

Abstract
The Ras–extracellular signal-regulated kinase 1 and 2 (ERK1/2) pathway appears to be important for the development, maintenance, aging, and pathology of mammalian skeletal muscle. Yet no gene targeting of Erk1/2 in muscle fibers in vivo has been reported to date. We combined a germ line Erk1 mutation with Cre-loxP Erk2 inactivation in skeletal muscle to produce, for the first time, mice lacking ERK1/2 selectively in skeletal myofibers. Animals lacking muscle ERK1/2 displayed stunted postnatal growth, muscle weakness, and a shorter life span. Their muscles examined in this study, sternomastoid and tibialis anterior, displayed fragmented neuromuscular synapses and a mixture of modest fiber atrophy and loss but failed to show major changes in fiber type composition or absence of cell surface dystrophin. Whereas the lack of only ERK1 had no effects on the phenotypes studied, the lack of myofiber ERK2 explained synaptic fragmentation in the sternomastoid but not the tibialis anterior and a decrease in the expression of the acetylcholine receptor (AChR) epsilon subunit gene mRNA in both muscles. A reduction in AChR protein was documented in line with the above mRNA results. Evidence of partial denervation was found in the sternomastoid but not the tibialis anterior. Thus, myofiber ERK1/2 are differentially required for the maintenance of myofibers and neuromuscular synapses in adult mice.
INTRODUCTION
Mitogen-activated protein kinases (MAPKs) are components of intracellular signaling modules that control a myriad of cellular processes. MAPK modules consist of 3 core protein kinase components. The most downstream is the actual MAPK, an S/T kinase that phosphorylates the transcription factors, cytoskeletal elements, or other kinases that are the targets of regulation by signaling cascades started at the cell surface. A MAPK is activated by an upstream MAPK kinase (MAP2K), which, in turn, is activated by a MAP2K kinase (MAP3K). MAP3Ks are usually at the receiving end of signals derived from small, monomeric GTPases such as the Ras family or by other more intricate mechanisms (1). In mammalian cells, the prototypical MAPK module is composed of the MAPKs extracellular signal-regulated kinases 1 and 2 (ERK1/2), the MAP2Ks MEK1/2, and the MAP3K Raf. ERK1/2 regulate normal cellular responses to multiple growth factors and cytokines in proliferation, differentiation, and apoptosis (2, 3).
Multiple studies suggest an important role for the Ras-ERK1/2 pathway in the development, normal maintenance, aging, and pathology of mammalian skeletal muscle. Thus, ERK1/2 activity has both stimulatory and inhibitory roles in the differentiation of cultured skeletal myotubes that vary with the stage of this protracted process (4–8). ERK1/2 have been implicated in the maintenance of adult skeletal muscle mass (9) and, seemingly paradoxically, in the control of both the fast-twitch (10) and the slow-twitch (11) fiber type phenotypes. Alterations in levels of ERK1/2 activity in aging rodent muscle correlate with sarcopenia (12), the loss of muscle mass and strength that occurs with aging (13). Ras-ERK1/2 pathway activity dysregulation underlies the pathology of neuromuscular diseases such as autosomal Emery-Dreifuss muscular dystrophy (14) and of the RASopathies, a group of rare genetic diseases with accompanying skeletal muscle abnormalities (15–17). Our own work with cultured myotubes (18) suggests a modulatory role for ERK1/2 on the activity of agrin (19), a key synaptogenic factor in the formation and maintenance of the neuromuscular junction (NMJ), the synapse between a motoneuron and a skeletal muscle fiber (20). In vitro and in vivo studies implicated ERK1/2 in the control of synapse-specific expression of acetylcholine receptor (AChR) subunit genes at the NMJ, particularly of Chrne, the gene coding the adult AChRε subunit (21–23).
Most of the experiments that have been carried out to characterize the role of ERK1/2 in skeletal muscle biology have been done with cultured cells, using either pharmacological tools, in particular MEK inhibitors, small interfering RNA (siRNA), and constitutively active or dominant negative constructs for the different components of the Ras-ERK1/2 pathway. However, no gene targeting investigations on the role of ERK1/2 in developing muscle fibers in vivo have been reported to date. We combined a germ line Erk1 mutant with Cre-loxP inactivation of Erk2 in skeletal muscle to produce, for the first time, mice lacking ERK1/2 selectively in skeletal myofibers. We report that ERK1/2 are required for the maintenance of myofibers and NMJs in adult animals.
MATERIALS AND METHODS
Ethics statement.
Care and treatment of all animals followed the National Research Council's Guide for the Care and Use of Laboratory Animals (24) and were approved by the Institutional Animal Care and Use Committee of Texas A&M University under animal use protocol 2012-168.
Mice and genotyping.
The Cre driver mice in which Cre is under the control of the human α-skeletal muscle actin promoter are represented as Hsa-Cre+/−. The Erk2 floxed allele is represented as Erk2f. Local colonies were established from breeders obtained initially as follows: Hsa-Cre+/− mice from The Jackson Laboratory (JAX stock number 006149) and Erk1+/; Erk2f/+ and Erk2f/f mice from The Landreth Lab, Case Western Reserve University. These crosses were used to generate experimental animals as follows. Erk1−/− mice came from Erk1+/− × Erk1−/−. Mice deficient in muscle ERK2 (referred to below as mErk2CKO) came from Hsa-Cre+/−; Erk2f/+ × Hsa-Cre−/−; Erk2f/f. Mice deficient in germ line ERK1 and muscle ERK2 (referred to below as DKO) came from Hsa-Cre+/−; Erk1+/−; Erk2f/f × Hsa-Cre−/−; Erk1−/−; Erk2f/f. Genotyping was done by PCR with the following primers: for Cre, 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and 5′-GTGAAACAGCATTGCTGTCACTT-3′; for Erk1 (detection of the wild type and null allele), 5′-GTATCTTGGGTTCCCCATCC-3′, 5′-GGGGAACTTCCTGACTAGGG-3′, and 5′-GCTCCATGTCGAAGGTGAAT-3′; and Erk2 (detection of the wild type and floxed allele), 5′-AGCCAACAATCCCAAACCTG-3′ and 5′-GGCTGCAACCATCTCACAAT-3′. Mice were housed at 25°C with a 12-h light/dark cycle, fed ad libitum, and monitored daily.
Western blotting.
Muscles were dissected, snapped frozen in liquid N2, and stored at −80°C until use. Most tissue homogenates were prepared in the following buffer: 25 mM Tris (pH 7.4), 95 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% SDS, 10% protease inhibitor cocktail (P8340; Sigma), 5 mM NaF, 2 mM Na3VO4, and 2.5 mM Na4P2O7. Lysates used to analyze p38 were made in 1% Triton X-100, 30 mM triethanolamine (pH 7.5), 50 mM NaCl, 5 mM EGTA, and 5 mM EDTA, with the same phosphatase and protein inhibitors as mentioned above. Total protein was measured using Bio-Rad protein assay, and 50 μg per sample was run on 10% acrylamide denaturing gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes using a semidry blotter (Bio-Rad). Suppliers of primary antibodies and dilutions were Epitomics for anti-ERK1 (1171-1, 1:1,000) and anti-ERK2 (1586-1, 1:1,000); Cell Signaling for anti-tERK1/2 (9102; 1:1,000), anti-p38 (9212, 1:1,000), anti-pp38 (4511, 1:1,000), and anti-c-jun NH2-terminal kinase (anti-JNK) (9252, 1:500); Santa Cruz Biotechnology for anti-pERK1/2 (SC7383, 1:200) and anti-pJNK (SC6254, 1:200); and Sigma for anti-α-tubulin (T6199, 1:4,000). Horseradish peroxidase-conjugated secondary antibodies (SC2020 and SC2031 from Santa Cruz Biotechnology and 111-035-003 and 315-035-003 from Jackson ImmunoResearch) were used at 1:1,000 to 1:3,000. Blots were visualized by chemiluminescence by following the manufacturer's instructions (PerkinElmer). Images were acquired and analyzed with an AlphaImager gel imaging system (Protein Simple).
AChR affinity isolation and probing.
AChRs were isolated and detected as previously described, with minor modifications (25). Briefly, muscles were lysed in 1% Triton X-100, 30 mM triethanolamine (pH 7.5), 50 mM NaCl, 5 mM EGTA, 5 mM EDTA, 10% protease inhibitor cocktail (P8340, Sigma), 5 mM NaF, 2 mM Na3VO4, and 2.5 mM Na4P2O7. One milligram of total protein per muscle lysate was incubated for 30 min with 200 nM biotin–α-bungarotoxin (BBTX) (B-1196; Life Technologies) at 4°C. Streptavidin-agarose (SA100-04; Life Technologies) was used to precipitate the BBTX-AChR complexes. After washing with lysis buffer, complexes were separated by SDS-PAGE, transferred to PVDF membranes, and probed with a goat polyclonal antibody to AChRε at 1:250 (ab166931; Abcam). Bands were visualized by chemiluminescence and quantified as described above.
Grip strength assay.
Forelimb grip strength was assessed with a grip strength meter with a single sensor and a standard pull bar and software (Columbus Instruments) as previously described (26). Briefly, a mouse was directed to grip the bar with forelimbs and then pulled off the bar by the tail, and peak force (in grams) was recorded for 3 consecutive trials. Trial averages were normalized to body weight (in grams).
Rotarod.
A Rotarod series 8 (IITC Life Science Inc.) was used for rotarod analysis. From 7 to 19 weeks of age, mice were subjected to two rotarod modes every other week. First, we used a constant speed of 4 rpm for 5 min, immediately followed by 0.1-rpm/s acceleration to a maximum speed of 30 rpm, ending the trial at 5 min (26). Time to fall (in seconds) was recorded for both modes. Mice were allowed to rest for 10 min, and this procedure was repeated twice more for a total of 3 trials. Time to fall per mode was averaged for the 3 trials. Data for week 7 were considered adaptation and are not presented in the results.
Whole-mount staining, confocal microscopy, and NMJ morphological characterization.
Whole-mount staining of sternomastoid (STN) and tibialis anterior (TA) was essentially done as previously described (27). Antisynaptophysin (anti-SYN) (180130; Life Technologies) was used at 1:200, and fluorescein–α-bungarotoxin (BTX) (F-1176; Life Technologies) was used at 1:1,000. Rhodamine-conjugated rabbit secondary antibody (111-025-144; Jackson ImmunoResearch) was used at 1:400. At the dilution of anti-SYN used, both nerve terminal and preterminal axon were visible in many cases. Vectashield (Vector Laboratories)-mounted muscle bundles from these preparations were imaged on an A1 confocal microscope (Nikon), with 40× (numerical aperture [NA], 1.30) and 60× (NA, 1.40) oil immersion objectives. Images were acquired and maximal intensity projections were generated with NIS Elements software (Nikon). On the maximal projections, NMJ morphology was studied by counting (i) AChR domains per end plate; (ii) faint or weakly BTX stained NMJs, defined as those end plates with noticeably weaker or dimmer BTX staining relative to others on the same field; and (iii) terminal sprouts, defined as extensions of any length and direction of nerve terminal staining beyond AChR stain at a synaptic site.
Central myonucleus quantification.
One slide with several 12- to 14-μm-thick frozen cross sections from the belly of a STN was stained with hematoxylin and eosin (H&E) by a standard procedure. Images from 5 20× fields per slide were acquired with an EC3 camera and software (Leica), mounted on an Eclipse E1000 microscope (Nikon). Total fibers and fibers with central nuclei were counted. Replicates within a genotype were averaged for final quantification.
Real-time quantitative PCR.
Total RNA extraction, reverse transcription, and real-time PCR were done essentially as previously reported (27). Two hundred nanograms of total RNA per sample was used to generate cDNA. All TaqMan primer sets and probes were from ABI/Life Technologies as follows: Chrna, Mm00431629_m1; Chrnb, Mm00680412_m1; Chrnd, Mm00445545_m1; Chrne, Mm00437411_m1; Chrng, Mm00437419_m1; Runx1, Mm01213404_m1; Myh3, Mm01332463_m1; and 18S rRNA, 4333760F.
Myofiber morphological analysis.
Dystrophin staining of transverse frozen sections was performed essentially as previously described (27), except that sections were not fixed and individual primary antibodies to myosin heavy chains (MyHCs) were coincubated with the antidystrophin antibody (15277; Abcam). All MyHC antibodies were from the Developmental Studies Hybridoma Bank as follows: type 1, A4.840; type 2A, SC-71; type 2B, 10F5; and type 2X, 6H1. All but SC-71, which was sometimes used at 1:600, were used undiluted. Fluorophore-conjugated secondary antibodies that matched the primary isotype (Jackson ImmunoResearch and Sigma) were used at 1:400 and 1:128, respectively. Fiber area analysis was carried out as previously described (27) on overlapping 10× images that covered an entire muscle cross section. Care was taken not to measure the same fibers more than once. For fiber typing, 10× overlapping images of individual MyHC type and dystrophin staining were assembled in Photoshop (Adobe) to reconstruct an entire muscle cross section. Total fibers were counted from the dystrophin staining, and MyHC type fibers were counted from the respective antibody staining. Replicates per muscle/genotype were averaged for final quantification.
Statistical analysis.
Quantitative data are expressed as means ± standard errors of the means (SEM). Kaplan-Meier survival curves were generated and tested for statistical significance using the log rank test with Prism5 (GraphPad Software). Analysis of variance (ANOVA) was performed at http://vassarstats.net/. One- and two-sample Student t tests were computed with Prism5 (GraphPad Software) and Microsoft Excel (Microsoft Corporation), respectively. Wilcoxon rank sum test probabilities were computed at http://socr.stat.ucla.edu. Significance was set at a P value of <0.05.
RESULTS
Generation of skeletal muscle-selective Erk2 conditional mice and of mice lacking ERK1/2 in skeletal muscle fibers.
While germ line Erk1−/− mice are viable and fertile but display defective thymocyte maturation (28, 29), Erk2−/− mice are embryonic lethal due to failure to form the ectoplacental cone and extraembryonic ectoderm (30, 31). Erk2−/− embryos do not form mesoderm (32). A conditional Erk2 allele (Erk2f/f) was generated by Samuels and colleagues (33). To delete Erk2 selectively in developing and adult skeletal muscle fibers, Erk2f/f mice were crossed to mice that express Cre under the control of the human α-skeletal muscle actin promoter (Hsa-Cre+/−) (34, 35). Expression of Cre under the control of this promoter was detected beginning at embryonic day 9.5 onwards (34), and in skeletal muscle, it is restricted to myofibers. Neither myoblasts nor satellite cells express Cre in these mice (36). Hsa-Cre+/−; Erk2f/f mice (here referred to as mErk2CKO) were viable and fertile. Western blots of muscle extracts showed an ∼90% reduction in ERK2 levels in mErk2CKO mice (Fig. 1A). Full removal of ERK2 from whole muscle tissue was not expected, as myofiber nuclei represent only ∼41% of the total nuclei in muscle tissue (37). ERK1 levels in mErk2CKO mutants were ∼20% lower than in controls (Fig. 1B). As expected, ERK1 was completely absent in muscle homogenates from germ line Erk1−/− mice (Fig. 1C, top blot). Mice lacking both ERK1 and ERK2 in skeletal muscle fibers were generated by crossing Erk1−/− and mErk2CKO animals (for details, see Materials and Methods). Skeletal muscle from Hsa-Cre+/−; Erk1−/−; Erk2f/f mice (here referred to as DKO) lacked ERK1 and had a great reduction of ERK2, as expected (Fig. 1C, top blot). Phosphorylated ERK2 (pERK2) was diminished to similar extents in both mErk2CKO and DKO mutants, in direct correspondence to the decrease in total ERK2 (Fig. 1C, bottom blot). The reduction in ERK2 was specific to skeletal muscle, as it was not observed in heart, spinal cord, or liver (Fig. 1D). However, an ∼50% reduction in kidney ERK2 levels was detected in DKO animals relative to levels in the control (Fig. 1D).
FIG 1.

Mouse generation and characterization of their ERK1/2 expression. (A) Sternomastoid (STN) and tibialis anterior (TA) extracts from 14-week-old mice were probed simultaneously with antibodies to ERK2 and α-tubulin (α-Tub). Genotypes were as follows: control, Hsa-Cre−/−; Erk2f/f; Het, Hsa-Cre+/−; Erk2f/+; and mErk2CKO, Hsa-Cre+/−; Erk2f/f. Histograms show normalized quantification relative to that of the control. A 90% reduction in ERK2 levels in both muscles in mErk2CKO animals (n = 3) relative to that of the control (n = 2) was observed (**, P < 0.01). (B) Same extracts as in panel A were probed simultaneously with antibodies to ERK1 and α-Tub. Histograms show normalized quantification relative to that of the control. A 20% reduction in ERK1 in mErk2CKO muscle was observed (*, P < 0.05). (C) TA extracts from 14-week-old control, Erk1−/−, mErk2CKO, and DKO mice were first probed with antibodies to phosphorylated ERK1/2 (pERK1/2) (bottom blot). The blot was stripped and reprobed simultaneously with antibodies to total ERK1/2 (tERK1/2) and α-Tub (top blot). ERK1 is totally absent in Erk1−/− and DKO muscle, and reduced pERK2 in mErk2CKO and DKO muscle is in line with reduced tERK2 levels. The same results were obtained with STN extracts (data not shown). (D) Spinal cord (SPC), heart, kidney, and liver extracts from 9-week-old control and DKO mice probed with antibodies to tERK1/2 and α-Tub. ERK2/α-Tub ratios are shown at the bottoms of the blots. Except for kidney, no reduction in ERK2 levels was observed in these tissues.
Mice lacking ERK1/2 in skeletal muscle fibers are viable but display stunted postnatal growth, muscle weakness, and shorter life spans.
DKO animals were born at the expected Mendelian ratios predicted from the crosses. Thus, from the cross Hsa-Cre−/−; Erk1−/−; Erk2f/f × Hsa-Cre+/−; Erk1+/−; Erk2f/f, 78/317 pups (i.e., the expected one-fourth) showed the Hsa-Cre+/−; Erk1−/−; Erk2f/f genotype when assayed postweaning. We followed their weight starting at week 4 after birth. Starting at week 7, we also assayed muscle strength and overall motor coordination and fatigue resistance by measuring forelimb grip strength and by subjecting mice to a rotarod protocol, respectively. Muscle ERK2-deficient and germ line ERK1-deficient mice showed no difference in weight progression and forelimb grip strength relative to those in control animals (Hsa-Cre−/−; Erk1+/+; Erk2f/f), whereas DKO mutants failed to gain weight and showed a progressive loss of grip strength as young adults (Fig. 2A and B). Interestingly, double mutants could be divided into two groups according to how fast they lost weight (Fig. 2A). “Severe” animals lost weight rapidly starting at about 7 weeks of age, while “mild” double mutants were able to keep their weight at that age but clearly failed to keep up with controls or single mutants (Fig. 2A). We currently do not understand the basis of this differential effect on weight. When their results were normalized to body weight, the two types of DKO mutants showed very similar reductions of grip strength (Fig. 2B). Severe mice were not tested with the rotarod due to their overall frailty. However, mild DKO mice displayed a clear tendency to fall earlier from an accelerating rotarod (Fig. 2C). DKO mutants do not survive as long as controls or single mutants. Their deaths either were sudden and unexplained or followed weight loss of such severity that demanded humane euthanasia according to protocol guidelines. The median life spans were 71 days (n = 23) for severe mice and 121 days (n = 7) for mild mice (Fig. 2D). Interestingly, all 5 registered deaths among the mild DKO animals were among males, while the 2 females remained alive. Kyphosis, a sign of muscle weakness, was clearly evident in the long-surviving animals (Fig. 2E) and in at least one of the males that lived for 120 days. Thus, these results suggest that muscle depletion of either ERK1 or ERK2 has no overt phenotypic effects on the mice and that ERK1 and ERK2 together are required for myofiber postnatal maintenance or growth.
FIG 2.

DKO animals displayed stunted postnatal growth, muscle weakness, and shorter life spans. (A) Male weight progression was similar to the control for Erk1−/− and mErk2CKO mice, whereas mild DKO animals failed to gain weight from around week 9 and severe DKO mice began losing weight around week 7. Similar weight progression was seen in females (data not shown). The following numbers of mice were assessed per time point: control, n ≥ 5; Erk1−/−, n ≥ 6; mErk2CKO, n ≥ 7; mild DKO, n ≥ 3 for weeks 4 to 16 and n = 2 for week 18; and severe DKO, n ≥ 4 for weeks 4 to 10 and n = 2 for week 11. DKO mild, P < 0.01 versus matching time point control, t test, starting at week 6 onwards. DKO severe, P < 0.01 starting at week 5 onwards. (B) Forelimb grip strength was measured every other week starting at week 7 in males and females. Peak tension was normalized to body weight. DKO mild and severe animals showed comparable declines in muscle strength, while Erk1−/− and mErk2CKO mice were similar to controls. The following numbers of mice were assessed per time point: control, n ≥ 4; Erk1−/−, n ≥ 4; mErk2CKO, n ≥ 5; DKO mild, n ≥ 4 for weeks 7 to 13, n = 2 for week 15, and n = 1 for week 17; and DKO severe, n ≥ 3 for weeks 7 to 9 and n = 1 for week 11. DKO mild and severe, P < 0.01, ANOVA, week 9 onwards. (C) Mice were subjected to an accelerating rotarod protocol, and the time(s) to fall was recorded. No significant differences between controls and Erk1−/− or mErk2CKO mice were resolved. DKO mild animals fell off the rotating drum consistently earlier than controls (P < 0.01 versus control; ANOVA). The following numbers of mice were assessed per time point: control, n ≥ 5; Erk1−/−, n ≥ 3 for week 9 to 17 and n = 2 for week 19; mErk2CKO, n ≥ 5; and DKO mild, n ≥ 3 for weeks 9 to 13, n = 2 for week 15, and n = 1 for week 17. (D) Kaplan-Meier survival curves for DKO mild and severe mice. Median life spans were 71 days (n = 23) for severe mice and 121 days (n = 7) for mild mice (P < 0.0001, log rank test). (E) A surviving female mild DKO mouse that developed kyphosis (arrowhead), absent in a matched control.
Extensive fragmentation of the mature NMJ in ERK1/2-deficient muscle.
These studies were prompted by our previous results with cultured myotubes. We found that agrin stimulated the transient activation of ERK1/2 in an low-density lipoprotein (LDL) receptor-related protein 4 (Lrp4)/muscle-specific kinase (MuSK)-dependent fashion. Pharmacological blockade of this activation failed to inhibit agrin-induced acetylcholine receptor (AChR) clustering. Instead, it potentiated it by ∼60%. These and other observations led us to propose that agrin-induced ERK1/2 activation is part of a feedback loop that keeps agrin's clustering activity in check, at least in vitro (18). The fact that the DKO mice are viable and appear normal well after NMJs have formed and matured suggests that this ERK-dependent feedback mechanism is dispensable for the formation of the NMJ in vivo. However, it was still possible that ERK signaling had some role in NMJ maintenance, especially in light of the overall postnatal muscle weakness of the DKO mice (Fig. 2). We stained whole mounts of the neck sternomastoid (STN) muscle in control and DKO young adults for pre- and postsynaptic markers and examined the samples under confocal microscopy. The STN has been used by others to study the effects of aging and amyotrophic lateral sclerosis (ALS) on the structure of the mature NMJ (38–40). In control muscle, the NMJs exhibited their characteristic “pretzel” shape with long, continuous domains of postsynaptic AChRs labeled by α-bungarotoxin (BTX) tightly apposed by nerve terminals labeled by synaptophysin (SYN) (Fig. 3A). In STN DKO muscles, many NMJs looked very fragmented with small, mostly round AChR domains variably apposed to nerve terminal staining (Fig. 3B and C). This synaptic fragmentation, reminiscent of NMJs in aged normal animals (38, 39, 41), mdx mice, and utrophin/dystrophin knockout mice (42, 43), was observed in both severe and mild DKO muscle from males and females (Fig. 3B and C).
FIG 3.

Fragmented NMJs in STN from young adult DKO mice. (A) An example of a control NMJ labeled with fluorescein-BTX to mark AChRs (A′) and with antibodies to synaptophysin (SYN), followed by rhodamine-conjugated secondary antibodies, to label nerve terminals (A″). Long, continuous domains of postsynaptic AChRs are tightly apposed by nerve terminals. (B and C) Examples of fragmented NMJs in severe and mild DKO STNs, respectively. Small, mostly round AChR domains are variably apposed by nerve terminal staining. Scale bars: 10 μm.
A lack of myofiber ERK2 is sufficient to observe extensive synaptic fragmentation in the sternomastoid muscle.
Next, we sought to explore whether synaptic fragmentation required the deficiency of both muscle ERK1 and ERK2, or whether the lack of only one of these two kinases was sufficient for this phenotype. STNs from control (Hsa-Cre−/−; Erk1+/+; Erk2f/f), Erk1−/−, mERK2CKO, and mild DKO young adults were dissected, stained, and imaged by confocal microscopy as described above. We chose to work with the mild DKO animals because they survive longer and displayed overall better health than the severe DKO mice. The number of AChR domains per end plate was quantified on NMJs viewed en face. For animals between 3 and 6 months of age, the average numbers of AChR domains per end plate were 7.26 ± 0.38 for control mice (n = 85 NMJs; 5 mice), 12.15 ± 1.69 for DKO mice (n = 52 NMJs; 3 mice), 8.38 ± 0.94 for Erk1−/− mice (n = 80 NMJs; 4 mice), and 12.93 ± 0.91 for mErk2CKO mice (n = 90 NMJs; 6 mice). Thus, end plates from DKO and mErk2CKO but not Erk1−/− STNs had significantly more AChR fragments on average than the control. We plotted the percentage of NMJs versus the number of AChR domains (in bins of 5) for each genotype (Fig. 4A). While a shift to NMJs with larger number of AChR fragments could be appreciated for all 3 mutants, the most striking observation was that NMJs with more than 20 AChR domains were found only in DKO and mErk2CKO STN, not in control or Erk1−/− muscle (Fig. 4A). These highly fragmented end plates constituted up to 20% of total NMJs in the mErk2CKO STN (Fig. 4A). Statistical comparison of the mutant distributions relative to the control using the Wilcoxon rank sum test showed that only the DKO and mErk2CKO distributions were significantly different than control (DKO versus control, P = 0.000003; mErk2CKO versus control, P = 0.000002; Erk1−/− versus control, P = 0.05). Furthermore, statistical comparison using the same test of the DKO versus the mErk2CKO distributions showed no difference between them (P = 0.88). Lastly, as an additional control, we quantified AChR domains/NMJ in STN from Hsa-Cre+/− driver mice. We found an average of 5.96 ± 0.57 (n = 77 NMJs; 2 mice), slightly lower than for our control. An example of a highly fragmented synapse in mERK2CKO STN is shown in Fig. 5A. Therefore, the lack of muscle ERK2 is sufficient to yield extensive synaptic fragmentation in the STN.
FIG 4.

Quantification of synaptic fragmentation in STN and TA. The AChR domains per end plate were counted from confocal maximal projections of NMJs in STN (A) and TA (B) from all 4 genotypes. Data were grouped in bins of 5 as represented in the x axis, and the percentages of NMJs in those bins were plotted in the y axis. For STN, numbers of NMJs and mice were as follows: for the control, 85 and 5, respectively; for Erk1−/− mice, 80 and 4, respectively; for mErk2CKO mice, 90 and 6, respectively; and for DKO mice, 52 and 3, respectively. For TA, numbers of NMJs and mice were as follows: for the control, 65 and 4, respectively; for Erk1−/− mice, 32 and 2, respectively; for mErk2CKO mice, 112 and 5, respectively; and for DKO mice, 37 and 3, respectively. The statistical analysis of the data is described in Results.
FIG 5.

Fragmented NMJs in mErk2CKO STN and DKO TA. Shown are examples of a highly fragmented NMJ from mErk2CKO STN (A), a normal NMJ from control TA (B), and a fragmented NMJ from DKO TA (C). Overexposure in the rhodamine channel explains the strong intensity of the SYN staining in panel C″. Scale bars: 10 μm.
Differential sensitivity to the lack of ERK2 between different muscles.
Next, we studied synapse morphology in the hind limb tibialis anterior (TA) muscle, which was also used recently to study effects of aging on the NMJ (41, 44). The TA showed a different response than the STN (Fig. 4B). First, control NMJs were much less fragmented in the TA than the STN to begin with. Average AChR domains per end plate in control TA were 3.85 ± 0.25 (n = 65; 4 mice). Second, NMJs in mErk2CKO and Erk1−/− TA muscles appeared, on average, as fragmented as the control. The former had a mean of 4.30 ± 0.25 AChR domains/end plate (n = 112 NMJs; 5 mice), and the latter had 4.91 ± 0.52 (n = 32 NMJs; 2 mice). Third, only DKO end plates displayed statistically significant fragmentation, as their average number of AChR domains/end plate rose to 6.46 ± 0.75 (n = 37 NMJs; 3 mice; P = 0.003, Wilcoxon rank sum test). Fourth, NMJs with more than 15 AChR domains were never seen in control or Erk1−/− muscle, were extremely rare in mERK2CKO TA, and reached less than 10% of the junctions in DKO muscle. Fifth, NMJs with more than 20 AChR fragments, which were easily detected in mErk2CKO and DKO STNs, were absent from the sampled TA junctions, at these ages and even at 9 months of age (data not shown). Examples of a control and a highly fragmented NMJ in DKO TA are shown in Fig. 5B and C, respectively. Thus, at the ages studied, the NMJs of STN and TA displayed differential intrinsic fragmentation and a differential sensitivity to the lack of myofiber ERK1/2.
In order to account for the inherent differences in fragmentation of control NMJs between STN and TA that our data reveal (Fig. 4), we propose that a fragmented NMJ could be defined as one having more AChR domains than the control median. The median numbers of AChR domains were 7 and 3 for control NMJs in STN and TA, respectively. Using this criterion, synaptic fragmentation becomes a predominant feature in the majority of NMJs from DKO muscle, as ∼70% of the sampled NMJs had more AChR domains than their respective control medians. Thus, 71.15% (37/52) and 67.57% (25/37) NMJs in DKO STN and TA showed more than 7 and 3 AChR domains, respectively. This high level of fragmentation above control median was also reached in the mErk2CKO STN (66.67%; 60/90).
Absence of central myonuclei in muscle ERK2-deficient sternomastoid.
Others have suggested that synaptic fragmentation in aging and dystrophic muscle is due to degeneration and regeneration cycles in the synaptic portion of the muscle fiber (39, 42). We sought to determine if the same was happening in mErk2CKO and DKO muscle. Nuclei within normal muscle fibers localize toward the periphery of the cell, adjacent to the sarcolemma. Muscle fiber damage induces degeneration of the old fibers and regeneration of new ones, which are derived from satellite cells, muscle stem cells present in the tissue (45). A hallmark of this process is the accumulation of myonuclei toward the center of the cell. We counted fibers with central nuclei in transverse sections of STN stained with hematoxylin and eosin (H&E). Numbers of myofibers with central nuclei in control, Erk1−/−, and mErk2CKO STNs did not differ from each other, hovering around 2% (Fig. 6). DKO STN had more fibers with central nuclei than the control, but they reached only about 4% of the total (Fig. 6). Experiments with Evans blue dye confirmed sarcolemma integrity in mErk2CKO muscle (data not shown). Thus, NMJ fragmentation was prominent in the mErk2CKO STN despite it having the same percentage of fibers with central nuclei as the control. Moreover, a doubling in the number of fibers with central nuclei in the DKO STN did not enhance NMJ fragmentation quantitatively (Fig. 4A).
FIG 6.
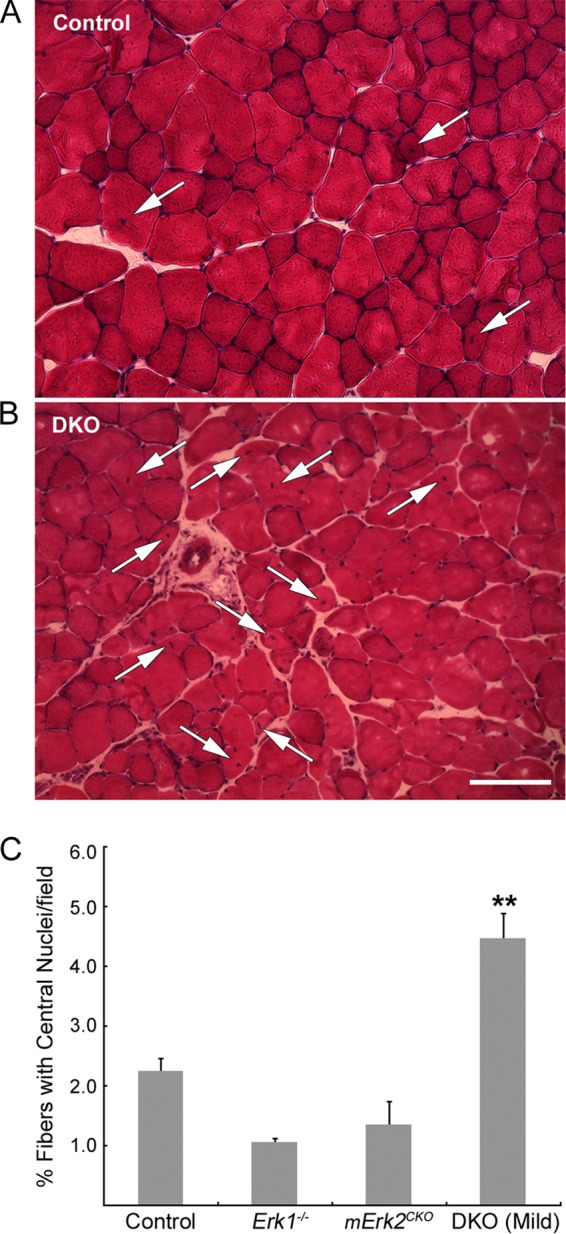
Quantification of central myonuclei in the STN. STN cross sections from 14-week-old mice were stained with H&E. Fields at 20× were selected, and total fibers and fibers with centrally located nuclei (arrows) were counted. Representative 20× fields from control (A) and from mild DKO (B) muscle are shown. Scale bar: 50 μm. (C) Quantification. Control, Erk1−/−, and mErk2CKO muscles had similar proportions of fibers with central nuclei, while DKO muscle had about twice as many. **, P < 0.01 versus control. For all genotypes, n = 3 muscles, except in the case of Erk1−/− (n = 2). Total fibers scored: for control muscle, 1,928; for Erk1−/− muscle, 1,234; for mErk2CKO muscle, 1,405; and for DKO muscle, 1,767.
ERK1/2 regulate AChR levels at the NMJ.
In addition to the synaptic fragmentation described above, we observed that about a third of NMJs in DKO muscles had relatively weak or sometimes faint BTX staining even in the presence of strong nerve terminal staining (Fig. 7A). Quantification showed that only ∼6% of control NMJs, but ∼29% and ∼38% of end plates in DKO STN and TA, respectively, showed dim AChR staining (Fig. 7B). While the fraction of end plates from Erk1−/− muscle with dim AChR staining was similar to that of the control, it tended to be higher in mErk2CKO muscles but reached statistical significance only in DKO samples (P = 0.03 for STN; P = 0.006 for TA) (Fig. 7B). To check if this apparent reduction in surface AChR protein levels was accompanied by a decrease in mRNA levels of its encoding subunit genes, we used real-time PCR to measure steady-state levels for the four genes that encode the adult AChR (46). Cycle threshold (CT) values obtained for 18S rRNA were used to equalize differences in total RNA per sample (27). Transcript level fold change was determined by the 2−ΔΔCT method (47), and values were normalized to the CT values obtained for control muscle for each gene (Fig. 7C). We found that Chrne mRNA was consistently reduced almost 2-fold in both mErk2CKO and DKO muscle. The reduction was statistically significant in DKO TA and mErk2CKO STN and TA but was borderline (P = 0.05) in the STN (Fig. 7C). Chrnd mRNA levels were essentially similar to those of the control regardless of genotype or muscle examined (Fig. 7C). Chrna and Chrnb mRNAs tended to be slightly lower than for the control in mErk2CKO and DKO TA, while they tended to increase particularly in the DKO STN (Fig. 7C). The lack of only ERK1 did not affect the mRNA levels for the subunits of the adult receptor (Fig. 7C), nor were Chrne mRNA levels different between control and Hsa-Cre+/− driver muscle (data not shown). Thus, a somewhat selective, muscle-ERK2-dependent reduction in mRNA Chrne expression was observed. We next checked if the expression of AChRε protein was also affected in DKO mice. We affinity purified AChRs from lysates of control and DKO TA muscle using biotinylated BTX and streptavidin-agarose and probed them for the AChRε subunit by Western blotting. In the DKO TA, there was a highly statistically significant decrease in Chrne mRNA (Fig. 7C). AChRε protein in DKO TA was ∼2- to 3-fold lower than in the control (P = 0.03) (Fig. 8). Chrne, the gene encoding the AChRε subunit indicative of the adult receptor, is transcribed almost exclusively at the synaptic site (48), so its transcription reflects the production of the synaptic AChR protein. Taken together, the weaker AChR staining at about a third of DKO NMJs, the decrease in AChRε protein demonstrated in the DKO TA, and the reduction in Chrne mRNA levels in mErk2CKO and DKO muscle suggest that ERK1/2 regulate expression of the synaptic AChR.
Fig 7.

Regulation of AChR expression by ERK1/2. (A) Examples of fields with DKO NMJs showing weaker (top row) or faint (bottom row) AChR staining (big single arrowheads) relative to those in the same field that show more normal levels of AChR staining (double arrowheads). Two examples of nerve terminal sprouts (arrows) in the DKO STN, labeled for SYN, are shown in the top row. The short sprout in the bottom right appears to induce or connect with two small AChR clusters in the next myofiber. Scale bars: 10 μm. (B) Quantification of weak or faint BTX-stained NMJs (n = 2 to 6 muscles/genotype). (C) Real-time PCR for the adult AChR subunit mRNAs in 9-week-old STN and TA muscle. Values were normalized to that for the control. A consistent decrease in Chrne mRNA was observed in mErk2CKO and DKO muscle. (D) Real-time PCR for the fetal AChRγ subunit gene (Chrng) mRNA in 9-week-old STN and TA. Values were normalized to that for the control. A 40-fold increase in Chrng mRNA was detected selectively in DKO STN. (E) Real-time PCR for two additional denervation markers, Runx-1 and Myh3. Values were normalized to that for the control. Significant induction for these markers was restricted to DKO STN. For all real-time PCR assays, n = 6 for both muscles and all genotypes except for Erk1−/− muscles (n = 5). Muscles from both male and female animals were combined because no significant gender differences were found in the CT values. *, P < 0.05; **, P < 0.01.
FIG 8.
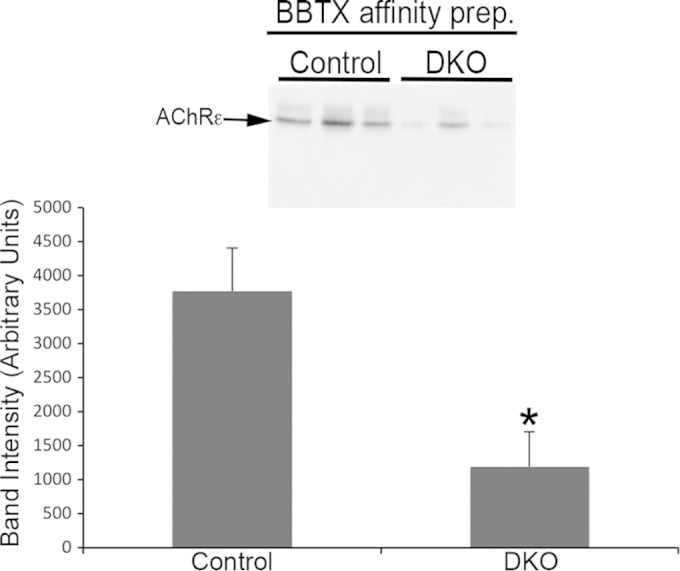
Reduced AChRε protein in DKO TA. AChRs were affinity purified from 1-mg lysates from 3 control and 3 DKO TA muscles using biotin-BTX (BBTX) and streptavidin-agarose. Precipitates were subjected to SDS-PAGE, transferred to a PVDF membrane, and probed for AChRε (arrow). The histogram shows averages ± SEMs. *, P < 0.05.
Most prominently in DKO STN, we observed NMJs with AChR patches without overlying SYN staining (i.e., aneural AChR patches) (Fig. 3) and evidence of nerve terminal sprouting (as illustrated in Fig. 7A, top row). Thus, 45% NMJs (18/40; 3 mice) in the DKO STN, but 16% in control STN (6/38; 4 mice), had terminal sprouts. Because these morphological features are hallmarks of partial denervation (49), we next used real-time PCR to check for mRNA levels for Chrng, the gene encoding the distinctive AChRγ subunit of the fetal AChR (46), which is strongly induced by functional denervation in the adult (50). Chrng mRNA levels were similar to that of the control in TA muscle from all three mutant genotypes. However, Chrng mRNA was moderately increased in mErk2CKO STN (2-fold) and reached 40-fold induction in the DKO STN (Fig. 7D). mRNA for Runx1 and the embryonic myosin heavy-chain gene Myh3, two other genes strongly induced by denervation (51), were also selectively increased in DKO STN (Fig. 7E). The sharp increases in these denervation markers in the DKO STN but not the DKO TA were consistent with the trend toward elevated mRNA levels for Chrna, Chrnb, and Chrnd in the DKO STN but not in the DKO TA (Fig. 7C), as these AChR genes are also induced by denervation (52). Thus, together, these results suggest that loss of myofiber ERK1/2 led to significant partial denervation selectively in the STN.
Myofiber number, size, and type effects.
Lastly, we studied myofiber number, size, and type in STN and TA muscles to determine whether there was a correlation between potential changes in these parameters and the synapse maintenance phenotypes described above. Cross sections from the belly of the muscles were costained for dystrophin, to mark the boundaries of individual myofibers, and for the 4 canonical myosin heavy-chain (MyHC) isoforms that define adult fiber types (1, 2A, 2B, and 2X) (53). Dystrophin was present on the surface of muscle fibers of these DKO muscles, just as it was in controls and single Erk mutants (Fig. 9A and B). Total numbers of fibers per TA cross section were similar across all four genotypes, while there was a statistically significant (21%) reduction in fiber numbers limited to the DKO STN (P = 0.02) (Fig. 9C). DKO STN and TA showed modest average fiber atrophy (∼14% and ∼11%, respectively) that was statistically significant for the DKO TA (P = 0.008) (Fig. 9D). The Erk1−/− and mErk2CKO muscles tended to have slightly hypertrophied fibers on average compared to controls (Fig. 9D). Thus, the modest fiber atrophy in both TA and STN together with the fiber loss in the STN is consistent with the lower weight of DKO animals relative to that of the controls (Fig. 2A). We sought to determine the fiber type composition of the STN and TA in our Erk1/2 mutant mice. In the adult mouse, STN and TA are predominantly fast-fiber muscles (54–56). Our experiments confirmed these published observations, as our control and 3 mutant STN and TA muscles had a maximum of 0.2% type 1 fibers (data not shown). Analysis of MyHC staining in STN showed that ERK1-lacking mice were similar to controls in the distribution of fast fibers (2A, 2B, and 2X) (Fig. 9E). There was a tendency toward fewer 2B and more 2A fibers than for the control in DKO STN that did not reach statistical significance (Fig. 9E). mErk2CKO and DKO STNs displayed a statistically significant (∼7%) increase in 2X fibers (P = 0.004 and P = 0.03, respectively) (Fig. 9E). Although the variability in the data prevents a clear-cut conclusion, this slight increase in 2X fibers seems to be at the expense of 2B fibers. Mutant TA was no different from the control regarding fast fibers (Fig. 9F). Thus, the analysis of the predominantly fast-twitch muscles STN and TA showed only modest changes in fiber type composition due to lack of muscle ERK1/2. Consistent with this conclusion, and with its proposed role in regulating fiber type composition (57), levels of active p38 MAPK were unaltered in DKO muscle relative to control (Fig. 10). Active c-jun NH2-terminal kinase (JNK) levels in DKO were also statistically similar to control levels. Values for fold change versus the control are as follows: STN p46JNK, 0.99 ± 0.10; STN p54JNK, 0.52 ± 0.05; TA p46JNK, 1.79 ± 0.76; and TA p54JNK, 0.55 ± 0.19.
Fig 9.

Effects on fiber number, size, and type. (A) Representative cross sections of STN muscles from 14-week-old female mice stained for dystrophin. Scale bar: 200 μm. (B) Representative cross sections of TA muscles from 14-week-old female mice stained for dystrophin. Scale bar: 400 μm. (C) Quantification of fiber numbers. A significant reduction in total fibers per cross section was detected in the DKO STN. n = 5 for controls, and n = 3 for all other genotypes. We combined data for males and females per genotype because no significant gender differences were found for this parameter. (D) Quantification of average fiber area. Because of gender differences in fiber area, raw data were normalized relative to those for the control of the same sex. Controls were set at 100%, and a one-sample t test was used to statistically compare the results relative to control. n = 3 for both muscles and all genotypes. DKO muscle had a smaller average fiber area that was statistically significant for the TA. (E) Distribution of fast-fiber types in STN. Small but significant increases in 2X fibers were observed in muscles from mErk2CKO and DKO mice. (F) Distribution of fast-fiber types in TA. No changes in fiber type distribution due to genotype were observed in this muscle. We combined data within each muscle for males and females per genotype because no significant gender differences were found for this parameter. n = 3 for both muscles and all genotypes. *, P < 0.05; **, P < 0.01.
FIG 10.
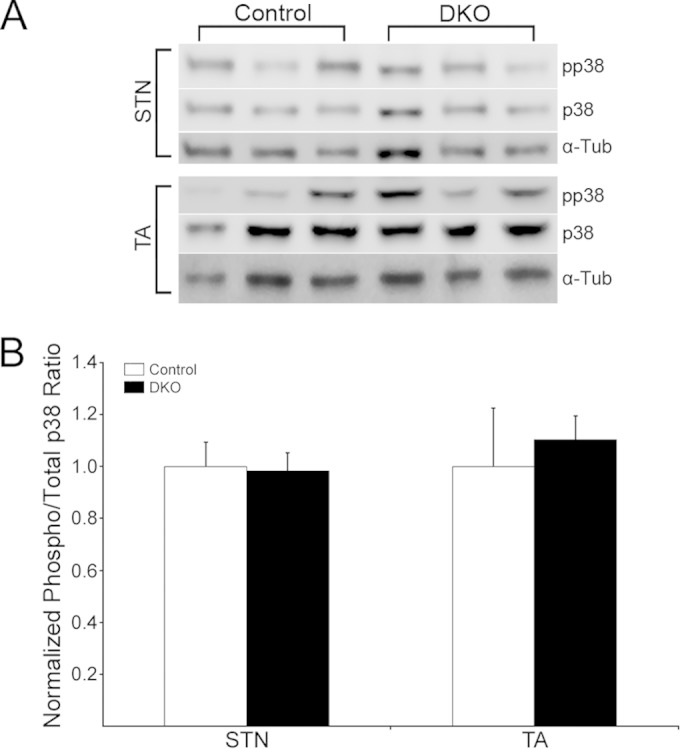
Activated p38 in DKO muscle. (A) Control and DKO STN and TA extracts, n = 3 per muscle/genotype, were probed with antibodies to phosphorylated p38 (pp38) and total p38 (p38). Variations in loading were checked by stripping membranes and probing for α-Tub. (B) Normalized phosphorylated/total protein ratios for p38 showed no statistically significant changes in the activation of this kinase in DKO muscle relative to the control.
DISCUSSION
We have for the first time selectively abrogated ERK1/2 in skeletal muscle fibers. We found that ERK1/2 are needed for the maintenance of myofibers and NMJs. DKO animals displayed stunted postnatal growth, muscle weakness, and shorter life spans. The muscles examined in this study, STN and TA, displayed a combination of modest fiber atrophy and loss without major changes in fiber type composition or absence of cell surface dystrophin. Loss of myofiber ERK1/2 yielded both overlapping and distinct changes in synaptic morphology and AChR gene expression that depended on the muscle studied. In contrast, the lack of only ERK1 had no apparent effects on the phenotypes studied, and the lack of myofiber ERK2 explained synaptic fragmentation in the STN but not the TA and a decrease in the expression of Chnre mRNA in both muscles. A corresponding reduction in AChR protein in the TA was documented. Evidence of partial denervation was also found in the STN but not the TA. Thus, myofiber ERK1/2 are differentially required for the maintenance of myofibers and neuromuscular synapses in adult mice.
The cause-effect relationship between muscle weakness and stunted growth in the DKO mice is unclear. One possibility is that muscles involved in mastication and swallowing become weak, leading to reduced food intake and weight loss. Although the weight loss of the DKO mice correlated with the combined muscle fiber loss and atrophy observed in their STN and TA muscles, it is possible that minor renal problems also contributed to this phenotype. Kidneys from DKO mice had about half ERK2 levels of the control (Fig. 1D). However, only a few isolated single cells in adult kidney tissue express a LacZ reporter that is driven by the same Hsa-Cre gene as in the mice used in this study (http://www.informatics.jax.org/recombinase/specificity?id=MGI:2447635&systemKey=4856358). It is unlikely that this low level of expression accounts for the 50% reduction in kidney ERK2 in DKO mice. This reduction, and perhaps any ensuing renal complications, may be secondary effects stemming from the overall muscle weakness. We do not know why the DKO mice died. All of the DKO mild animals that died were males, while the surviving ones were females. One of the male DKO mild animals that died at 16 weeks of age displayed kyphosis and showed compromised respiration before humane euthanasia. However, other animals died younger and failed to exhibit respiratory distress or kyphosis that was evident to the naked eye. Moreover, the surviving DKO mild females showed clear kyphosis (Fig. 2E). Future experiments will be needed to clarify these issues.
There was an intrinsic difference in fragmented NMJs between control STN and TA muscles. The average NMJ in control STN had about twice the number of AChR domains than the average NMJ in control TA. This correlated with the sensitivity to NMJ fragmentation in these muscles following the reduction in ERK1/2. Even after abrogation of myofiber ERK1/2, this difference was maintained, as the most fragmented synapses occurred in the STN and not the TA. It remains possible that these differences disappear in older animals. Wild-type TA and STN muscles showed no difference in NMJ fragmentation when examined at 2 years of age (40). However, synaptic fragmentation in that study was defined differently than in this study. To account for the intrinsic differences in fragmentation of control NMJs in various muscles, we propose that a fragmented NMJ could be defined as one having more AChR domains than the control median. In control muscle, the frequency of fragmented synapses using this criterion will always be by definition 50% at most. Two other groups previously used a threshold of AChR fragments to define a fragmented NMJ. Valdez and colleagues (40, 41) defined a fragmented NMJ as one having 5 or more AChR islands or a segment of the postsynaptic apparatus with severe abnormalities such as a small or irregularly shaped AChR cluster. Li and coworkers (39) defined a fragmented NMJ as one having 10 or more AChR domains. When applied to our data, the criterion of Valdez et al. suggests the puzzling conclusion that most of the NMJs in control STN are fragmented. In this regard, our control genotype was not wild type for Erk2; it was Erk2f/f. Because the average AChR patches/NMJ in the STN from the Cre driver mice (genotype Erk1+/+; Erk2+/+) was a bit lower than in our control STN, it is possible that the Erk2f allele has a small effect on fragmentation. Li and colleagues studied the STN, and their more stringent criterion perhaps resulted from studying this muscle, whose synapses appear to be particularly prone to fragmentation. This criterion was rather uninformative for our experiments with the TA because it sets the threshold for fragmentation (≥10 fragments) even above the average AChR domains per NMJ obtained in the DKO TA (∼6.46). Thus, we believe that our control median-based criterion, in combination with the statistical analysis in Fig. 4, is more fitting to define a fragmented NMJ and to account for the intrinsic differences in fragmentation that may occur among synapses in various muscles.
Unlike in the TA, loss of myofiber ERK2 in the STN was sufficient to yield the same levels of synaptic fragmentation as in the DKO (Fig. 4). These results were unexplained by higher Hsa-Cre-driven ERK2 reduction or a higher ERK1 decrease as a consequence of Erk2f/f recombination in STN than TA (Fig. 1). Nor were endogenous levels of ERK2 higher in control TA than in control STN by Western blotting (data not shown). Although ERK1 and ERK2 are generally viewed as functionally redundant, the differential embryonic lethality between their germ line mutants and other evidence (58–60) suggest specific roles for these two kinases in some physiological contexts. Alternatively, the more dramatic phenotypes we find in mErk2CKO animals may simply reflect the higher levels of expression of ERK2 relative to ERK1 in skeletal muscle fibers in general.
The extensive synaptic fragmentation in the mErk2CKO STN failed to correlate to changes in animal weight, forelimb grip strength, survival, fiber number, or fiber size, because none of these parameters was different than for the control. We measured a small increase (∼7%) in 2X fibers in the mErk2CKO STN relative to those in the control. However, it is unlikely that this accounts for the synaptic fragmentation in this muscle, because 67% of the NMJs in the mErk2CKO STN were fragmented according to our control median-based criterion. Although other general changes in the fibers might be linked to synaptic fragmentation, notably changes in metabolic capacity and generation of reactive oxygen species (ROS) (61), our results suggest that ERK2 regulates mechanisms that locally control synapse maintenance. In this context, NMJ fragmentation by excess ROS needs them produced in motoneurons and not in muscle fibers (62, 63). Hence, mechanisms underlying NMJ fragmentation by excess ROS and by muscle ERK1/2 deficiency may be fundamentally distinct. There was no correlation between the accumulation of central myonuclei (Fig. 6) and the synaptic fragmentation phenotype in mErk2CKO and DKO STN (Fig. 4A). Postsynaptic mechanisms other than local fiber degeneration and regeneration may account for NMJ fragmentation (64). This does not exclude that some of the fragmented NMJs in our mutant muscles result from degeneration and regeneration in the subsynaptic area. Our measurements of central nuclei were from extrasynaptic regions of the fibers. The relatively small subsynaptic portion of the muscle fiber in mutant muscle could be more prone to damage than the rest of the fiber and/or central nuclei could be transient and migrate quickly to the periphery of the fiber, so we would miss them in our experiments (39).
NMJs with dimly BTX-stained AChRs were present both in DKO STN and TA, although they were more easily detected in the latter (Fig. 7). Fewer AChRs at the synaptic site could be due to one or a combination of the following processes: lower rate of synthesis and/or multimeric assembly, higher rate of degradation, lower rate of insertion of the receptors in the synaptic sarcolemma, or higher retrieval from it. Synthesis depends highly on the rate of local transcription. We found a consistent reduction of the AChRε subunit gene mRNA, which was myofiber ERK2 dependent (Fig. 7C). We also documented a corresponding reduction in AChRε protein in the DKO TA (Fig. 8). Transcription of all AChR subunit genes is highly enriched at the subsynaptic myonuclei (65–67); however, Chrne's is perhaps the most synaptic of them all (48). Thus, a reduction in Chrne mRNA is expected to affect the synaptic AChR selectively. This reduction was not due to general fiber atrophy, as it was detected in mErk2CKO muscle, which was no different from the control regarding fiber morphology (Fig. 9). The ∼2-fold reduction in whole mErk2CKO and DKO muscle Chrne mRNA may underestimate the reduction at those NMJs with faint AChR staining (Fig. 7B). Dominant negative mutants for Ras, Raf, and MEK1 selectively inhibited synapse-specific expression of Chrne-luciferase reporters that were expressed in adult TA muscle following DNA injection (23). These experiments suggested that ERK signaling regulates Chrne expression at the transcriptional level. Our data showing parallel reductions in both AChRε protein and mRNA are consistent with this mechanism for the role of ERK in controlling Chrne expression.
The presence of terminal sprouts and of AChR patches lacking apposing nerve terminal staining in some NMJs from DKO STN is consistent with the significant increase in Chrng mRNA and suggests that a relevant proportion of NMJs in this muscle are at least functionally denervated. This effect explains the tendency toward increased levels of Chrna, Chrnb, and Chrnd mRNA in the DKO STN (Fig. 7C), as denervation is well known to induce expression of these AChR subunit genes along the entire muscle fiber (52). It might also explain why Chrne mRNA levels in the DKO STN were just borderline different from in the control (P = 0.05), while those in mErk2CKO STN were significantly lower (P < 0.05, Fig. 8C). This was not observed in the DKO TA, which highlights another important distinction in the response to the lack of myofiber ERK1/2 between these two muscles. Unlike synaptic fragmentation in the STN, full expression of this partial denervation-like effect required removal of both ERK1 and ERK2. This suggests that synaptic fragmentation and the apparent denervation are not tightly correlated. The fiber loss and the increased proportion of regenerating fibers detected in the DKO STN (Fig. 6 and 9) could account, at least in part, for the upregulation of Chrng in this muscle, as increased AChR expression was observed in regenerating muscle (68). In this context, complete denervation is rare in NMJs from aged normal STN; however, partial denervation, terminal sprouts, and aneural AChRs were detected (39, 40). Changes in Chrng in aged normal STN have not been studied; however, increases in expression of Chrng and other denervation markers were reported to occur in aged normal quadriceps (69) and seemed unaccompanied by motoneuron loss (70).
The significant fiber loss that we observed in the DKO STN, together with the mild fiber atrophy in DKO STN and TA, are consistent with a role for ERK1/2 in maintaining skeletal muscle mass and with prior studies that suggested so in C2C12 cells and rats (9). On the other hand, our analysis of fiber type composition in the STN and TA disagrees with the notion that ERK1/2 are essential to preserve the fast-twitch fiber phenotype, as previously proposed (10). To inactivate ERK1/2 signaling in vivo, that study used overexpression of MAPK phosphatase 1 (MKP-1) by electroporation of adult mouse muscle. However, MKP-1 not only inactivates ERK1/2 but actually shows substrate preference for other MAPKs such as JNK and p38 (57). Thus, the in vivo effects on fast-fiber expression reported in this study could have been unspecific to the inactivation of pERK1/2. Others have posited that ERK1/2 are critical to promote slow-fiber differentiation (11). Because the STN and TA muscles studied in this work bear such a low fraction of type 1 fibers, compelling conclusions about the role of ERK1/2 on the slow-fiber phenotype from our animals will have to wait until we examine muscles with significant content of type 1 fibers, such as the soleus.
Intrinsic distinctions in the normal development and maintenance of NMJs among different muscles have been described previously (71) and might underlie the differences in the response of the NMJs in STN and TA to the lack of myofiber ERK1/2. Thus, the STN is a delayed-synapsing muscle, while the TA is a fast-synapsing one (71). Muscles display differential susceptibility to sarcopenia and to neuromuscular diseases. Some are highly affected, while others appear to be resistant. In many cases, these muscle-selective effects include how their NMJs react to these conditions (40, 72, 73). The mechanisms underlying these unique sensitivities remain elusive and are likely to be complex and condition specific. Reductions in active ERK1/2 levels with aging were reported in specific muscles of the rat (13). Significant skeletal muscle abnormalities (16, 17) were found in patients suffering from a group of genetic conditions collectively known as RASopathies, in which different components of the Ras/MAPK pathway are anomalously activated (15). Recently, patients with deletions encompassing MEK2 were shown to have overlapping features with RASopathies, which suggests that haploinsufficiency of Ras-Erk1/2 pathway components is a potential novel mechanism underlying these disorders (74). Our results showing that myofiber-derived ERK1/2 are necessary for the maintenance and growth of adult muscle fibers and for the stability of their NMJs in a muscle-specific fashion further support an important role for this signaling pathway in muscle-selective sarcopenia and are informative as to relevant neuromuscular phenotypes that may be affected by the dysregulation of Ras-ERK signaling in RASopathies.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant NS077177 from the National Institute of Neurological Disorders and Stroke.
We thank J. Samuel for access to a confocal microscope, R. Miranda for access to a real-time PCR machine, S. Balaraman for help with quantitative real-time PCR experiments, R. Hastings for making us aware of the AChRε antibody ultimately used in this study, and W. Thompson for critically reading the manuscript. The MyHC monoclonal antibodies developed by H. Blau, C. Lucas, and S. Schiaffino were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA.
REFERENCES
- 1.Yoon S, Seger R. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 2.Osborne JK, Zaganjor E, Cobb MH. 2012. Signal control through Raf: in sickness and in health. Cell Res 22:14–22. doi: 10.1038/cr.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rimer M. 2011. Emerging roles for MAP kinases in agrin signaling. Commun Integr Biol 4:143–146. doi: 10.4161/cib.4.2.14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. 2001. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol 186:104–115. doi:. [DOI] [PubMed] [Google Scholar]
- 5.Yokoyama T, Takano K, Yoshida A, Katada F, Sun P, Takenawa T, Andoh T, Endo T. 2007. DA-Raf1, a competent intrinsic dominant-negative antagonist of the Ras-ERK pathway, is required for myogenic differentiation. J Cell Biol 177:781–793. doi: 10.1083/jcb.200703195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Johnson SE. 2006. ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem Biophys Res Commun 345:1425–1433. doi: 10.1016/j.bbrc.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 7.Cho YY, Yao K, Bode AM, Bergen HR III, Madden BJ, Oh SM, Ermakova S, Kang BS, Choi HS, Shim JH, Dong Z. 2007. RSK2 mediates muscle cell differentiation through regulation of NFAT3. J Biol Chem 282:8380–8392. doi: 10.1074/jbc.M611322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett AM, Tonks NK. 1997. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 9.Shi H, Scheffler JM, Zeng C, Pleitner JM, Hannon KM, Grant AL, Gerrard DE. 2009. Mitogen-activated protein kinase signaling is necessary for the maintenance of skeletal muscle mass. Am J Physiol Cell Physiol 296:C1040–C1048. doi: 10.1152/ajpcell.00475.2008. [DOI] [PubMed] [Google Scholar]
- 10.Shi H, Scheffler JM, Pleitner JM, Zeng C, Park S, Hannon KM, Grant AL, Gerrard DE. 2008. Modulation of skeletal muscle fiber type by mitogen-activated protein kinase signaling. FASEB J 22:2990–3000. doi: 10.1096/fj.07-097600. [DOI] [PubMed] [Google Scholar]
- 11.Murgia M, Serrano AL, Calabria E, Pallafacchina G, Lomo T, Schiaffino S. 2000. Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat Cell Biol 2:142–147. doi: 10.1038/35004013. [DOI] [PubMed] [Google Scholar]
- 12.Roubenoff R, Hughes VA. 2000. Sarcopenia: current concepts. J Gerontol A Biol Sci Med Sci 55:M716–M724. doi: 10.1093/gerona/55.12.M716. [DOI] [PubMed] [Google Scholar]
- 13.Rahnert JA, Luo Q, Balog EM, Sokoloff AJ, Burkholder TJ. 2011. Changes in growth-related kinases in head, neck and limb muscles with age. Exp Gerontol 46:282–291. doi: 10.1016/j.exger.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muchir A, Kim YJ, Reilly SA, Wu W, Choi JC, Worman HJ. 2013. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet Muscle 3:17. doi: 10.1186/2044-5040-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tidyman WE, Rauen KA. 2009. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tidyman WE, Lee HS, Rauen KA. 2011. Skeletal muscle pathology in Costello and cardio-facio-cutaneous syndromes: developmental consequences of germline Ras/MAPK activation on myogenesis. Am J Med Genet C Semin Med Genet 157:104–114. doi: 10.1002/ajmg.c.30298. [DOI] [PubMed] [Google Scholar]
- 17.Stevenson DA, Allen S, Tidyman WE, Carey JC, Viskochil DH, Stevens A, Hanson H, Sheng X, Thompson BA, Okumura JM, Reinker K, Johnson B, Rauen KA. 2012. Peripheral muscle weakness in RASopathies. Muscle Nerve 46:394–399. doi: 10.1002/mus.23324. [DOI] [PubMed] [Google Scholar]
- 18.Rimer M. 2010. Modulation of agrin-induced acetylcholine receptor clustering by extracellular signal-regulated kinases 1 and 2 in cultured myotubes. J Biol Chem 285:32370–32377. doi: 10.1074/jbc.M110.144774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMahan UJ. 1990. The agrin hypothesis. Cold Spring Harbor Symp Quant Biol 55:407–418. doi: 10.1101/SQB.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Xiong WC, Mei L. 2010. To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tansey MG, Chu GC, Merlie JP. 1996. ARIA/HRG regulates AChR epsilon subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway. J Cell Biol 134:465–476. doi: 10.1083/jcb.134.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altiok N, Altiok S, Changeux JP. 1997. Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO J 16:717–725. doi: 10.1093/emboj/16.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si J, Mei L. 1999. ERK MAP kinase activation is required for acetylcholine receptor inducing activity-induced increase in all five acetylcholine receptor subunit mRNAs as well as synapse-specific expression of acetylcholine receptor epsilon-transgene. Brain Res Mol Brain Res 67:18–27. doi: 10.1016/S0169-328X(99)00028-5. [DOI] [PubMed] [Google Scholar]
- 24.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 25.Ponomareva ON, Ma H, Vock VM, Ellerton EL, Moody SE, Dakour R, Chodosh LA, Rimer M. 2006. Defective neuromuscular synaptogenesis in mice expressing constitutively active ErbB2 in skeletal muscle fibers. Mol Cell Neurosci 31:334–345. doi: 10.1016/j.mcn.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK, Pellizzoni L, Rojas J, Valenzuela DM, Murphy AJ, Winberg ML, Monani UR. 2011. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest 121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paez-Colasante X, Seaberg B, Martinez TL, Kong L, Sumner CJ, Rimer M. 2013. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS One 8:e75866. doi: 10.1371/journal.pone.0075866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagès G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. 1999. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 29.Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. 2001. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M. 2003. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8:847–856. doi: 10.1046/j.1365-2443.2003.00680.x. [DOI] [PubMed] [Google Scholar]
- 31.Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S. 2003. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep 4:964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. 2003. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A 100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. 2008. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci 28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J. 1999. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res 27:e27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cifuentes-Diaz C, Frugier T, Tiziano FD, Lacene E, Roblot N, Joshi V, Moreau MH, Melki J. 2001. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol 152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicole S, Desforges B, Millet G, Lesbordes J, Cifuentes-Diaz C, Vertes D, Cao ML, De Backer F, Languille L, Roblot N, Joshi V, Gillis JM, Melki J. 2003. Intact satellite cells lead to remarkable protection against Smn gene defect in differentiated skeletal muscle. J Cell Biol 161:571–582. doi: 10.1083/jcb.200210117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Escher P, Lacazette E, Courtet M, Blindenbacher A, Landmann L, Bezakova G, Lloyd KC, Mueller U, Brenner HR. 2005. Synapses form in skeletal muscles lacking neuregulin receptors. Science 308:1920–1923. doi: 10.1126/science.1108258. [DOI] [PubMed] [Google Scholar]
- 38.Balice-Gordon RJ. 1997. Age-related chages in neuromuscular innervation. Muscle Nerve Suppl 5:S83–S87. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Lee YI, Thompson WJ. 2011. Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J Neurosci 31:14910–14919. doi: 10.1523/JNEUROSCI.3590-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. 2012. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. doi: 10.1371/journal.pone.0034640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valdez G, Tapia JC, Kang H, Clemenson GD, Gage FH, Lichtman JW, Sanes JR. 2010. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci U S A 107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyons PR, Slater CR. 1991. Structure and function of the neuromuscular junction in young adult mdx mice. J Neurocytol 20:969–981. doi: 10.1007/BF01187915. [DOI] [PubMed] [Google Scholar]
- 43.Rafael JA, Townsend ER, Squire SE, Potter AC, Chamberlain JS, Davies KE. 2000. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum Mol Genet 9:1357–1367. doi: 10.1093/hmg/9.9.1357. [DOI] [PubMed] [Google Scholar]
- 44.Cheng A, Morsch M, Murata Y, Ghazanfari N, Reddel SW, Phillips WD. 2013. Sequence of age-associated changes to the mouse neuromuscular junction and the protective effects of voluntary exercise. PLoS One 8:e67970. doi: 10.1371/journal.pone.0067970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauro A. 1961. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B. 1986. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature 321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 48.Gundersen K, Sanes JR, Merlie JP. 1993. Neural regulation of muscle acetylcholine receptor epsilon- and alpha-subunit gene promoters in transgenic mice. J Cell Biol 123:1535–1544. doi: 10.1083/jcb.123.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Son YJ, Thompson WJ. 1995. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- 50.Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S. 1987. Differential regulation of muscle acetylcholine receptor gamma- and epsilon-subunit mRNAs. FEBS Lett 223:104–112. doi: 10.1016/0014-5793(87)80518-5. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. 2005. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 19:1715–1722. doi: 10.1101/gad.1318305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kues WA, Brenner HR, Sakmann B, Witzemann V. 1995. Local neurotrophic repression of gene transcripts encoding fetal AChRs at rat neuromuscular synapses. J Cell Biol 130:949–957. doi: 10.1083/jcb.130.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schiaffino S, Reggiani C. 2011. Fiber types in mammalian skeletal muscles. Physiol Rev 91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 54.Guido AN, Campos GER, Neto HS, Marques MJ, Minatel E. 2010. Fiber type composition of the sternomastoid and diaphragm muscles of dystrophin-deficient mdx mice. Anat Rec 293:1722–1728. doi: 10.1002/ar.21224. [DOI] [PubMed] [Google Scholar]
- 55.Bloemberg D, Quadrilatero J. 2012. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Augusto V, Padovani CR, Campos GER. 2004. Skeletal muscle fiber types in C57BL6J mice. Braz J Morphol Sci 21:89–94. [Google Scholar]
- 57.Flach RJR, Bennett AM. 2010. MAP kinase phosphatase-1—a new player at the nexus between sarcopenia and metabolic disease. Aging (Albany NY) 2:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vantaggiato C, Formentini I, Bondanza A, Naldini L, Brambilla R. 2006. Central ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol 5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin S, Dimitri CA, Yoon S-O, Dowdle W, Blenis J. 2010. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38:114–127. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, von Kriegsheim A, Norman JC. 2012. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-β2. J Cell Sci 125:1465–1477. doi: 10.1242/jcs.092916. [DOI] [PubMed] [Google Scholar]
- 61.Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H. 2010. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A, Brooks SV. 2014. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J 28:1666–1681. doi: 10.1096/fj.13-240390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV, Van Remmen H. 2013. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J 27:3536–3548. doi: 10.1096/fj.13-228130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banks GB, Chamberlain JS, Froehner SC. 2009. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci 40:433–441. doi: 10.1016/j.mcn.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merlie JP, Sanes JR. 1985. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature 317:66–68. doi: 10.1038/317066a0. [DOI] [PubMed] [Google Scholar]
- 66.Fontaine B, Sassoon D, Buckingham M, Changeux JP. 1988. Detection of the nicotinic acetylcholine receptor alpha-subunit mRNA by in situ hybridization at neuromuscular junctions of 15-day-old chick striated muscles. EMBO J 7:603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simon AM, Hoppe P, Burden SJ. 1992. Spatial restriction of AChR gene expression to subsynaptic nuclei. Development 114:545–553. [DOI] [PubMed] [Google Scholar]
- 68.Brenner HR, Herczeg A, Slater CR. 1992. Synapse-specific expression of acetylcholine receptor genes and their products at original synaptic sites in rat soleus muscle fibres regenerating in the absence of innervation. Development 116:41–53. [DOI] [PubMed] [Google Scholar]
- 69.Barns M, Gondro C, Tellam RL, Radley-Crabb HG, Grounds MD, Shavlakadze T. 2014. Molecular analyses provide insight into mechanisms underlying sarcopenia and myofibre denervation in old skeletal muscles of mice. Int J Biochem Cell Biol 53:174–185. doi: 10.1016/j.biocel.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 70.Chai RJ, Vukovic J, Dunlop S, Grounds MD, Shavlakadze T. 2011. Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One 6:e28090. doi: 10.1371/journal.pone.0028090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. 2002. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron 34:357–370. doi: 10.1016/S0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 72.Punga AR, Lin S, Oliveri F, Meinen S, Rüegg MA. 2011. Muscle-selective synaptic disassembly and reorganization in MuSK antibody positive MG mice. Exp Neurol 230:207–217. doi: 10.1016/j.expneurol.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 73.Ling KKY, Gibbs RM, Feng Z, Ko C-P. 2012. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet 21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nowaczyk MJM, Thompson BA, Zeesman S, Moog U, Sanchez-Lara PA, Magoulas PL, Falk RE, Hoover-Fong JE, Batista DAS, Amudhavalli SM, White SM, Graham GE, Rauen KA. 2014. Deletion of MAP2K2/MEK2: a novel mechanism for a RASopathy? Clin Genet 85:138–146. doi: 10.1111/cge.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]