

Abstract
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that regulates a diverse array of cellular processes, including cell growth, survival, metabolism, and cytoskeleton dynamics. mTOR functions in two distinct complexes, mTORC1 and mTORC2, whose activities and substrate specificities are regulated by complex specific cofactors, including Raptor and Rictor, respectively. Little is known regarding the relative contribution of mTORC1 versus mTORC2 in vascular endothelial cells. Using mouse models of Raptor or Rictor gene targeting, we discovered that Rictor ablation inhibited vascular endothelial growth factor (VEGF)-induced endothelial cell proliferation and assembly in vitro and angiogenesis in vivo, whereas the loss of Raptor had only a modest effect on endothelial cells (ECs). Mechanistically, the loss of Rictor reduced the phosphorylation of AKT, protein kinase Cα (PKCα), and NDRG1 without affecting the mTORC1 pathway. In contrast, the loss of Raptor increased the phosphorylation of AKT despite inhibiting the phosphorylation of S6K1, a direct target of mTORC1. Reconstitution of Rictor-null cells with myristoylated AKT (Myr-AKT) rescued vascular assembly in Rictor-deficient endothelial cells, whereas PKCα rescued proliferation defects. Furthermore, tumor neovascularization in vivo was significantly decreased upon EC-specific Rictor deletion in mice. These data indicate that mTORC2 is a critical signaling node required for VEGF-mediated angiogenesis through the regulation of AKT and PKCα in vascular endothelial cells.
INTRODUCTION
Blood vessels supply oxygen and nutrients for tissue growth and repair. In response to hypoxia, ischemia, or developmental cues, new capillary sprouts are formed from preexisting vessels in a complex process called angiogenesis. Critical steps for angiogenesis include endothelial tip cell migration, stalk cell proliferation, vascular sprout coalescence into tubular structures, stabilization of newly formed vessels by deposition of basement membrane, recruitment of perivascular supporting cells, and initiation of blood flow (reviewed in references 1–3). Each of these events is tightly regulated at the molecular level during normal development and tissue maintenance, and these same molecular regulators are often exploited during angiogenesis-dependent diseases such as cancer, inflammatory disorders, and retinopathy.
Angiogenesis is regulated by a complex interplay between proangiogenic and antiangiogenic factors. A major signaling event downstream of proangiogenic factors such as vascular endothelial growth factor (VEGF) is the activation of AKT (4–6), which is regulated by phosphoinositide-dependent kinase 1 (PDK1) and mammalian target of rapamycin (mTOR) complex 2 (mTORC2). mTOR is a serine/threonine kinase that regulates a diverse array of cellular processes, including cell growth, survival, metabolism, and cytoskeleton dynamics (reviewed in references 7 and 8). mTOR functions in two distinct complexes, mTORC1 and mTORC2, whose activities and substrate specificities are regulated by complex specific cofactors, including Raptor and Rictor, respectively. Targets downstream of mTORC1 regulate protein and lipid synthesis as well as energy metabolism. Key molecular targets of mTORC1 include 4E-BP1, p70 S6K1, and mediators of lipid synthesis (8). In contrast, much less is known about the mTORC2 signaling pathway. mTORC2 phosphorylates a conserved hydrophobic motif (HM) in each AKT isoform, serving as an AKT “S473” kinase (9). mTORC2 also activates additional members of the AGC subfamily of kinases, including SGK1 and protein kinase Cα (PKCα), regulating cell viability and cytoskeletal organization (10, 11).
Signaling of mTORC1 and, to a lesser extent, mTORC2 has been extensively studied in metabolic diseases and cancer. However, very little is known regarding the relative contributions of mTORC1 and mTORC2 signaling in vasculature. Phung et al. showed previously that pathological angiogenesis induced by sustained AKT signaling can be inhibited by rapamycin (12), demonstrating the importance of mTOR signaling in neovascularization. Moreover, hypoxia induces transient mTORC1 activity but sustained mTORC2 activity in vascular endothelial cells (ECs), further suggesting the relevance of mTORC2 activity in angiogenesis (13). Accordingly, activated vasculature represents a good target for mTOR inhibition. Rapamycin and its analogues (rapalogues) have been associated with limited efficacy in cancer and other diseases due to a relief of negative-feedback inhibition of several oncogenic pathways (11, 14). As a result, mTOR kinase inhibitors that inhibit both mTORC1 and mTORC2 have been developed. These compounds have been shown to reduce VEGF production and angiogenesis in several animal models (15). However, the specific impact of these agents on tumor vasculature cannot be determined due to their simultaneous effects on both complexes in both tumor and endothelial cells.
To understand the relative contributions of mTORC1 and mTORC2 function to angiogenesis, we analyzed conditional loss-of-function models harboring floxed alleles encoding either the essential mTORC1 subunit Raptor or the mTORC2 subunit Rictor (16, 17). Rictor ablation inhibited endothelial cell proliferation and assembly in vitro as well as subcutaneous angiogenesis and tumor neovascularization in vivo. In contrast, the loss of Raptor had only a modest effect on endothelial cells. Biochemical analysis revealed that mTORC2 deficiency affected both AKT activity and the phosphorylation of PKCα. Interestingly, constitutively active AKT rescued vascular assembly but not proliferation defects in Rictor-deficient endothelial cells, whereas PKCα led to a reciprocal outcome. Thus, we have identified that distinct mTORC2 targets can selectively regulate unique steps in angiogenic remodeling, revealing an important branch point downstream of this signal-transducing complex in vascular endothelial cells.
MATERIALS AND METHODS
Mice, adenoviruses, and reagents.
Rictorfl/fl mice were provided by Mark Magnuson (Vanderbilt University, Nashville, TN) (18). Raptorfl/fl mice were generated in Sabatini laboratory and purchased from Jackson Laboratories (Bar Harbor, ME) (19). Endothelial SCL-CreERT2 transgenic mice were provided by Joachim Gothert (University Hospital Essen, Essen, Germany) (20).
Ad5-CMV-AKT1 (myristoylated) (catalogue number 1020) (1 × 1010 PFU ml−1) and Ad5-CMV-PKCα (catalogue number JMAd-68) (1 × 108 PFU ml−1) adenoviruses were purchased from Vector Biolabs and Seven Hills Bioreagents, respectively. Cre adenovirus was generously provided by Rebecca Cook (Vanderbilt University, Nashville, TN). Mouse recombinant VEGF protein (catalogue number 493-MV), basic fibroblast growth factor (bFGF) (catalogue number 3139-FB-025), and human angiopoietin 1 (Ang1) (catalogue number 923-AN-025) were purchased from R&D Systems and used at 20 to 200 ng/ml, as indicated for different assays. The AKT inhibitor 5J8/0360263-1 was a kind gift from Craig Lindsley (Vanderbilt University, Nashville, TN) and was described previously (21). The mTOR inhibitor rapamycin was purchased from Calbiochem (catalogue number 553211) and used at 5 nM. A phosphatase inhibitor, calyculin A (catalogue number CAS 101932-71-2), was purchased from Calbiochem and used at 50 ng ml−1. Tamoxifen (catalogue number T5648) (15 mg ml−1) and fluorescein isothiocyanate (FITC)-labeled dextran (catalogue number 74817) (150 kDa; 2%) were obtained from Sigma and reconstituted in sunflower seed oil and 1× phosphate-buffered saline (PBS), respectively.
Mouse monoclonal anti-Rictor (catalogue number 05-1471; Millipore) and anti-β-tubulin (catalogue number T4026; Sigma) and rabbit polyclonal anti-phosphorylated PKCα (anti-p-PKCα) (S657) (catalogue number ab23513; Abcam), anti-p-PKCε (S729) (catalogue number SC-12355; Santa Cruz), and anti-phospholipase C-γ1 (anti-PLC-γ1) (catalogue number SC-81; Santa Cruz) antibodies were purchased and used for Western blotting at 1:1,000, 1:2,500, 1:1,000, 1:1,000, and 1:5,000 dilutions, respectively. Rabbit polyclonal antibodies against Raptor (catalogue number 2280), p-AKT (S473) (catalogue number 4060), total AKT (catalogue number 2920), p-NDRG1 (T346) (catalogue number 5482), p-S6K1 (T389) (catalogue number 9234), total S6K1 (catalogue number 9202), p-PLC-γ1 (Y783) (catalogue number 14008), p-PKCδ (T505) (catalogue number 9374), p-p38 (T180/Y182) (catalogue number 9211), total p38 mitogen-activated protein kinase (MAPK) (catalogue number 9212), total PKCα (catalogue number 2056), p-PDK1 (S241) (catalogue number 3061), total PDK1 (catalogue number 5662), phosphorylated endothelial nitric oxide synthase (p-eNOS) (S1177) (catalogue number 9571), and total eNOS (catalogue number 9572) and rabbit monoclonal antibodies against p-AKT (T308) (catalogue number 4056), p-4E-BP1 (T37/46) (catalogue number 2855), total 4E-BP1 (catalogue number 9644), and total NDRG1 (catalogue number 9408) were purchased from Cell Signaling Technology and used according to the manufacturer's protocols.
Cell culture, viral transduction, and siRNA transfection.
Murine pulmonary microvascular endothelial cells (MPMECs) were isolated from 1- to 3-month-old Rictorfl/fl or Raptorfl/fl mice and maintained in EGM-2 medium (Lonza) for <5 passages, as described in our previous studies (22–25). For adenovirus-mediated expression of Cre (Ad-Cre) or Ad-LacZ, MPMECs were seeded into 10-cm dishes at 70 to 80% confluence and infected with 107 PFU ml−1 virus for 48 h prior to biological assays and Western blot analysis.
For assays involving PKCα silencing, 2 × 105 MPMECs were plated onto a 6-cm plate at ∼50% confluence and transfected with 50 pmol of ON-TARGETplus SMARTpool small interfering RNA (siRNA) against mouse PKCα (catalogue number L-040348-00-0005) or ON-TARGETplus Non-Targeting pool siRNA (catalogue number D-001810-10-05) (Dharmacon), using Lipofectamine RNAiMAX transfection reagent (Invitrogen). Forty-eight hours after transfection, cells were serum starved with EBM-2 medium containing 0.2% fetal bovine serum (FBS) overnight and subjected to proliferation or vascular assembly assays as described below.
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza and maintained in EGM-2 medium (Lonza) for <10 passages. For signaling analysis, 2 × 105 HUVECs on 6-cm plates were transfected with 50 pmol of siRNAs against Raptor (catalogue number D-004107-03-0005; Dharmacon), Rictor (catalogue number D-016984-01-0005; Dharmacon), or the no-target control by using Lipofectamine RNAiMAX reagent. Forty-eight hours after transfection, cells were serum starved overnight, stimulated with VEGF (20 ng ml−1), and harvested for Western blotting. For signaling analysis involving adenovirus infection, HUVECs were treated with control siRNA or siRNA against Rictor (siRictor) for 24 h, followed by viral transduction (Ad-myristoylated AKT [Myr-AKT] or Ad-PKCα) for another 24 h. Cells were then serum starved and stimulated with VEGF for signaling analysis.
Western blot analysis.
MPMECs transduced with either Ad-Cre or Ad-LacZ were serum starved in EBM-2 medium containing 0.2% FBS overnight and stimulated with recombinant VEGF (20 ng ml−1) following a time course. Cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, protease inhibitor cocktail [Sigma], and phosphatase inhibitor cocktail tablet [Roche]) for Western blot analysis. Thirty to fifty micrograms of proteins from clarified lysates was separated by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked in 5% nonfat dry milk in TBS-T buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween 20) for 30 min, followed by incubation with primary antibody in blocking buffer from 1 h to overnight at room temperature. Blots were washed three times with TBS-T buffer and then incubated with horseradish peroxidase-conjugated secondary antibodies (Promega) for 1 h. Signals were detected by using Clarity Western ECLSubstrate (Bio-Rad).
BrdU-labeled cell proliferation assay.
MPMECs transduced with either Ad-Cre or Ad-LacZ were grown on coverslips in EBM-2 medium containing 0.2% FBS for 24 h, followed by incubation with the same medium containing bromodeoxyuridine (BrdU) (10 ng ml−1) in the presence or absence of recombinant VEGF (20 ng ml−1) for an additional 16 h. The incorporation of BrdU into proliferating cells was detected by using the In Situ Cell Proliferation kit (Roche) according to the manufacturer's instructions. Briefly, cells were fixed with 2% paraformaldehyde for 20 min and permeabilized with 1% Triton X-100 in PBS for 5 min. DNA was denatured by incubation with 2 N HCl for 30 min at 37°C, followed by five rinses in PBS to bring the pH back to 7.0. Cells on coverslips were blocked for 30 min by using 3% bovine serum albumin (BSA) in PBS and then probed with a fluorochrome-conjugated anti-BrdU antibody (1:15; Roche) in a humidified chamber at 37°C for 60 min. After several washes with PBS, coverslips were mounted onto slides by using ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Images were taken by using a 40× objective on an Olympus inverted fluorescence microscope and processed by using the Cellsens Dimension software program. BrdU-positive cells were quantified by counting >200 cells per sample, and the proliferation index was calculated as the percentage of BrdU-positive nuclei/total nuclei.
In vitro vascular assembly assay.
In vitro vascular assembly assays were performed as described previously (22, 23). Briefly, 24-well plates were coated with 100 μl of growth factor-reduced Matrigel (Becton-Dickinson) for 30 min at 37°C. MPMECs transduced with either Ad-Cre or Ad-LacZ were serum starved in EBM-2 medium containing 0.2% FBS overnight. A total of 3.5 × 104 cells were plated into each well of a 24-well plate in the presence or absence of VEGF (20 ng ml−1). Vascular assembly into capillary-like structures was documented after 16 h. Images were acquired on an Olympus CK40 inverted microscope through an Optronics DEI-750C charge-coupled-device (CCD) video camera using the Cellsens Dimension software program. The degree of assembly was quantified by counting the number of intersections between branches in assembled endothelial cell networks per 10× field in 3 independent fields in each well, with triplicate samples per condition. In some experiments, MPMECs were transduced with Myr-AKT (0.25 × 107 PFU ml−1)- or PKCα (0.25 × 105 PFU ml−1)-expressing adenoviruses for 48 h prior to assembly assays.
Endothelial cell in vitro permeability assay.
An endothelial cell monolayer permeability assay was performed as described previously (26, 27). Briefly, 1 × 105 MPMECs were plated onto one insert of a transwell plate with 3.0-μm pores (catalogue number 354575; Corning) in EGM-2 growth medium. Seeding of endothelial cells was repeated 24 h later. Forty-eight hours after the first seeding, cells were serum starved with EBM-2 medium containing 0.2% FBS for 4 h. VEGF (20 ng ml−1) and FITC-labeled dextran (70 kDa; 250 μg ml−1) (catalogue number 46945-100MG-F; Sigma-Aldrich) were simultaneously added into the lower chamber of each well. Twenty-microliter aliquots of medium were removed from the upper inserts every 30 min and diluted in 80 μl water/well in a 96-well assay plate. The fluorescence intensity of FITC-labeled dextran that passed through the inserts was measured by using a Synergy HT multiwell plate reader (Bio-TEK Instrument Inc.) with fluorescence settings (485 nm for excitation and 520 nm for emission).
In vivo sponge angiogenesis assay.
All animals were housed under pathogen-free conditions, and experiments were performed in accordance with AAALAC guidelines and with Vanderbilt University Institutional Animal Care and Use Committee approval. Rictorfl/fl or Raptorfl/fl animals were crossed to endothelial SCL-CreERT2 transgenic mice. Raptorfl/fl (wild type [WT]), SCL-CreERT2 Raptorfl/fl (Raptor knockout [KO]), Rictorfl/fl (WT), or SCL-CreERT2 Rictorfl/fl (Rictor KO) mice were given tamoxifen (3 mg/animal) every other day via intraperitoneal injection for 14 days. At day 7, each recipient animal received one VEGF (100 ng ml−1)-impregnated surgical sponge and one PBS control sponge implanted in the contralateral flank, as described previously (22). A week after implantation, the mice were injected with a 2% FITC–dextran–phosphate-buffered saline solution to visualize host blood vessels (22, 23), after which the sponges were collected and analyzed. Whole-mount images were acquired on an Olympus CK40 inverted microscope with an Optronics DEI-750C charge-coupled-device video camera using the Cellsens Dimension software program. Blood vessels within the sponges were quantified by the FITC-dextran fluorescence pixel area (×10 magnification). Data represent results from five independent sponges under each condition. Statistical significance was determined by a two-tailed, paired Student t test. Vessel identity was confirmed in paraffin sections prepared from sponges by costaining with the endothelial cell marker von Willebrand factor (vWF) (28–30).
Tumor allograft studies.
To evaluate endothelial cell-specific roles of Rictor in tumor growth, 8-week-old Rictorfl/fl (wild-type control) or SCL-CreERT2 Rictorfl/fl mice were given tamoxifen (3 mg/animal) every other day via intraperitoneal injection for 14 days. At day 10, 2 × 105 Lewis lung carcinoma (LLC) cells were injected subcutaneously into SCL-CreERT2 Rictorfl/fl or wild-type control recipient mice. Tumor progression was monitored by palpation, and tumor size was measured by a digital caliper. Tumor volume was calculated by using the following formula: volume = length × width2 × 0.52. At the end of the experiments, tumors were harvested, and tumor sections were prepared for histological analyses. Tumor cell proliferation was quantified by calculating the average percentage of PCNA-positive (PCNA+) nuclei relative to total nuclei (four fields of at least five tumors per genotype or treatment condition were assessed). Apoptosis assays were performed by using the Apoptag Red in situ apoptosis detection kit according to the manufacturer's protocol (Chemicon International). Apoptosis was measured as the percentage of terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL)-positive nuclei relative to the total number of nuclei (four fields from at least five independent tumors per genotype or treatment condition were assessed). Immunofluorescence staining for von Willebrand factor was performed as described previously (28). Tumor vessel density was determined by assessing the vWF+ vessels/pixels in four fields per sample of at least five independent tumors per genotype or treatment condition. Antibodies against the following proteins were used: biotin-conjugated anti-PCNA (catalogue number 555567; Pharmingen), vWF (catalogue number A0082; Dako Cytomation), and anti-rabbit antibody–Alexa Fluor 594 (catalogue number A-110-12; Life Technologies). Additionally, Retrievagen A (pH 6.0) (catalogue number 550524; BD Pharmingen), streptavidin peroxidase reagents (catalogue number 51-75477E; BD Pharmingen), and a liquid 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate kit (Zymed Laboratories) were used. Cytoseal XYL (Richard Allan Scientific) or ProLong Gold antifade reagent with DAPI (Life Technologies) was used to mount slides.
RESULTS
Rictor is critical for endothelial cell proliferation and vascular assembly.
To investigate the relative contributions of Raptor and Rictor in vascular endothelial cells, murine primary pulmonary microvascular endothelial cells (MPMECs) were isolated from Raptorfl/fl or Rictorfl/fl mice and transduced with either LacZ- or Cre-expressing viruses to generate Rictor- or Raptor-deficient endothelial cells. To analyze the role of mTORC2 versus mTORC1 in angiogenic responses, we measured endothelial cell proliferation and vascular assembly in Rictor-deficient and Raptor-deficient MPMECs. The loss of Rictor inhibited VEGF-induced proliferation, whereas the loss of Raptor had only a modest effect on cell proliferation (Fig. 1A). Next, we measured the effects of Rictor or Raptor loss on endothelial cell assembly into interconnected tubelike structures on a matrix (vascular assembly), a key step in the angiogenic process. As shown in Fig. 1B, vascular assembly in response to VEGF was significantly impaired in Rictor-deficient MPMECs relative to wild-type control cells. In contrast, VEGF-induced assembly was increased in Raptor-deficient endothelial cells. To test the effects of Raptor and Rictor on the regulation of the flow of small molecules through endothelial monolayers, we performed an in vitro vascular permeability assay. The loss of either Raptor or Rictor reduced VEGF-induced cell permeability (Fig. 1C). Collectively, these results indicate that mTORC2 function is required for VEGF-induced angiogenic responses, whereas mTORC1 has a relatively modest effect on vascular endothelial cell function.
FIG 1.

Rictor is required for endothelial cell proliferation, vascular assembly, and permeability. Primary microvascular endothelial cells were isolated from Rictorfl/fl or Raptorfl/fl mice, and targeted deletion of the Rictor or Raptor allele was induced by transducing cells with Cre- or control LacZ-expressing adenoviruses. Cells were serum starved for 24 h and stimulated with VEGF (20 ng/ml). Data from three independent experiments were pooled and are presented as means ± standard errors of the means. P values of <0.05 were considered to be statistically significant (two-tailed Student t test). WT, wild type; KO, knockout. (A) Cell proliferation was assessed by BrdU incorporation. Proliferation indices were calculated as the percentage of BrdU+ nuclei/total nuclei. Arrowheads indicate BrdU-positive cells. (B) Vascular assembly on Matrigel was measured as described in Materials and Methods. The degree of assembly was quantified by enumerating branching points of the assembled vascular network. (C) Cell permeability was assayed as described in Materials and Methods. Representative kinetics of dextran-FITC that passed through the monolayer of endothelial cells with a Raptor or Rictor deletion are shown. *, P < 0.05; **, P < 0.01.
mTORC1 and mTORC2 signaling in wild-type, Rictor-deficient, or Raptor-deficient primary endothelial cells in response to VEGF stimulation was assessed by Western blot analyses. The loss of Rictor did not affect the expression of Raptor, a key component of mTORC1, but significantly decreased the phosphorylation levels of AKT at both residues S473 and T308 (Fig. 2B). S473 of AKT is a major downstream target of mTORC2, and decreased phosphorylation levels at S473 validate that the loss of Rictor impairs mTORC2 signaling. Although T308 is not a direct target of mTORC2, reduced phosphorylation at T308 upon Rictor ablation suggests that it can be indirectly regulated by mTORC2. However, despite the decreased phosphorylation of AKT at T308, phosphorylation of S6K1 (T389), a key signaling component downstream of mTORC1, was not affected significantly (Fig. 2B). In contrast, the phosphorylation of NDRG1 (T346), a downstream target of SGK1, and the phosphorylation of PKCα (S657) were markedly decreased (Fig. 2B), suggesting that the loss of Rictor attenuates mTORC2 signaling without affecting mTORC1 signaling. In comparison, the loss of Raptor reduced the phosphorylation of S6K1 without affecting the phosphorylation of PKCα and NDRG1, indicating an inhibition of mTORC1 but not mTORC2 signaling (Fig. 2A). However, the phosphorylation of AKT at both the S473 and T308 sites was increased upon the loss of Raptor, consistent with previously reported findings that an inhibition of mTORC1 signaling alone leads to a relief of negative-feedback inhibition (11). Other signaling molecules known to be regulated by VEGF, such as PKCδ, PKCε, extracellular signal-regulated kinase 1/2 (ERK1/2), PLC-γ, and p38 MAPK, were not affected by Rictor or Raptor deficiency in endothelial cells (Fig. 2).
FIG 2.

Cell signaling in Rictor- or Raptor-deficient primary endothelial cells. Primary microvascular endothelial cells were isolated from Rictorfl/fl or Raptorfl/fl mice, and targeted deletion of the Rictor or Raptor allele was induced by transducing cells with Cre- or control LacZ-expressing adenoviruses, as described in the legend to Fig. 1. Raptor KO (A), Rictor KO (B), or WT control cells were serum starved overnight and stimulated with VEGF (20 ng/ml) for 0, 5, 15, or 30 min, and lysates were harvested for signaling analysis using the indicated antibodies. Experiments were repeated 2 to 3 times, and representative blots for each signaling component are shown.
To address whether the mTORC2 signaling changes upon Rictor ablation in angiogenesis are specific to VEGF stimulation, we also treated cells with bFGF or angiopoietin 1 (Ang1) (Fig. 3). Stimulation with either bFGF or Ang1 induced largely similar results for endothelial cell signaling in Rictor-null and Raptor-null cells, relative to VEGF stimulation, suggesting that mTOR is a common signaling node for proangiogenic factors.
FIG 3.

bFGF- or Ang1-mediated cell signaling in Raptor- and Rictor-deficient primary endothelial cells. Primary microvascular endothelial cells were isolated from Rictorfl/fl or Raptorfl/fl mice, and targeted deletion of the Rictor or Raptor allele was induced by transducing cells with Cre- or control LacZ-expressing adenoviruses, as described in the legend to Fig. 2. Cells were serum starved overnight and stimulated with bFGF (10 ng ml−1) (A and B) or angiopoietin 1 (200 ng ml−1) (C and D). Cell lysates were harvested at 0, 5, 15, or 30 min for signaling analysis using the indicated antibodies.
Myr-AKT rescues vascular assembly in Rictor-deficient endothelial cells without affecting cell proliferation.
AKT is known to regulate cell growth, survival, and motility in many cell types. Because AKT (S473) phosphorylation levels were markedly decreased in Rictor knockout endothelial cells (Fig. 2B), we investigated whether activated AKT can rescue growth and/or assembly phenotypes in Rictor-deficient endothelial cells. We used constitutively activated myristoylated AKT (Myr-AKT), which bypasses the physiological role of PH domain-mediated binding to PIP3 (31). While Myr-AKT rescued the defects in vascular assembly in Rictor KO cells (Fig. 4B), surprisingly, it had no effect on cell proliferation (Fig. 4A), indicating that activated AKT alone is not sufficient for endothelial cell proliferation. Accordingly, we examined two other downstream targets of mTORC2. As shown in Fig. 4C, the expression of Myr-AKT rescued phosphorylation levels of NDRG1 but not PKCα in Rictor-deficient endothelial cells, suggesting that PKCα may represent a distinct signaling arm downstream of mTORC2 that may be required for proliferation in vascular endothelial cells.
FIG 4.

Myr-AKT rescues vascular assembly in Rictor-deficient endothelial cells without affecting cell proliferation. (A and B) WT or Rictor KO endothelial cells were transduced with control LacZ- or Myr-AKT-expressing adenovirus. Two days after infection, cells were subjected to BrdU incorporation (A) and assembly (B) assays to measure cell proliferation and the formation of a tubule-like vascular network, respectively. (C) Expression of Rictor, Myr-AKT, and signaling molecules was analyzed by Western blotting using the indicated antibodies. Experiments were repeated 3 times, and data were pooled for statistical analyses. Arrowheads in panel A indicate BrdU-positive nuclei. In the case of Western blot analysis, representative blots for each signaling component are shown in panel C. **, P < 0.01.
Because increased vascular assembly in Raptor-null endothelial cells was associated with increased phosphorylation levels of AKT at both the T308 and S473 sites (Fig. 2A), we investigated if upregulated AKT activity was responsible for the increased vascular network morphogenesis in Raptor knockout cells using an allosteric AKT inhibitor, 5J8/0360263-1 (21). As expected, the inhibitor significantly abrogated the phosphorylation of AKT in endothelial cells (Fig. 5A). It also abolished vascular assembly in both WT and Raptor knockout endothelial cells (Fig. 5B). In addition, treatment of endothelial cells with rapamycin decreased AKT phosphorylation at S473 (Fig. 5A) and rescued the assembly phenotype in Raptor-null cells (Fig. 5C). Collectively, these data suggest that AKT plays a central role in vascular network formation in endothelial cells.
FIG 5.

Increased vascular assembly in Raptor-deficient endothelial cells is suppressed by an AKT inhibitor and the mTOR inhibitor rapamycin. (A) The effects of an AKT inhibitor, 5J8/0360263-1, and rapamycin on cell signaling were measured by Western blot analysis. Representative blots for each signaling component are shown. (B) WT or Raptor KO endothelial cells were treated with 1 μM an AKT inhibitor (AKT-in) or the vehicle control for the duration of the assembly assay. The degree of assembly was quantified by measuring branching points of the assembled vascular network. Experiments were repeated 3 times, and data were pooled for statistical analyses. (C) WT or Raptor KO endothelial cells were treated with 5 nM rapamycin (Rapa) or the vehicle control (dimethyl sulfoxide) for the duration of the assembly assay, as described above for panel B. **, P < 0.01.
Expression of PKCα rescues proliferation defects in Rictor-null cells.
To test if PKCα, alone or in combination with AKT, is required for proliferation in vascular endothelial cells, Rictor-deficient endothelial cells were transduced with viruses expressing PKCα either alone or in combination with Myr-AKT. Overexpression of PKCα alone did not affect phosphorylation levels of AKT or another mTORC2 downstream target, NDRG1, but did increase the phosphorylation of PKCα at S657. The expression of both PKCα and Myr-AKT upregulated the phosphorylation of AKT (S473), NDRG1 (T473), and PKCα (S657) (Fig. 6C). In contrast to Myr-AKT, PKCα alone had no effect on vascular assembly, but the addition of Myr-AKT restored vascular assembly in Rictor-deficient cells (Fig. 6B), consistent with our above-described finding that AKT plays an important role in vascular network formation (Fig. 4). Interestingly, PKCα alone or with Myr-AKT partially rescued the proliferation defects in Rictor-deficient endothelial cells (Fig. 6A). To complement our PKCα expression studies in Rictor-null cells, we knocked down PKCα via siRNA in wild-type endothelial cells. The depletion of PKCα significantly inhibited cell proliferation but had no effect on vascular assembly (Fig. 6D). Together, these results suggest that PKCα is critical for endothelial cell proliferation downstream of mTORC2.
FIG 6.

PKCα partially rescues endothelial cell proliferation in Rictor-deficient endothelial cells without affecting vascular assembly. (A and B) WT or Rictor KO endothelial cells were transduced with control LacZ-, PKCα-, or PKCα- and Myr-AKT-expressing adenoviruses. Two days after infection, cells were assayed to measure proliferation (A) or vascular assembly (B). Experiments were repeated 3 times, and data were pooled for statistical analyses (Student t test). Arrowheads indicate BrdU-positive nuclei. (C) The effects of PKCα and Myr-AKT on cell signaling were measured by Western blot analysis. Representative blots for each signaling component are shown. (D) PKCα was knocked down in primary endothelial cells by siRNA-mediated silencing. Knockdown of PKCα was confirmed by Western blot analysis. Forty-eight hours after transfection, cells were assayed for proliferation and vascular assembly. *, P < 0.05; **, P < 0.01; N.S., not significant.
Rictor is required for angiogenesis in vivo.
To analyze the role of mTORC1 and mTORC2 in angiogenesis in vivo, we initially crossed Rictor or Raptor floxed mice with transgenic animals expressing endothelial cell-specific Tie2-Cre. Analysis of ∼100 pups failed to identify Rictor-null animals, indicating embryonic lethality (Table 1). Likewise, the loss of Raptor also appeared to result in embryonic death (Table 1). These results suggest that both Rictor and Raptor play critical roles in embryonic angiogenesis.
TABLE 1.
Raptor or Rictor deletion in endothelial cells causes embryonic lethality in micea
| Genotype | No. of mice genotyped | % of mice with genotype |
|---|---|---|
| Raptor conditional knockout | ||
| Raptorfl/fl/Tie2-Cre | 0 | 0 |
| Raptorfl/fl | 29 | 44.6 |
| Raptorfl/+/Tie2 | 15 | 23.1 |
| Raptorfl/+ | 21 | 32.3 |
| Total | 65 | 100 |
| Rictor conditional knockout | ||
| Rictorfl/fl/Tie2-Cre | 0 | 0 |
| Rictorfl/fl | 31 | 29.5 |
| Rictorfl/+/Tie2-Cre | 36 | 34.3 |
| Rictorfl/+ | 38 | 36.2 |
| Total | 105 | 100 |
Wild-type female mice (Raptorfl/fl or Rictorfl/fl) were mated with male transgenic mice (Raptorfl/+/Tie2-Cre or Rictorfl/+/Tie2-Cre) to generate offspring with the Raptor or Rictor allele deleted in endothelial cells. Genotypes of pups were determined by PCR analysis, and the distribution of each genotype was calculated as a percentage of the total cohort.
To assess the role of Rictor or Raptor in adult tissue angiogenesis, we crossed Rictor floxed mice with inducible endothelial SCL-CreERT2 transgenic animals (20). Tamoxifen was administered to Rictorfl/fl or SCL-CreERT2; Rictorfl/fl mice to induce LoxP recombination. After 7 days of tamoxifen treatment, surgical sponges were impregnated with either VEGF or the PBS vehicle control into the opposing dorsal flanks of Rictorfl/fl (WT) or SCL-CreERT2; Rictorfl/fl (KO) recipient mice. One week following implantation, FITC-dextran was injected intravenously to visualize and quantify blood vessels in the sponge. As shown in Fig. 7A, tamoxifen treatment abolished Rictor expression in primary endothelial cells isolated from KO mice relative to WT animals. Sponges harboring VEGF stimulated a robust angiogenic response in WT mice, with a significant increase in the number of FITC-positive surface blood vessels (green), relative to sponges containing the vehicle alone. In contrast, VEGF failed to induce robust angiogenesis in EC-specific Rictor KO mice. To analyze vessels that infiltrated the sponges, paraffin sections prepared from sponges were stained with von Willebrand factor (vWF) (28–30), an endothelial cell marker, confirming that the FITC-positive structures observed were functional blood vessels. Collectively, these data suggest that mTORC2 function is required for adult tissue angiogenesis in vivo.
FIG 7.

Rictor is required for angiogenesis in vivo. (A) Rictorfl/fl mice were crossed to endothelial SCL-CreERT2 transgenic animals. Tamoxifen (3 mg/mouse) was administered by intraperitoneal injection for 14 days to induce LoxP recombination. Deletion of Rictor alleles in isolated primary vascular endothelial cells was verified by Western blot analysis. Surgical sponges impregnated with either VEGF or the vehicle were implanted in WT or EC-specific Rictor-deficient hosts subcutaneously in the dorsal flanks. Seven days after transplantation, FITC-dextran was injected via the tail vein, and sponges were harvested. Angiogenesis was quantified by measuring the fluorescent pixel area on the surface the sponge. Internal vessels were assessed by vWF staining in paraffin sections derived from sponges. Experiments were repeated 2 times, and data were pooled and analyzed statistically. Arrows indicate dextran-perfused functional blood vessels, and arrowheads indicate vWF-positive endothelial cells in the sponges. AU, arbitrary units. (B) Raptorfl/fl mice were crossed to endothelial-SCL-CreERT2 transgenic animals as described above for panel A. Raptor-deficient or control wild-type mice were subjected to a sponge angiogenesis assay as described above for panel A (n = 7/genotype). **, P < 0.01; N.S., not significant.
To compare the roles of Rictor and Raptor in adult angiogenesis, we also crossed Raptor floxed mice with inducible endothelial SCL-CreERT2 transgenic animals. However, Raptor expression was only modestly reduced in endothelial cells, as judged by Western blot analysis (Fig. 7B). Accordingly, we did not observe significant changes in VEGF-induced angiogenesis in vivo (Fig. 7B), suggesting that further studies are required to determine the role of Raptor in adult tissue angiogenesis.
Rictor deficiency impairs tumor growth and tumor vessel density in vivo.
To directly determine the in vivo role of Rictor in pathological angiogenesis, we transplanted Lewis lung carcinoma (LCC) cells into Rictor-deficient animals. Compared to wild-type mice, tumor growth in Rictor-deficient mice was greatly reduced (Fig. 8A). Tumor cell proliferation, as measured by PCNA immunohistochemistry, was significantly decreased in the endothelial cell-specific Rictor-deficient host (Fig. 8B), whereas tumor cell apoptosis, as measured by TUNEL staining, was unchanged (Fig. 8C). There was also a significant decrease in the number of tumor microvessels in situ, as judged by immunofluorescence detection of vWF to visualize endothelial cells (Fig. 8D). Collectively, these results show that endothelial Rictor is required for tumor growth and tumor neovascularization.
FIG 8.

Loss of Rictor in host vasculature inhibits tumor growth and angiogenesis in vivo. (A) SCL-CreERT2 Rictorfl/fl (KO) or WT control mice were given tamoxifen (3 mg/animal) every other day via intraperitoneal injection for 14 days. At day 10, 2 × 105 LLC cells were injected subcutaneously into Rictor KO or WT control recipient mice. Tumor progression was monitored by palpation, and tumor size was measured by a digital caliper. (B) Tumor cell proliferation assessed by PCNA immunohistochemistry. The proliferation index is presented as the percentage of PCNA-positive nuclei/total nuclei with standard errors of the means (n = 5 to 10 per genotype). Arrowheads indicate representative proliferating nuclei. (C) Apoptosis assessed by TUNEL staining. The apoptosis index is presented as the percentage of TUNEL-positive nuclei/total nuclei ± standard errors of the means (n = 5 to 10 per genotype). (D) Microvascular density was quantified and is presented as mean numbers of vWF-positive pixels (arrowhead) per section with standard errors of the means (n = 5 to 10 per genotype). *, P < 0.05; NS, not significant.
DISCUSSION
The serine/threonine kinase mTOR is a major signaling target downstream of growth factors and cytokines. However, the role of mTOR signaling in the vascular endothelium during angiogenic responses had remained undefined. Surprisingly, in this report, we have shown that mTORC2 is a major regulator of angiogenic processes in adult primary endothelial cells, whereas mTORC1 has only a modest effect. Dissection of the mTORC2 effects on proliferation and vascular assembly revealed an unexpected signaling hierarchy of this complex. Rictor-deficient primary endothelial cells display decreased cell proliferation and vascular assembly when stimulated with VEGF in vitro. Phosphorylation of AKT at S473, a direct target of mTORC2, and at T308, a target of PDK1, was attenuated in Rictor-deficient endothelial cells. In addition, the loss of Rictor inhibited the phosphorylation of PKCα and NDRG1, an SGK1 target. Activated AKT restored the phosphorylation of NDRG1 and vascular assembly but did not restore PKCα activity or proliferation. In contrast, activated PKCα resulted in reciprocal effects, restoring proliferation without significantly affecting vascular assembly. These data suggest that the primary role of mTORC2 in endothelial cells is to regulate proliferation and vascular assembly through two distinct signaling effectors, PKCα and AKT, respectively.
mTORC1 was originally identified as the key target of rapamycin, an immune suppressant that was subsequently found to inhibit tumor growth and angiogenesis (32). Although mTORC2 does not bind to rapamycin, prolonged treatment with rapamycin also inhibits mTORC2 (33, 34), probably due to the sequestration of mTOR in rapamycin-binding mTORC1, thereby reducing the availability of mTOR for mTORC2. Thus, the chronic effects of rapamycin or its analogues (rapalogues) on angiogenesis might be attributable to either mTORC1, mTORC2, or both. In addition, the recent development of mTOR kinase inhibitors that block both mTORC1 and mTORC2 (15) makes it difficult to distinguish the relative contributions of these distinct complexes in vascular endothelial cells. Using a mouse model of endothelial cell-specific gene targeting, we show that mTORC2 is critical for both cell proliferation and vascular assembly. Rictor-dependent angiogenic responses appear to be mediated dichotomously by the mTORC2 targets AKT and PKCα. While this study focuses on the role and signaling mechanism of mTORC2, mTORC1 likely contributes to VEGF-dependent angiogenesis independently. Indeed, cell proliferation is moderately decreased in Raptor-deficient endothelial cells (Fig. 1A), as is VEGF-induced cell permeability (Fig. 1C). Further studies are required to determine Raptor's role in adult tissue angiogenesis, as we were unable to achieve a complete deletion of Raptor in vascular endothelial cells in our conditional knockout model (Fig. 7B). In addition, knockdown of Raptor has also been shown to affect the viability of rat aortic endothelial cells in endothelial spheroids (13), and knockdown of S6K1 inhibited VEGF-induced angiogenesis in vivo (35). However, our studies provide a direct comparison between Rictor- and Raptor-deficient cells in vitro, where deletion is complete (Fig. 1), or between Rictor and Raptor knockdown in HUVECs (36) and demonstrated the essential role of mTORC2 in primary vascular endothelial cells.
Our data support a critical role of AKT downstream of mTORC2 in VEGF-induced endothelial cell assembly. The loss of Rictor markedly decreased vascular assembly in response to VEGF stimulation, accompanied by a decrease in the phosphorylation of AKT at both residues S473 and T308. These defects were rescued by constitutively activated Myr-AKT (Fig. 4B), suggesting that AKT activity is necessary and sufficient for VEGF-induced endothelial cell assembly downstream of Rictor-dependent mTORC2. This model is supported by data derived from direct targeting of AKT1 in endothelial cells, which showed that a loss of AKT1 impairs VEGF-induced endothelial cell migration and angiogenesis (6). Furthermore, increased vascular assembly in Raptor-null endothelial cells was inhibited by an AKT kinase inhibitor (Fig. 5B), suggesting a central role of AKT in vascular network formation. Interestingly, despite its known role in regulating cell growth, constitutively activated Myr-AKT alone does not appear to be sufficient to rescue proliferation defects in Rictor-null cells (Fig. 4A). Another mTORC2 target, PKCα, is required to rescue proliferation in Rictor-deficient endothelial cells (Fig. 6A).
It has been reported that critical roles of AKT and PKCα in angiogenesis can be mediated by their downstream effector eNOS (37, 38). Accordingly, impaired nitric oxide (NO) synthesis could account for the defect in proliferation and vascular assembly observed upon Rictor deletion. We investigated the effects of the Rictor or Raptor deficiency on eNOS phosphorylation in response to VEGF stimulation. As shown in Fig. 9, the loss of Rictor or Raptor markedly inhibited the phosphorylation of eNOS, which could be rescued by the reexpression of Myr-AKT or PKCα in Rictor-null cells, confirming that eNOS is one of the downstream effectors of AKT and PKCα. However, because p-eNOS levels are decreased in both Rictor- and Raptor-deficient cells, which have very different phenotypes, it is unlikely that the change in eNOS alone accounts for the observed defects in endothelial cell proliferation and vascular assembly in Rictor KO cells.
FIG 9.

Raptor and Rictor are required for eNOS activity in endothelial cells. (A) HUVECs were transfected with siRNAs against Raptor (Raptor knockdown [KD]) or Rictor (Rictor KD) or with scrambled siRNA (WT control). Forty-eight hours after transfection, cells were serum starved in EBM-2 medium containing 0.2% FBS overnight and stimulated with VEGF (20 ng/ml) for 0, 5, 15, and 30 min. VEGF-mediated signaling in cell lysates was analyzed by using the indicated antibodies. (B and C) WT or Rictor knockdown HUVECs were transduced with adenoviruses expressing control LacZ, Myr-AKT (B), or PKCα (C). Twenty-four hours after infection, cells were serum starved overnight, stimulated with VEGF, and subjected to Western blot analysis.
It is well established that S473 of AKT is a direct substrate of mTORC2, whereas T308 is phosphorylated by PDK1. Therefore, it is surprising that the loss of Rictor also reduced the phosphorylation of the T308 site on AKT in endothelial cells. Similar results, however, were reported previously, demonstrating that the phosphorylations of S473 and T308 are often coregulated (39–41). We did not find significant changes in levels of activated PDK1, suggesting that upstream signaling was not altered. Both protein phosphatase 1 (PP1) and PP2A are known to associate with AKT and have been implicated in its dephosphorylation (42–47). Treatment of Rictor KO cells with a PP1/PP2A phosphatase inhibitor, calyculin A, markedly increased the phosphorylation of AKT at T308 and S473 (Fig. 10). While these data confirmed that PP1/PP2A can dephosphorylate AKT, it remains to be determined whether PP1/PP2A is regulated by Rictor.
FIG 10.
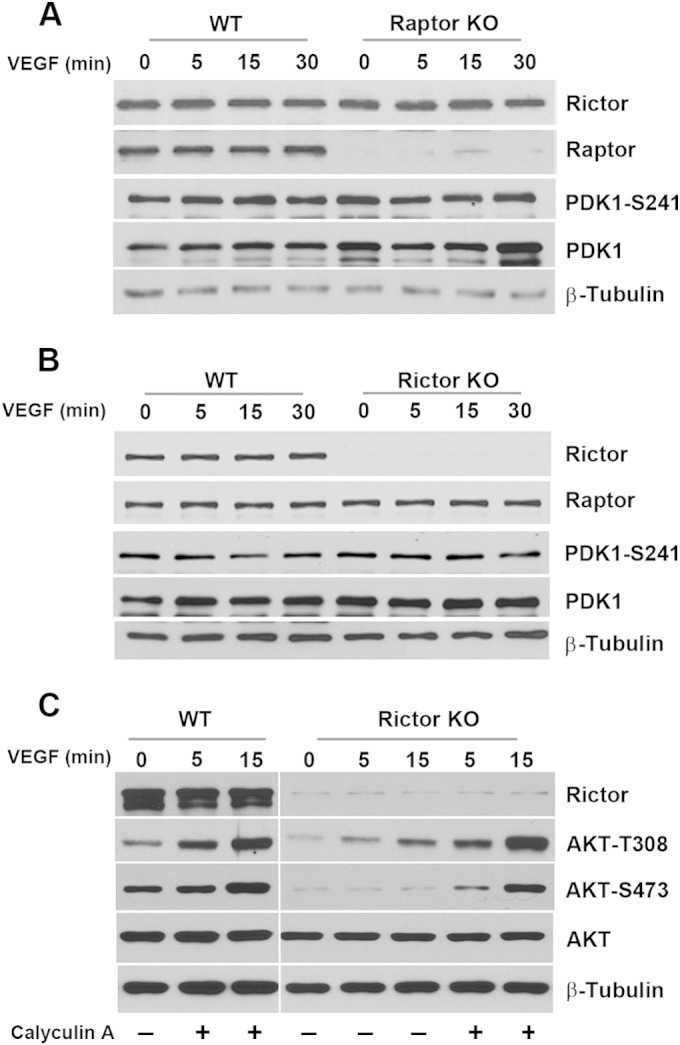
Regulation of AKT phosphorylation at residue T308 in endothelial cells. (A and B) Rictor- or Raptor-deficient primary vascular endothelial cells were serum starved and stimulated with VEGF. Phosphorylation of PDK1 was assessed by Western blot analysis. (C) WT or Rictor KO endothelial cells were pretreated with a phosphatase inhibitor, calyculin A (50 ng ml−1), or control dimethyl sulfoxide for 15 min, followed by stimulation with VEGF for the indicated times. Cells were harvested for Western blot analysis using the indicated antibodies.
The data presented in this study showed that a loss of Rictor in vascular endothelial cells inhibits mTORC2 signaling and endothelial cell function. However, we cannot exclude a role of Rictor in mTORC2-independent function in endothelial cells. For example, Rictor can interact with integrin-linked kinase (ILK) to regulate AKT phosphorylation and cancer cell survival (48). Moreover, a second mTOR-independent Rictor function has been discovered in Dictyostelium, where Rictor regulates cell migration by suppressing Rho-GDP dissociation inhibitor 2 (RhoGDI2) (49). Further studies are needed to determine the mTORC2-dependent and -independent functions of Rictor in vascular endothelial cells.
Our study on the mTORC2 function in vascular endothelial cells and VEGF-induced angiogenesis has broad implications for vascular diseases and cancer therapeutics. Allosteric inhibitors of mTORC1, rapamycin and rapalogues, have been developed. However, these drugs have limited efficacy due to the relief of negative-feedback inhibition of several receptor tyrosine kinase pathways (14, 50). Understanding the role of mTORC1 versus mTORC2 will allow more precise strategies to maximize efficacy and minimize toxicity while selectively “fine-tuning” vascular response characteristics. Furthermore, despite the promise of mTOR kinase inhibitors and mTOR-AKT dual inhibitors, these agents affect diverse signaling networks as well as energy metabolism in the body, which may have long-term and currently unknown effects on human health. Targeting mTORC2 specifically may offer an independent therapeutic approach with potentially fewer inherent toxicities. Such inhibitors could have an enhanced therapeutic window while avoiding perturbation of the mTORC1-dependent negative-feedback loops. Because there are currently no specific inhibitors of mTORC2 that do not affect mTORC1, genetic ablation of Rictor allows us to model mTORC2 inhibition specifically in the vascular endothelium during tumorigenesis and progression. These studies provide a rationale for the development of mTORC2-specific inhibitors.
ACKNOWLEDGMENTS
We thank Yoonha Hwang and Tammy Sobolik for helping to manage the mouse colonies, teaching techniques, and assisting in various aspects of experiments. We also acknowledge Mark Magnuson (Vanderbilt University, Nashville, TN), Joachim Gothert (University Hospital Essen, Germany), and Rong Wang (UCSF, San Francisco, CA) for providing Rictorfl/fl, endothelial SCL-CreERT2, and Tie2-Cre mice, respectively, and Rebecca Cook (Vanderbilt University, Nashville, TN) for providing Cre-expressing adenovirus.
This work was supported by the Department of Veterans Affairs through a VA merit award (J.C.) and by NIH grants R01 CA95004, CA177681 (J.C.), and CA148934 (D.M.B.-S.). This work was also supported in part by NCI Cancer Center support grant P30 CA068485, utilizing the Translational Pathology Shared Resources.
REFERENCES
- 1.Potente M, Gerhardt H, Carmeliet P. 2011. Basic and therapeutic aspects of angiogenesis. Cell 146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Eilken HM, Adams RH. 2010. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22:617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Geudens I, Gerhardt H. 2011. Coordinating cell behaviour during blood vessel formation. Development 138:4569–4583. doi: 10.1242/dev.062323. [DOI] [PubMed] [Google Scholar]
- 4.Zachary I, Gliki G. 2001. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res 49:568–581. doi: 10.1016/S0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 5.Jiang BH, Liu LZ. 2008. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets 8:19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 6.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. 2005. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest 115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laplante M, Sabatini DM. 2012. mTOR signaling. Cold Spring Harb Perspect Biol 4:a011593. doi: 10.1101/cshperspect.a011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 10.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. 2004. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 11.Sparks CA, Guertin DA. 2010. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 29:3733–3744. doi: 10.1038/onc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, Dvorak HF, Benjamin LE. 2006. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Petrimpol M, Molle KD, Hall MN, Battegay EJ, Humar R. 2007. Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ Res 100:79–87. doi: 10.1161/01.RES.0000253094.03023.3f. [DOI] [PubMed] [Google Scholar]
- 14.Wander SA, Hennessy BT, Slingerland JM. 2011. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. 2011. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71:1573–1583. doi: 10.1158/0008-5472.CAN-10-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. 2006. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. 2006. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. 2011. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 60:827–837. doi: 10.2337/db10-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. 2011. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, Green AR, Gottgens B, Izon DJ, Begley CG. 2004. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood 104:1769–1777. doi: 10.1182/blood-2003-11-3952. [DOI] [PubMed] [Google Scholar]
- 21.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. 2005. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett 15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Brantley-Sieders D, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. 2004. EphA2 receptor tyrosine kinase regulates endothelial cell migration and assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci 117:2037–2049. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 23.Hunter SG, Zhuang G, Brantley-Sieders DM, Swatt W, Cowan CW, Chen J. 2006. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol 26:4830–4842. doi: 10.1128/MCB.02215-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang WB, Brantley-Sieders DM, Hwang Y, Ham AJ, Chen J. 2008. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J Biol Chem 283:16017–16026. doi: 10.1074/jbc.M709934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brantley-Sieders DM, Dunaway CM, Rao M, Short S, Hwang Y, Gao Y, Li D, Jiang A, Shyr Y, Wu JY, Chen J. 2011. Angiocrine factors modulate tumor proliferation and motility through EphA2 repression of Slit2 tumor suppressor function in endothelium. Cancer Res 71:976–987. doi: 10.1158/0008-5472.CAN-10-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martins-Green M, Petreaca M, Yao M. 2008. An assay system for in vitro detection of permeability in human “endothelium.” Methods Enzymol 443:137–153. doi: 10.1016/S0076-6879(08)02008-9. [DOI] [PubMed] [Google Scholar]
- 27.Romon R, Adriaenssens E, Lagadec C, Germain E, Hondermarck H, Le Bourhis X. 2010. Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Mol Cancer 9:157. doi: 10.1186/1476-4598-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. 2005. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J 19:1884–1886. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- 29.Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, Coffman K, Jackson D, Bruckheimer E, Muraoka-Cook RS, Chen J. 2008. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest 118:64–78. doi: 10.1172/JCI33154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brantley-Sieders DM, Zhuang G, Vaught D, Freeman T, Hwang Y, Hicks D, Chen J. 2009. Host deficiency in Vav2/3 guanine nucleotide exchange factors impairs tumor growth, survival, and angiogenesis in vivo. Mol Cancer Res 7:615–623. doi: 10.1158/1541-7786.MCR-08-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohn AD, Takeuchi F, Roth RA. 1996. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem 271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 32.Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK. 2002. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 33.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Barilli A, Visigalli R, Sala R, Gazzola GC, Parolari A, Tremoli E, Bonomini S, Simon A, Closs EI, Dall'Asta V, Bussolati O. 2008. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc Res 78:563–571. doi: 10.1093/cvr/cvn024. [DOI] [PubMed] [Google Scholar]
- 35.Xue Q, Nagy JA, Manseau EJ, Phung TL, Dvorak HF, Benjamin LE. 2009. Rapamycin inhibition of the Akt/mTOR pathway blocks select stages of VEGF-A164-driven angiogenesis, in part by blocking S6Kinase. Arterioscler Thromb Vasc Biol 29:1172–1178. doi: 10.1161/ATVBAHA.109.185918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang G, Yu K, Jiang Z, Chung A, Yao J, Ha C, Toy K, Soriano R, Haley B, Blackwood E, Sampath D, Bais C, Lill JR, Ferrara N. 2013. Phosphoproteomic analysis implicates the mTORC2-FoxO1 axis in VEGF signaling and feedback activation of receptor tyrosine kinases. Sci Signal 6:ra25. doi: 10.1126/scisignal.2003572. [DOI] [PubMed] [Google Scholar]
- 37.Partovian C, Zhuang Z, Moodie K, Lin M, Ouchi N, Sessa WC, Walsh K, Simons M. 2005. PKCalpha activates eNOS and increases arterial blood flow in vivo. Circ Res 97:482–487. doi: 10.1161/01.RES.0000179775.04114.45. [DOI] [PubMed] [Google Scholar]
- 38.Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, Simons M, Kyriakides TR, Morales-Ruiz M, Sessa WC. 2014. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A 111:12865–12870. doi: 10.1073/pnas.1408472111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan M, Pino E, Wu L, Kacergis M, Soukas AA. 2012. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287:29579–29588. doi: 10.1074/jbc.M112.386854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomanetz V, Angliker N, Cloetta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Ruegg MA. 2013. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol 201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carson RP, Fu C, Winzenburger P, Ess KC. 2013. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum Mol Genet 22:140–152. doi: 10.1093/hmg/dds414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janssens V, Longin S, Goris J. 2008. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci 33:113–121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 43.Westermarck J, Hahn WC. 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Narasimhan SD, Mukhopadhyay A, Tissenbaum HA. 2009. InAKTivation of insulin/IGF-1 signaling by dephosphorylation. Cell Cycle 8:3878–3884. doi: 10.4161/cc.8.23.10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CS, Weng SC, Tseng PH, Lin HP, Chen CS. 2005. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem 280:38879–38887. doi: 10.1074/jbc.M505733200. [DOI] [PubMed] [Google Scholar]
- 46.Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. 2003. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res 63:7777–7784. [PubMed] [Google Scholar]
- 47.Li L, Ren CH, Tahir SA, Ren C, Thompson TC. 2003. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Mol Cell Biol 23:9389–9404. doi: 10.1128/MCB.23.24.9389-9404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ, Dedhar S. 2008. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res 68:1618–1624. doi: 10.1158/0008-5472.CAN-07-5869. [DOI] [PubMed] [Google Scholar]
- 49.Agarwal NK, Chen CH, Cho H, Boulbes DR, Spooner E, Sarbassov DD. 2013. Rictor regulates cell migration by suppressing RhoGDI2. Oncogene 32:2521–2526. doi: 10.1038/onc.2012.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vilar E, Perez-Garcia J, Tabernero J. 2011. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther 10:395–403. doi: 10.1158/1535-7163.MCT-10-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]