-expression experiments in HEK cells: Wild-type Nav1.8/Nav1.5 channels increased the whole-cell current compared to Nav1.5 alone, whereas co-expression of dysfunctional Nav1.8 channel variants led to a significant reduction of the Nav1.5-mediated current. It remains to be elucidated whether the proposed protein-protein interaction indeed occurs in vivo. Neither up-regulation of Nav1.5 by wild-type Nav1.8, nor Nav1.5 down-regulation by mutant Nav1.8 in cardiomyocytes or in intracardiac neurons has been demonstrated yet. In murine Scn10a − / − cardiomyocytes, changes in peak current amplitude were not reported.41
In conclusion, it seems that the discovery of SCN10A/Nav1.8 as a functionally and clinically relevant player in the heart is one of the most important milestones towards the understanding of cardiac excitability, after establishing the role of Nav1.5 in inherited cardiac diseases in the mid-90s. Despite all the fascinating efforts, however, the actual role of SCN10A/Nav1.8 in the heart and the mechanisms by which this gene and/or its gene product affects cardiac function remains at least partially obscure. For example, the Nav1.8 blocker A-803467 shortened action potentials in cardiomyocytes, and PR intervals were shorter in Scn10a − / − mice, suggesting that Nav1.8 acts to lengthen cardiac conduction.41,64 However, in mice treated with A-803467, QRS and PR were longer, and mutations in SCN10A were associated with BrS, suggesting acceleration of impulse propagation by Nav1.8.67,74 It is possible that numerous factors have to be considered in future experiments to solve this fascinating puzzle, like a possible alternative splicing in the heart, a species-dependent regulation of expression, the turn-over of the protein, a possible inducible expression under certain conditions, the cell-specificity of expression in different cardiac cells of higher mammals, essential interactions of Nav1.8 with other cardiac proteins, or the role of SCN10A/Nav1.8 in the diseased myocardium using animal models.
Conclusions
Nav1.5 is the most prominent and most important cardiac Na+ channel. It determines important features of cardiac excitability. More recently, SCN10A/Nav1.8 was linked in genome-wide association studies with abnormal cardiac conduction parameters. It seems that SCN10A/Nav1.8 is of great clinical importance despite its low expression in the mammalian heart. The TTX-sensitive Na+ channels are most likely not functionally expressed in the heart of higher mammals, and there are no indications for a clinically relevant cardiac expression of neuronal Nav1.9.
Acknowledgements
The authors would like to thank Karin Schoknecht for excellent technical assistance and Prof. Dr. Klaus Benndorf for critical reading the manuscript and for helpful comments.
References
- 1.Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 2.Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12–23. doi: 10.1093/cvr/cvr252. [DOI] [PubMed] [Google Scholar]
- 3.Patton DE, Isom LL, Catterall WA, Goldin AL. The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J Biol Chem. 1994;269:17649–17655. [PubMed] [Google Scholar]
- 4.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. Beta 3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci U S A. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmer T, Biskup C, Bollensdorff C, Benndorf K. The beta1 subunit but not the beta2 subunit colocalizes with the human heart Na+ channel (hH1) already within the endoplasmic reticulum. J Membr Biol. 2002;186:13–21. doi: 10.1007/s00232-001-0131-0. [DOI] [PubMed] [Google Scholar]
- 6.Zimmer T, Benndorf K. The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta1 subunit. J Gen Physiol. 2002;120:887–895. doi: 10.1085/jgp.20028703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheets MF, Hanck DA. Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J Gen Physiol. 1995;106:617–640. doi: 10.1085/jgp.106.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 10.Shah VN, Wingo TL, Weiss KL, Williams CK, Balser JR, Chazin WJ. Calcium-dependent regulation of the voltage-gated sodium channel hH1: Intrinsic and extrinsic sensors use a common molecular switch. Proc Natl Acad Sci U S A. 2006;103:3592–3597. doi: 10.1073/pnas.0507397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potet F, Chagot B, Anghelescu M, Viswanathan PC, Stepanovic SZ, Kupershmidt S, Chazin WJ, Balser JR. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009;284:8846–8854. doi: 10.1074/jbc.M806871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cormier JW, Rivolta I, Tateyama M, Yang AS, Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- 13.Chagot B, Potet F, Balser JR, Chazin WJ. Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J Biol Chem. 2009;284:6436–6445. doi: 10.1074/jbc.M807747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas S, DiSilvestre D, Tian Y, Halperin VL, Tomaselli GF. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ Res. 2009;104:870–878. doi: 10.1161/CIRCRESAHA.108.193565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmer T, Biskup C, Dugarmaa S, Vogel F, Steinbis M, Bohle T, Wu YS, Dumaine R, Benndorf K. Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J Membr Biol. 2002;186:1–12. doi: 10.1007/s00232-001-0130-1. [DOI] [PubMed] [Google Scholar]
- 16.Zimmer T, Bollensdorff C, Haufe V, Birch-Hirschfeld E, Benndorf K. Mouse heart Na+ channels: Primary structure and function of two isoforms and alternatively spliced variants. Am J Physiol Heart Circ Physiol. 2002;282:H1007–H1017. doi: 10.1152/ajpheart.00644.2001. [DOI] [PubMed] [Google Scholar]
- 17.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, Takahashi H, Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320:188–192. doi: 10.1038/320188a0. [DOI] [PubMed] [Google Scholar]
- 19.Kayano T, Noda M, Flockerzi V, Takahashi H, Numa S. Primary structure of rat brain sodium channel III deduced from the cDNA sequence. FEBS Lett. 1988;228:187–194. doi: 10.1016/0014-5793(88)80614-8. [DOI] [PubMed] [Google Scholar]
- 20.Trimmer JS, Cooperman SS, Tomiko SA, Zhou JY, Crean SM, Boyle MB, Kallen RG, Sheng ZH, Barchi RL, Sigworth FJ, Goodman RH, Agnew WS, Mandel G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- 21.George AL, Jr., Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann Neurol. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- 22.Rogart RB, Cribbs LL, Muglia LK, Kephart DD, Kaiser MW. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci U S A. 1989;86:8170–8174. doi: 10.1073/pnas.86.20.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stodberg T, Hennings JC, Bergmann M, Altmuller J, Thiele H, Wetzel A, Nurnberg P, Timmerman V, De Jonghe P, Blum R, Schaible HG, Weis J, Heinemann SH, Hubner CA, Kurth I. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45:1399–1404. doi: 10.1038/ng.2767. [DOI] [PubMed] [Google Scholar]
- 25.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 26.Noda M, Hiyama TY. The Nax channel: What it is and what it does. The Neuroscientist. 2014 pii: 1073858414541009. [Google Scholar]
- 27.Fozzard HA, Lipkind GM. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Marine Drugs. 2010;8:219–234. doi: 10.3390/md8020219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen R, Chung SH. Mechanism of tetrodotoxin block and resistance in sodium channels. Biochem Biophys Res Commun. 2014;446:370–374. doi: 10.1016/j.bbrc.2014.02.115. [DOI] [PubMed] [Google Scholar]
- 29.Satin J, Kyle JW, Chen M, Bell P, Cribbs LL, Fozzard HA, Rogart RB. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- 30.Dudel J, Peper K, Rudel R, Trautwein W. Effect of tetrodotoxin on membrane currents in mammalian cardiac fibres. Nature. 1967;213:296–297. doi: 10.1038/213296a0. [DOI] [PubMed] [Google Scholar]
- 31.Renaud JF, Kazazoglou T, Lombet A, Chicheportiche R, Jaimovich E, Romey G, Lazdunski M. The Na+ channel in mammalian cardiac cells: Two kinds of tetrodotoxin receptors in rat heart membranes. J Biol Chem. 1983;258:8799–8805. [PubMed] [Google Scholar]
- 32.Lombet A, Renaud JF, Chicheportiche R, Lazdunski M. A cardiac tetrodotoxin binding component: Biochemical identification, characterization, and properties. Biochemistry. 1981;20:1279–1285. doi: 10.1021/bi00508a036. [DOI] [PubMed] [Google Scholar]
- 33.Rogart RB, Regan LJ, Dziekan LC, Galper JB. Identification of two sodium channel subtypes in chick heart and brain. Proc Natl Acad Sci U S A. 1983;80:1106–1110. doi: 10.1073/pnas.80.4.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lombet A, Frelin C, Renaud JF, Lazdunski M. Na+ channels with binding sites of high and low affinity for tetrodotoxin in different excitable and non-excitable cells. Eur J Biochem. 1982;124:199–203. doi: 10.1111/j.1432-1033.1982.tb05925.x. [DOI] [PubMed] [Google Scholar]
- 35.Benndorf K, Boldt W, Nilius B. Sodium current in single myocardial mouse cells. Pflugers Arch. 1985;404:190–196. doi: 10.1007/BF00585418. [DOI] [PubMed] [Google Scholar]
- 36.Follmer CH, ten Eick RE, Yeh JZ. Sodium current kinetics in cat atrial myocytes. J Physiol. 1987;384:169–197. doi: 10.1113/jphysiol.1987.sp016449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol. 2005;564:683–696. doi: 10.1113/jphysiol.2004.079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satin J, Kyle JW, Chen M, Rogart RB, Fozzard HA. The cloned cardiac Na channel alpha-subunit expressed in Xenopus oocytes show gating and blocking properties of native channels. J Membr Biol. 1992;130:11–22. doi: 10.1007/BF00233735. [DOI] [PubMed] [Google Scholar]
- 39.Gellens ME, George AL, Jr., Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nuss HB, Chiamvimonvat N, Perez Garcia MT, Tomaselli GF, Marban E. Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu 1) sodium channel alpha subunits expressed in Xenopus oocytes. J Gen Physiol. 1995;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, Dumaine R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–127. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Yoo S, Dobrzynski H, Fedorov VV, Xu SZ, Yamanushi TT, Jones SA, Yamamoto M, Nikolski VP, Efimov IR, Boyett MR. Localization of Na+ channel isoforms at the atrioventricular junction and atrioventricular node in the rat. Circulation. 2006;114:1360–1371. doi: 10.1161/CIRCULATIONAHA.106.613182. [DOI] [PubMed] [Google Scholar]
- 44.Lei M, Jones SA, Liu J, Lancaster MK, Fung SS, Dobrzynski H, Camelliti P, Maier SK, Noble D, Boyett MR. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J Physiol. 2004;559:835–848. doi: 10.1113/jphysiol.2004.068643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation. 1996;94:3083–3086. doi: 10.1161/01.cir.94.12.3083. [DOI] [PubMed] [Google Scholar]
- 46.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmer T, Surber R. SCN5A channelopathies – an update on mutations and mechanisms. Progress in Biophysics and Molecular Biology. 2008;98:120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Shang LL, Gao G, Dudley SC., Jr The tail of the cardiac sodium channel. Channels. 2008;2:161–162. doi: 10.4161/chan.2.3.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wahbi K, Algalarrondo V, Becane HM, Fressart V, Beldjord C, Azibi K, Lazarus A, Berber N, Radvanyi-Hoffman H, Stojkovic T, Behin A, Laforet P, Eymard B, Hatem S, Duboc D. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Archives of Cardiovascular Diseases. 2013;106:635–643. doi: 10.1016/j.acvd.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Schroeter A, Walzik S, Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J Mol Cell Cardiol. 2010;49:16–24. doi: 10.1016/j.yjmcc.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Haufe V, Chamberland C, Dumaine R. The promiscuous nature of the cardiac sodium current. J Mol Cell Cardiol. 2007;42:469–477. doi: 10.1016/j.yjmcc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 52.Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–1310. doi: 10.1161/01.cir.103.9.1303. [DOI] [PubMed] [Google Scholar]
- 54.Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol. 1979;236:H561–H567. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- 55.Maier SKG, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109:1421–1427. doi: 10.1161/01.CIR.0000121421.61896.24. [DOI] [PubMed] [Google Scholar]
- 57.Maier SK, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, Catterall WA, Scheuer T. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci U S A. 2003;100:3507–3512. doi: 10.1073/pnas.2627986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Progress in Biophysics and Molecular Biology. 2008;98:309–318. doi: 10.1016/j.pbiomolbio.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 59.Kullmann DM, Hanna MG. Neurological disorders caused by inherited ion-channel mutations. Lancet Neurology. 2002;1:157–166. doi: 10.1016/s1474-4422(02)00071-6. [DOI] [PubMed] [Google Scholar]
- 60.Bernard G, Shevell MI. Channelopathies: A review. Pediatric Neurology. 2008;38:73–85. doi: 10.1016/j.pediatrneurol.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Shi X, Yasumoto S, Kurahashi H, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Clinical spectrum of SCN2A mutations. Brain & Development. 2012;34:541–545. doi: 10.1016/j.braindev.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 62.Surges R, Sander JW. Sudden unexpected death in epilepsy: Mechanisms, prevalence, and prevention. Current Opinion in Neurology. 2012;25:201–207. doi: 10.1097/WCO.0b013e3283506714. [DOI] [PubMed] [Google Scholar]
- 63.Zimmer T. Effects of tetrodotoxin on the mammalian cardiovascular system. Marine Drugs. 2010;8:741–762. doi: 10.3390/md8030741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El-Hamamsy I, Wass MN, Dekker LR, de Jong JS, Sternberg MJ, McKenna W, Severs NJ, de Silva R, Wilde AA, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 65.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Lochen ML, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 66.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton-Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller-Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, Nolte IM, van der Harst P, Muller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Kottgen A, Johnson T, Rice K, Sie MP, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo Leach I, Estrada K, Volker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, de Boer RA, Uitterlinden AG, Volzke H, Spector TD, Liu FY, Boerwinkle E, Dominiczak AF, Rotter JI, van Herpen G, Levy D, Wichmann HE, van Gilst WH, Witteman JC, Kroemer HK, Kao WH, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, van Veldhuisen DJ, Schwienbacher C, O'Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RS, te Meerman G, Groen HJ, Weersma RK, van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, van Duijn CM, Felix SB, Fishman GI, Jamshidi Y, Stricker BH, Samani NJ, Kaab S, Arking DE. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rabert DK, Koch BD, Ilnicka M, Obernolte RA, Naylor SL, Herman RC, Eglen RM, Hunter JC, Sangameswaran L. A tetrodotoxin-resistant voltage-gated sodium channel from human dorsal root ganglia, hPN3/SCN10A. Pain. 1998;78:107–114. doi: 10.1016/S0304-3959(98)00120-1. [DOI] [PubMed] [Google Scholar]
- 69.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 70.Facer P, Punjabi PP, Abrari A, Kaba RA, Severs NJ, Chambers J, Kooner JS, Anand P. Localisation of SCN10A gene product Na(v)1.8 and novel pain-related ion channels in human heart. International Heart Journal. 2011;52:146–152. doi: 10.1536/ihj.52.146. [DOI] [PubMed] [Google Scholar]
- 71.Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, Bezzina CR, Veldkamp MW. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111:333–343. doi: 10.1161/CIRCRESAHA.112.274035. [DOI] [PubMed] [Google Scholar]
- 72.Dun W, Boyden P. Does SCN10A gene product play a role in canine Purkinje cell electrophysiology? Heart Rhythm. 2012;9:S394. [Google Scholar]
- 73.van den Boogaard M, Smemo S, Burnicka-Turek O, Arnolds DE, van de Werken HJ, Klous P, McKean D, Muehlschlegel JD, Moosmann J, Toka O, Yang XH, Koopmann TT, Adriaens ME, Bezzina CR, de Laat W, Seidman C, Seidman JG, Christoffels VM, Nobrega MA, Barnett P, Moskowitz IP. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124:1844–1852. doi: 10.1172/JCI73140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu D, Barajas-Martinez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM, Rajamani S, De Antonio HJ, Myerburg RJ, Ito H, Deshmukh P, Marieb M, Nam GB, Bhatia A, Hasdemir C, Haissaguerre M, Veltmann C, Schimpf R, Borggrefe M, Viskin S, Antzelevitch C. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. Journal of the American College of Cardiology. 2014;64:66–79. doi: 10.1016/j.jacc.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baruscotti M, Di Francesco D, Robinson RB. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. J Physiol. 1996;492(Pt 1):21–30. doi: 10.1113/jphysiol.1996.sp021285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baruscotti M, Westenbroek R, Catterall WA, Di Francesco D, Robinson RB. The newborn rabbit sino-atrial node expresses a neuronal type I-like Na+ channel. J Physiol. 1997;498:641–648. doi: 10.1113/jphysiol.1997.sp021889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikmaram MR, Liu J, Abdelrahman M, Dobrzynski H, Boyett MR, Lei M. Characterization of the effects of ryanodine, TTX, E-4031 and 4-AP on the sinoatrial and atrioventricular nodes. Progress in Biophysics and Molecular Biology. 2008;96:452–464. doi: 10.1016/j.pbiomolbio.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Qu Y, Karnabi E, Chahine M, Vassalle M, Boutjdir M. Expression of skeletal muscle Na(V)1.4 Na channel isoform in canine cardiac Purkinje myocytes. Biochem Biophys Res Commun. 2007;355:28–33. doi: 10.1016/j.bbrc.2007.01.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verkerk AO, van Ginneken AC, van Veen TA, Tan HL. Effects of heart failure on brain-type Na+ channels in rabbit ventricular myocytes. Europace: European Pacing, Arrhythmias, and Cardiac Electrophysiology: Journal of the Working Groups on Cardiac Pacing, Arrhythmias, and Cardiac Cellular Electrophysiology of the European Society of Cardiology. 2007;9:571–577. doi: 10.1093/europace/eum121. [DOI] [PubMed] [Google Scholar]
- 80.Huang B, El Sherif T, Gidh Jain M, Qin D, El Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol. 2001;12:218–225. doi: 10.1046/j.1540-8167.2001.00218.x. [DOI] [PubMed] [Google Scholar]
- 81.Camacho JA, Hensellek S, Rougier JS, Blechschmidt S, Abriel H, Benndorf K, Zimmer T. Modulation of Nav1.5 channel function by an alternatively spliced sequence in the DII/DIII linker region. J Biol Chem. 2006;281:9498–9506. doi: 10.1074/jbc.M509716200. [DOI] [PubMed] [Google Scholar]
- 82.Fukuda A, Tani A. Records of puffer poisonings. Report 3. Nippon Igaku Oyobi Kenko Hoken. 1941;3528:7–13. [Google Scholar]