

Abstract
Study Objectives:
Sleep fragmentation (SF) is an integral feature of sleep apnea and other prevalent sleep disorders. Although the effect of repetitive arousals on cognitive performance is well documented, the effects of long-term SF on electroencephalography (EEG) and molecular markers of sleep homeostasis remain poorly investigated. To address this question, we developed a mouse model of chronic SF and characterized its effect on EEG spectral frequencies and the expression of genes previously linked to sleep homeostasis including clock genes, heat shock proteins, and plasticity-related genes.
Design:
N/A.
Setting:
Animal sleep research laboratory.
Participants :
Sixty-six C57BL6/J adult mice.
Interventions:
Instrumental sleep disruption at a rate of 60/h during 14 days
Measurements and Results:
Locomotor activity and EEG were recorded during 14 days of SF followed by recovery for 2 days. Despite a dramatic number of arousals and decreased sleep bout duration, SF minimally reduced total quantity of sleep and did not significantly alter its circadian distribution. Spectral analysis during SF revealed a homeostatic drive for slow wave activity (SWA; 1–4 Hz) and other frequencies as well (4–40 Hz). Recordings during recovery revealed slow wave sleep consolidation and a transient rebound in SWA, and paradoxical sleep duration. The expression of selected genes was not induced following chronic SF.
Conclusions:
Chronic sleep fragmentation (SF) increased sleep pressure confirming that altered quality with preserved quantity triggers core sleep homeostasis mechanisms. However, it did not induce the expression of genes induced by sleep loss, suggesting that these molecular pathways are not sustainably activated in chronic diseases involving SF.
Citation:
Baud MO, Magistretti PJ, Petit JM. Sustained sleep fragmentation induces sleep homeostasis in mice. SLEEP 2015;38(4):567–579.
Keywords: BDNF, heat shock proteins, paradoxical sleep, sleep apneas, sleep fragmentation, slow waves, spindles, SWA
INTRODUCTION
Sleep fragmentation (SF) is a periodic sleep disruption observed in highly prevalent sleep disorders such as obstructive sleep apnea (OSA, 2–4% prevalence in the population)1 and restless legs syndrome (RLS, > 5% prevalence)2 as well as in narcolepsy and chronic pain. In severe sleep apneas, individuals may arouse several hundred times per night, leading to a dramatic loss of sleep quality and excessive daytime sleepiness.3 Patients' lack of self-awareness, overlooked cases, and insufficient recognition of the widespread problematic contribute to a major socioeconomic burden because of the condition's broad effect on productivity,2 motor vehicle accidents,4 and medical complications such as diabetes and obesity.5
At the cognitive level, recurrent interference with the natural architecture of sleep dramatically affects reaction times and working memory in humans.3,6 These effects may conceivably be the consequence of the repeated interruption of brain restoration and memory processing during sleep.7 Indeed, the arousal index (AI, number of arousals per hour) correlates with sleepiness in patients with OSA,8,9 RLS,10 and narcolepsy.11 Models of chronic SF recently developed in rodents12–14 confirmed an effect on cognition, in particular memory consolidation, suggesting that preserved sleep quality in addition to duration is essential for normal cognitive performance. We recently developed a device that automatically enforces chronic SF in mice during 14 days without interruption13 with the aim of identifying electrophysiological and molecular markers of sleep homeostasis during prolonged SF.
The homeostatic regulation of sleep consists in a compensatory increase in sleep depth and duration after a period of sleep loss. We showed using our device that 6 h of sleep deprivation is followed by a 50% rebound in slow wave activity (SWA, 1–4 Hz) in the subsequent 4-h sleep.14 Supporting the view that sleep promotes memory consolidation, SWA homeostasis is locally upregulated in the motor cortex after motor learning and correlates with subsequent behavioral improvement.15 Therefore, SWA is thought to reflect the global strength and synchronization of an underlying cortical circuit16 as well as a mechanism for network refinement through synaptic downscaling.17 On the one hand, at the molecular level, transcripts related to brain plasticity such as Homer1a, c-Fos, Egr1 (also known as zif268) and BDNF, mirror net potentiation of synaptic connections during enforced wakefulness.17–19 Specifically, BDNF peptide has been proposed as a causal agent for synaptic potentiation during prolonged wakefulness and subsequent increase in SWA.20,21 On the other hand, heat shock protein (Hsp) transcripts and Bip1 in particular are induced after a few hours of sleep loss, reflecting cellular stress response and suggesting a core role for sleep in preventing cellular damage through energy sparing.22,23 A third set of transcripts, the clock-genes including NPas2, Per1 and Per2, and Clock, are induced by sleep loss in the forebrain in addition to their role in the circa-dian regulation of sleep.24–26
In this study, we report the detailed electrophysiological characterization of sleep homeostasis during chronic SF and analyze the expression of the aforementioned genes and of the BDNF peptide in structures known to undergo synaptic plasticity including the cerebral cortex, the hippocampus, and the thalamus.
METHODS
Animals
Sixty-six adult male C57BL6/J mice (7–9 w old, 22–25 g, Charles River, France) were single housed under a 12 h light/ dark cycle (lights on at 05:00) in standard conditions (temperature 23 ± 1°C) during at least 7 days prior to experimentation, and for the duration of the studies described here. All procedures were carried out according to the European Community's Council directives (86/609/EEC) and with the authorization of the veterinary service of the canton of Vaud. For electrophysiological measures, 21 animals (fragmented [F], n = 7; motor control [MC], n = 8; and quiet control [QC], n = 6) were implanted with electroencephalography/electromyography (EEG/EMG) 3 w before undergoing 14 days of SF and a subset of 12 animals were recorded during a 2-day recovery period (F, n = 6 and MC, n = 6). For molecular and biochemical measures, 21 and 24 animals that did not undergo surgery were sacrificed at the end of the light period after 1 and 14 days of SF, respectively.
Sleep Fragmentation
A new homemade device named ‘CaResS’ (French acronym for “Cage pour la Restriction de Sommeil”) was developed to perform automatic instrumental SF.13 Briefly, the device consists of a cylindrical cage partitioned with fixed dividing walls and a rotating floor covered with sawdust. Upon rotation of the floor, the mouse is forced to move by the vertical walls (Figure 1). Specific attention was paid to develop a system that maximally diminished stressful stimulations for the animal known to interfere with gene expression.27 From this standpoint, CaResS differs from systems using water surroundings, limited space for movements, or human intervention. During the whole protocol, mice were left undisturbed except for automatic cage rotation and a single change of sawdust on day 8. SF was enforced by intermittent cage rotation at 1.5 revolutions/ min for 20 sec every min 24/24 h for 1 or 14 days (F group). Two control groups were time-matched in all procedures for comparison. One group was left undisturbed in the CaResS device for the duration of the experiment (QC group). The MC group underwent enforced locomotion 40 min/h during the active period only (dark, 17:00 to 05:00) to match locomotor activity of the F group while not interfering with sleep. Accordingly, the total cage rotation time was 8 h/day for the F and MC group. EEG/EMG and actimetry were analyzed for a baseline day before SF (BSL), for days 1, 7, and 14 of SF (D1, D7, and D14) and two recovery days following the end of SF (R1 and R2).
Figure 1.
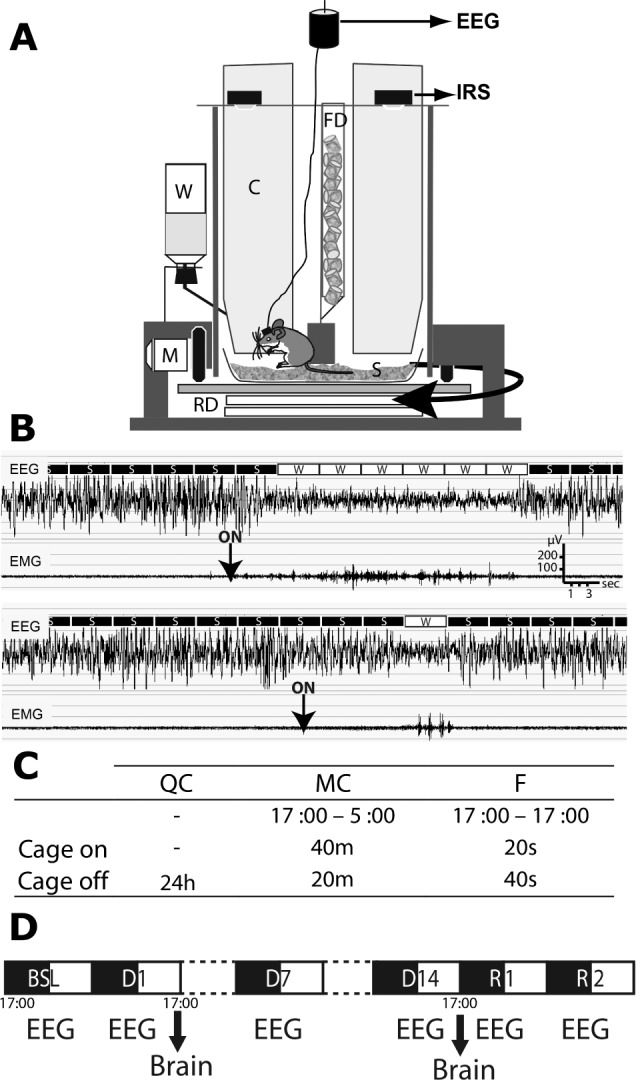
Overview of chronic sleep fragmentation methods. (A) Details of the CaResS device: Cylindrical plexiglas cage (diameter 30 cm) equipped with inner dividing walls (C), rotating circular floor (RD) activated by two electrical motors (M, 1.5 rpm), sawdust (S), free access to water (W) and food (FD) and infrared sensors for locomotor activity recording (IRS). Electroencephalographic/electromyographic (EEG/ EMG) electrodes are connected to swivel on top of the cage. (B) Parallel EEG/EMG traces and example of visual scoring of 4-sec epochs of slow wave sleep (S) and wake (W). Note that the slow wave sleep episode is interrupted by an awakening just after the device activation (ON). Upper trace: maximal length brief awakening (24 sec). Lower trace: minimal length brief awakening (> 4 sec). (C) Summary of the cage rotation schedule for the three groups (m, minutes; s, seconds). (D) Timeline of EEG recordings and tissue collection.
Locomotor Activity
Homemade infrared sensors placed above each cage recorded locomotor activity during the 14 days of the SF protocol (F, n = 8; QC, n = 7; MC, n = 10). These sensors detected rear and lateral movements with a magnitude greater than 1–2 cm; neither grooming nor small movements were detected.14 Number of movements was summed over a period of 15 min and analyzed with a free web-based software to identify the period, the acrophase, the mesor, and the amplitude of the locomotor rhythms using the cosine fitting method28 (cosinor.exe 2.3 and acro.exe 3.4, available online at http://www.circadian.org/softwar.html). To rule out muscular fatigue the grip strength of the forelimbs was assessed in all EEG implanted mice. The device used (Griptest, Bioseb, Vitrolles, France) is composed of a metallic grid connected to an isometric dynamometer. The maximal value in grams of two successive trials of grip strength was used for group means calculation.
EEG Recording and Signal Processing
Briefly, anesthesia was induced with isoflurane 4% and maintained with a mix of xylazine 8% and ketamine 9% (10 μL/g intraperitoneally). After shaving and skin disinfection, the scalp was incised longitudinally and the skull was drilled to insert two gold covered stainless screws (1 mm diameter) over the frontal cortex (bregma + 1.5 mm and 1 mm laterally) and over the parietal cortex (bregma −3 mm and 1 mm laterally), respectively. Two additional screws were inserted on the contralateral side for implant anchorage. Two gold wire electrodes (500 μm diameter) were placed into the nuchal muscles. Skull and muscular electrodes were then stuck to the bone with resin (Relyx, 3M, Switzerland), soldered to a connector and covered with dental cement (Paladur, Heraeus-Kulzer, Germany). After 1 w of recovery, a swivel-connected wire was plugged into this connector to allow free movements of the animal.
The EEG/EMG signals were recorded and digitalized with an Embla A10 amplifier (Medcare, USA) sampled at 100 Hz and filtered between 0.5 and 50 Hz. Using the Somnologica software (Medcare, USA), 4-sec epochs of the EEG/EMG were visually scored in three vigilance states (wakefulness [W], slow wave sleep [SWS), and paradoxical sleep [PS]) according to classic criteria.29 Brief episodes of wakefulness were scored for EEG desynchronization between 4–24 sec with or without concomitant EMG activation immediately preceded or followed by SWS and excluding transitions to PS.
Spectral Analysis
After discarding epochs with lead motion artifacts, as well as one epoch preceding and one epoch following each state transition, EEG power spectra of remaining SWS epochs were calculated by a fast Fourier transform (FFT) using MATLAB embedded FFT function and personalized routines (Math-Works Inc., Natick, MA USA). For each mouse, individual FFT values of 0.25-Hz bins were averaged over 12 h for a given vigilance state and divided by corresponding values obtained during the baseline day (BSL) expressed in percent from 0.5 to 40 Hz. Power expressed in percent was collapsed into 1-Hz bins for visual rendering and grouped in six frequency bands classically defined as delta: 0.5–4 Hz, theta: 4–10 Hz, alpha: 10–13 Hz, beta1: 13–20 Hz, beta2: 20–35 Hz, and gamma: > 35 Hz (Figure 5A–C). Total SWA in the dark or light period of R1 was individually calculated relative to baseline values and averaged (Figure 5C).
Figure 5.

Spectral analysis of sleep electroencephalography during sleep fragmentation (SF) and recovery. (A,B) Frequency spectrum of slow wave sleep (SWS) during dark (upper panel) and light phase (lower panel) of the SF protocol. Power is expressed in percent of baseline for each individual and averaged (n for quiet control [QC] = 6, motor control [MC] = 8, fragmented [F] = 7). Horizontal gray bars indicate significant difference (P < 0.05 on Tukey post-significant one-way analysis of variance). Note the consistent increase in power in theta to gamma bands (4–40 Hz). (C) Note normalization of theta-gamma effect during the dark phase on R1 but significant increase in slow wave activity (SWA; insert). (D–I) Dynamics of the power changes at transitions from wake (W) to SWS (t = 0) and from paradoxical sleep (PS) to SWS (t = 0) during light phase of D1 (n for QC = 6, MC = 8, F = 7), D14 (n = 6 per group) and R1 (n = 6 per group). Power is expressed in percent of SWS values during normalizing day. 1 Hz FFT values (displayed in A–C) were summed up in classic frequency bands, α and β were grouped. Horizontal gray bars indicate significant difference between F compared to both MC and QC (Tukey possignificant one-way analysis of variance, P < 0.05 for both). Note the overshoot of theta to gamma frequencies in the F group following SWS onset during SF (D,E) and its disappearance on R1 (F). Note also the increase in SWA buildup rate on D14 (E). Note that α and β power transitory increase before PS onset is slightly delayed in F group (G,H).
To study W-SWS and SWS-PS stage transition dynamics (Figure 5D–5I), 4-sec FFTs in defined frequency bands were summed for each animal, normalized by the mean power of the band during SWS of the light or dark phase of the normalizing day, aligned in a time-series of epochs preceding stage transition and epochs following stage transition, averaged for the group and plotted along time axis (SWS or PS onset, t = 0). To calculate kinetics in the delta band (SWA) at the W-SWS transition, we fitted a saturating exponential function according to the formula30: y = a(1−et/τ) + y0 where y is the SWA at time t, a is the saturating asymptote, τ the time constant of the rate of SWA buildup and y0 the SWA value at t = 0 set to be the SWA value of the preceding waking epoch. Average time constant and asymptote were calculated for each group. Because of a slight drift in the EEG power in our chronic recordings, the normalizing day was either the baseline recording for D1 analysis or the second recovery day (R2) for D14 and R1. SWA decay during recovery was calculated for intervals of equal SWS time (four in dark phase, eight in light phase) by normalizing individual values by the last 2 h of sleep of R2.
Wave-Triggered Analysis
Only the F group was analyzed during the light phase and compared to individually matched baseline (BSL) recording in the case of D1 and recording after recovery (R2) in the case of D14 and R1. As a global approach to features of the EEG in the time domain, we calculated the envelope of the filtered EEG (band-pass Butterworth digital filter > 10 Hz) starting at sleep onset (t = 0). For each mouse of the F group, an average upper and lower envelope (respectively joining local maxima and local minima) was determined over all sleep episodes. Group means were plotted against the time axes (Figure S3).
To more precisely describe the effect of SF on two cardinal waves of SWS, we studied wave-triggered averages of slow waves (SW, high amplitude waves in 1–4 Hz range) and spindles (spindle bursts in 10–15 Hz range) using a method already described.20 SW were defined as unique negative deflections between two positive deflections separated by at least 0.15 sec in the 1–4 Hz (delta) band-pass filtered SWS EEG.20 To maintain their global shape, SW detected in the 1–4 Hz EEG were further studied in the 1–15 Hz band-pass filtered EEG after adjustment for the exact occurrence of the negative deflection on the time axis (Figure 6A). Incidence, amplitude, and negative deflection triggered average were first averaged in the early (E, first third of light phase SWS) and late (L, third third of light phase SWS) SWS for each mouse, normalized by reference values (means of 12 h of light phase SWS during BSL or R2) and averaged. Peak-to-peak amplitudes of the SW (from maximal positive deflection to negative deflection) were sorted along seven percentiles (0.025, 0.1, 0.25, 0.5, 0.75, 0.9, 0.95) and plotted as a distribution histogram (Figure S4A and S4B, supplemental material). The slopes of the first and second segment were obtained by dividing the amplitude by time.
Figure 6.

Sleep waves and spindles architecture. Wave triggered average of slow waves (SW) and spindles during the light phase slow wave sleep (SWS). Representative raw electroencephalographic data with corresponding band-pass filtered traces and typical shape and amplitude (A) of SW (1–15 Hz) and spindles (10–15 Hz) is shown in panel A. (B,E) Wave-triggered average of down deflections showing a significant decrease in minima reached on D1 and D14 (P < 0.05, paired t test, inserts). (C,F) Histograms of SW amplitude in relative values for early (E, first third of light phase SWS) and late (L, third third) occurrence (P < 0.001, two-way analysis of variance [ANOVA]). (D,G) Increase in slope 1 (P < 0.005 on D1 and D14) and slope 2 (P < 0.05 on D1) for SW of equal amplitude (percentile 0.4 to 0.6). (H,I) Wave-triggered average where the maximum of spindle is centered on t = 0. Spindle envelope underlines the difference in amplitude for each peak (black squares, P < 0.05, paired t test, inserts). (J,K) Histograms showing increase in the amplitude of spindles on D1 and D14 (P < 0.001, two-way ANOVA for all) with maintenance of circadian effects when considering L versus E (P < 0.005, two-way ANOVA for all).
Individual spindles were defined as local maxima greater than the mean 10–15 Hz root mean square SWS signal and spaced by at least 0.25 sec. After transposition and adjustment of the local maxima on the 10–15 Hz band-pass filtered signal, buildup and decay rates were calculated as the slopes from half height to full height and inversely (Figure 6 and Figure S5, supplemental material). As for SW, all parameters were normalized before group averaging.
Corticosterone Plasma Levels
Corticosterone levels were quantified in trunk blood plasma of animals used for the polymerase chain reaction (PCR) and biochemical study and sacrificed (end of light phase) on D1 (n = 7 per group) and D14 (n = 8 per group). Immunoassay was performed according to manufacturer's instructions (Corticosterone Enzyme Immunoassay Kit, Assay Designs, LuBioScience, Lucerne, Switzerland). Briefly, samples and standards were incubated for 2 h in separate wells of an antibody-coated plate along with corticosterone coupled to an alkaline phosphatase (competitive assay). After incubation (1 h) with the enzyme substrate, the yellow color generated was read at 405 nm. A standard curve was established using a four-parameter logistic curve fitting and was used to determine corticosterone levels in each sample.
Quantitative Reverse Transcription-PCR
Brains and livers were rapidly dissected out and frozen at -80°C after 1 day (n = 7 per group) and 14 days of SF (n = 8 per group). The somatosensory cortex, the dorsal hippocampus, the thalamus, and the cerebellum were microdissected from frozen slices (300 μm, −15°C) using a microscope. These samples were homogenized in the lysis buffer of a commercially available kit (Nucleospin II, Macherey-Nagel, Switzerland) with addition of β-mercaptoethanol 1% and kept at -80°C. Total ribonucleic acid (RNA) was extracted using columns provided with the kit (Macherey-Nagel), treated with deoxyribonuclease (DNAse) and checked for RNA quality (RNA Integrity Number ranging from 7.5 to 9.5) on Agilent 2100 bio-analyzer chips (Agilent Technologies, Waldbronn, Germany). After reverse transcription with the high-capacity RNA to complementary DNA (cDNA) kit (Applied Biosystems, Foster City, CA, USA), 1.5 μL of cDNA (1:10) from each individual sample underwent amplification (ABI Prism 7900 machine, Applied Biosystems) on a 384-well plate where each well contained 3.5 μL of forward and reverse primers (300 nM) and 5 μL of Power SYBR Green MasterMix (Applied Biosystems). Data were computed using the sequence detector software SDS 2.3 (Applied Biosystems). All samples belonging to the same experiment (D1 or D14) and brain structure were processed in triplicates in the same PCR run. For each gene, the Ct values were normalized to the Ct of β-actin and averaged individually (GeNorm, Frontiers in Genetics, UNIGE, Geneva, Switzerland). For each structure and experiment, we reported β-actin levels normalized by other housekeeping genes commonly used in sleep experiments31 to ensure its stability. The normalization factor was the geometric mean of Cyclophilin-A Ct and Hypoxanthine-guanine phosphoribosyltransferase (Hprt) Ct except for the thalamus where only Hprt Ct was used, given that cyclophilin-A expression had a high intragroup variance in that particular structure. Primers reported in Table S1 (supplemental material) were designed using the web-based software Primer Blast (National Center for Biotechnology Information) and synthesized by Microsynth (Belgach, Switzerland). Whenever possible, we chose amplicons extending over an exon/exon junction to avoid amplification of any residual genomic DNA contamination (Table S1).
BDNF Peptide Quantification.
We quantified BDNF in the somatosensory cortex controlateral to that used for the RT-PCR experiment. Samples were shortly sonicated in a Triton containing lysis buffer at pH 7.0 without Bovine Serum Albumin (BSA, piperazine-N,N′-bis(2-ethanesulfonic acid) 100 mM, NaCl 500 mM, TritonX100 0.2%, sodium azide 0.1%, EDTA 2 mM, antiprotease complete mix [Roche, Switzerland]). Then a 5-μL sample diluted in water (100×) was taken to quantify proteins using bicinchoninic acid assay (Sigma-Aldrich, Buchs, Switzerland). Thereafter, we added equivalent volume of identical buffer plus BSA 4% to the samples to obtain a final lysis buffer with BSA 2% shown to improve BDNF recovery32. We used a commercially available sandwich enzyme-linked immunosorbent assay kit (BDNF Emax Immunoassay, Promega, Madison, USA) according to the manufacturer's instructions. Briefly, 96-well Spectraplate (Perkin-Elmer, Waltham, USA) were coated with anti-BDNF monoclonal antibody overnight at 4°C. The plate was washed once with Tris-buffered saline containing Tween 20 0.1% (TBS-T) and incubated with block and sample buffer at room temperature (RT) for 1 h. Dilutions of samples and standard curve were made in duplicate using final lysis buffer added to the plate after centrifugation (20 min, 13,000 g, 4°C) and incubated for 2 h at RT. After washing five times the plate was incubated 2 h with anti-human BDNF polyclonal antibody, washed again and incubated with anti-immunoglobulin Y antibody conjugated to horseradish peroxidase for 1 h. Finally, tetramethylebenzidine was added and after 10 min, the reaction was stopped with 1 M HCl and the absorbance was immediately measured on a plate reader at 450-nm.
Statistical Analysis
For all studies including the three groups (F, MC, QC), group means were compared using either parametric one-way analysis of variance (ANOVA) or nonparametric Kruskal-Wallis test according to the sample size and the result of Bartlett test of variance. When group differences were significant on variance analyses (P < 0.05), post hoc tests for multiple comparisons were performed using Tukey or Dunn post hoc test when variance was homogenous or not, respectively. For wave-triggered analysis, values were compared to individually matched controls by repeated-measures two-way ANOVA for D14 versus R1 and R2, or paired t test for D1 versus BSL. For all tests, significance was accepted when P < 0.05. All values are expressed as mean ± standard deviation (mean ± SD).
RESULTS
Control Parameters
As expected with our method, the F and MC groups showed an increase in the mean locomotor activity over 24 h compared to the QC group (respectively, +80% and +92% on D1, +37% and +68% on D14, P < 0.001 for all, ANOVA, Figure 2A). Weight of the animals was unchanged on D14 (QC, 31.9 ± 1.6, MC, 31.5 ± 1.8, and F, 30.9 ± 1.3 g) compared to baseline (QC, 31.6 ± 1.7, MC, 31.1 ± 1.8, and F, 30.7 ± 1.5 g). A grip test attested to the absence of any muscular exhaustion on D14 (106 ± 15 g for QC, 117 ± 10 g for MC, and 107 ± 27 g for F, mean ± SD, P > 0.05, ANOVA, not shown). When measured at the end of the light period, the stress hormone corticosterone reached similar levels in trunk blood in all three groups on D1 and D14 (P > 0.05, ANOVA, Figure 2B). Moreover, daily visual inspection of our animals did not reveal behavioral signs of stress such as prostration and stereotypy or physical alterations such as skin lesions contrary to chronic total sleep deprivation experiments.33
Figure 2.
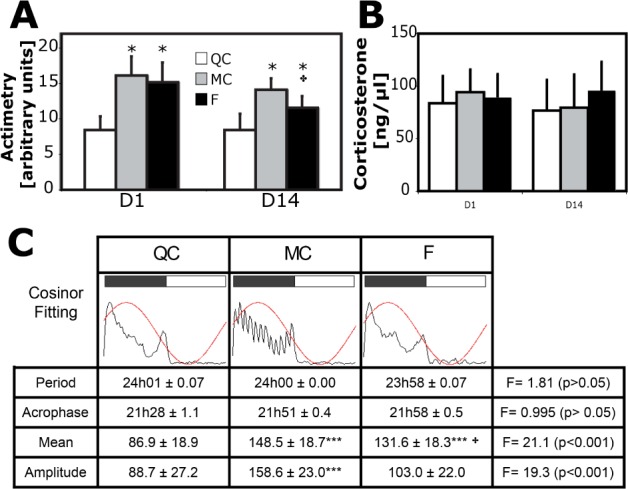
Locomotor activity and plasma corticosterone. (A) Locomotor activity cumulated over 24 h on D1 and D14 (n for quiet control [QC] = 7, motor control [MC] = 10 and F = 8). * MC and fragmented group (F) have significantly higher values than QC (P < 0.001). Significant difference between MC and F (P < 0.01). (B) Corticosterone in trunk plasma of animals sacrificed at the end of the light phase on D1 (n = 7 per group) and D14 (n = 8 per group). (C) Circadian rhythms of locomotor activity determined by cosinus fit of locomotor activity over 14 days for each group. The three figures represent group means during the dark and light phase (gray and white rectangles, respectively) without standard deviation. Note that the saw-tooth ultradian rhythm in MC is artificially caused by the specific pattern of enforced locomotion (40 min/h, dark phase).
Sleep Fragmentation Model Validation
As a result of intermittent cage rotation in the F group, we observed short wake intrusions lasting 18 ± 2 sec into sleep episodes and the AI increased by approximately 60 awakenings per hour (as targeted) compared to controls on D1 (QC 33 ± 6, MC 38 ± 7, and F 94 ± 1, P < 0.001, ANOVA) and D14 (QC = 31 ± 7, MC = 32 ± 3, F = 87 ± 5, P < 0.001, ANOVA). Accordingly, the reduction in the mean SWS bout duration enforced by our device (−65% to −74% for D1, D7, and D14, P < 0.001, ANOVA, Figure 3A) was accompanied by a compensatory increase in the number of bouts throughout the protocol (+110% to +150% on D1, D7, and D14, P < 0.001, ANOVA, Figure 3A). PS mean bout duration was also decreased throughout the protocol when compared to QC and MC (−60% to −63% on D1, P < 0.001, −40% to −45% on D7, P < 0.001, −20% to −27% on D14, P < 0.001, ANOVA) but contrary to SWS, this did not lead to any compensatory increase in the number of bouts per 24 h (P > 0.05 for all comparisons). These results showed that SWS and PS were fragmented throughout the 14-day protocol.
Figure 3.

Vigilance states distribution during sleep fragmentation. (A) Slow wave sleep (SWS) bout mean duration, bout number, and cumulated SWS time over 24 h (mean ± standard deviation, n for QC = 6, MC = 8, F = 7). *F is different from QC and MC. #F is different from QC only. Note effective shortening of sleep bouts on D1 and D14 along with minimal total sleep time restriction. (B) Circadian distribution of time of sleep in minutes cumulated over 2 h on D1 and D14. Upper panel: SWS; lower panel: paradoxical sleep. Black horizontal bars indicate significant difference for the F group compared to both QC and MC groups (Tukey post-analysis of variance, P < 0.05 for both). (C) Consolidated wake (> 10 min without interruption) expressed in percentage of total wake during the 12 h of the active phase on D1, D7, and D14. Note the decreased ability to maintain prolonged wakefulness in the F group. (D,E) Spontaneous sleep latencies measured after any awakening > 12 sec on D1 and D14 showed decrease in the F group. F, fragmented; MC, motor control; QC, quiet control.
Circadian Distribution of Vigilance States and Locomotor Activity During SF
There was no significant difference in circadian rhythm of the sleep-wake cycle (Figure 3B) and locomotor activity (Figure 2C). SF led to decreased spontaneous sleep latencies, defined as the time from any awakening > 12 sec to sleep (Figure 3D and 3E). Time spent in the different vigilance states is reported below and in full details in Table S2 (supplemental material).
Slow Wave Sleep
On D1, SWS total time over 24 h was slightly but significantly decreased in the F group compared to both QC and MC (−14% to −19%, P < 0.001, ANOVA, Figure 3A and 3B).
On D7 and D14, 24-h SWS time almost normalized being only slightly decreased versus QC (−15% on D7, P = 0.01, −12% on D14, P = 0.04, Tukey post-ANOVA, Figure 3A) but not significantly when compared to MC (P > 0.05 on D7 and D14, Tukey post-ANOVA, Figure 3A).
Paradoxical Sleep
PS loss was more important than SWS and sustained throughout our protocol. It was significantly decreased compared to both controls on D1 (−66% to −71%, P < 0.001, Figure 3B), D7 (−24% to −38%, P < 0.001, ANOVA), and D14, (−18% to −25%, P < 0.05, ANOVA, Figure 3B). There was however a significant trend toward increase from D1 to D14 without reaching controls values (P < 0.05 for day effect, ANOVA, Table S2).
Wake and Locomotor Activity
Total wake time over 24 h was increased in the F group throughout the protocol (+6% to +18% on D1, D7, and D14, P < 0.01 for all, ANOVA) reflecting a decrease in both SWS and PS. Interestingly, on D7 and D14, consolidated wake, defined as more than 10 min of uninterrupted wake was decreased in the F group (P < 0.05 on both days, ANOVA, Figure 3C) showing decreased ability to maintain prolonged wakefulness during SF.
Circadian Distribution of Vigilance States During Recovery
Slow Wave Sleep
On R1, total SWS time slightly increased over the first 12 h (+18% to +24%, P < 0.05, ANOVA, Table S3, supplemental material, and Figure 4B) consolidated as reflected by an increase in bout mean duration (+25% to +56%, P < 0.04, ANOVA, Table S3). Interestingly, whereas SWS time normalized during the 12-h light phase of R1, it remained consolidated compared to controls, suggesting that bout duration may be a more sensitive assessment of SWS pressure (SWS bout mean duration, +23% to +33%, P < 0.01, ANOVA, Table S3). On R2, SWA, SWS total time and bout duration returned to control values (P > 0.05, ANOVA, Table S3). This SWS rebound during recovery showed that sustained SF increased SWS pressure throughout the protocol.
Figure 4.

Slow wave sleep (SWS) and paradoxical sleep (PS) rebound during recovery. (A,B) Upper (SWS) and lower panel (PS) show sleep time in minutes cumulated over 2 h intervals during recovery day R1 and R2 (cage off at t = 17:00 on R1, n = 6 per group, values for quiet control [QC] reported from D14). (C,D) Slow wave activity (SWA) normalized by last 2 h of sleep of R2 calculated for equal sleep intervals (4 in dark phase, 8 in light phase). (E–H) SWS and PS total and bout time cumulated over 12 h. Note significant quantitative SWS and PS rebound during the dark phase around 23:00 and significant qualitative rebound including SWA and bout consolidation throughout R1 with complete resolution on R2. Horizontal black bars and asterisks indicate significance (P < 0.05, Tukey post-analysis of variance versus QC and motor control).
Paradoxical Sleep
On R1, we observed a large rebound of PS during the 12-h dark phase, reflected by an increase in PS total time (+218% to +290 %, P < 0.001, ANOVA, versus QC and MC, Table S3 and Figure 4C). Similarly to SWS, PS total time returned to control values during the 12-h light phase of R1 but not PS bout mean duration (+18%, P < 0.01, ANOVA, Table S3). On R2, PS total time and bout duration returned to control values (P > 0.05, ANOVA, Table S3). This massive quantitative PS rebound during recovery is in agreement with PS loss observed throughout the SF protocol.
Wake
Consolidated wake was decreased during the dark phase of R1 compared to QC and MC (−14% to −21%, P < 0.05, ANOVA), reflecting residual sleep pressure. Total wake time also decreased as a sleep rebound took place (Table S3). Wake parameters returned to control values during the light phase of R1 and R2.
EEG Spectral Analysis During SF
Artifact free epochs (> 98%) underwent FFT to study frequency distribution in the three vigilance states over 12-h periods across the protocol. To explore the dynamics of the spectral effects of SF, we also analyzed the frequency changes at vigilance state transitions. Results for wake, SWS, and PS are detailed in Figure 5 (as well as Figure S1 for wake, supplemental material) and summarized in the following paragraphs.
Slow Wave Sleep
Throughout the protocol, a significant increase in theta to gamma power (4–40 Hz) was observed in the F group (+20% to +25%, P < 0.05, ANOVA, Figure 5A, 5B, 5D, and 5E). In contrast, total SWA (delta power) remained unchanged compared to controls, except for a transient decrease during the light phase of D1 (−10% to −15%, P < 0.05, ANOVA, Figure 5A and 5B). However, spectral analyses of sleep immediately following wake-SWS transition, revealed that SWA builds up at a faster rate in the F group indicating that these mice entered deep sleep earlier after sleep onset than their matched controls (60% decrease in time constant of fitted curves, P < 0.05, ANOVA, Figure S2, supplemental material, Figure 5D and 5E). Corroborating the power increase above 10 Hz, we observed an increase in the global amplitude of the sleep EEG filtered above 10 Hz on D1 and D14 (+15% to +25%, P < 0.05, paired t test, Figure S3).
Paradoxical Sleep
No change in the spectral composition of PS was observed. In rodents, the transition from SWS to PS is naturally preceded by a transient shift in SWS frequencies 30 sec before PS appearance including a progressive decrease in delta power and a simultaneous increase in alpha and beta power.30,34 In the F group, we observed a slight reduction of this transition time in the alpha-beta band, notably on D14 (Figure 5H; P < 0.05).
Wake
There was a transient significant increase in theta power during the D1 light phase in the F group (+10%, P < 0.05 versus QC and MC, ANOVA, Figure S1A) but no change in spectral composition of wakefulness thereafter including that of short awakenings between two sleep episodes (P > 0.05, ANOVA, Figure 5D and 5E).
These spectral analysis results revealed that SF induced sustained qualitative changes during SWS only and not during PS or wakefulness beyond D1.
EEG Spectral Analysis During Recovery
On R1, we observed a significant SWA rebound during dark and light phase SWS in the F group compared to QC and MC (approximately +20%, P < 0.04, Figure 4C and Figure S2B) and an immediate and complete normalization of the theta-gamma bands (Figure 5C). SWA returned progressively to control values toward the end of R1 light phase and remained at control values during R2 (Figure 4C and 4D). Of note, SWA was found to be elevated in the same range when normalizing by baseline day (Figure 5C) or R2 values (Figure 4C and Figure S2B). There were no changes of the PS spectrum during the quantitative rebound.
Sleep Waves Architecture During SF
To corroborate the effects observed during SF in the frequency domain, we calculated several parameters of the EEG in the time domain for SW and spindles. For the sake of clarity, only the F group was analyzed during the light phase and individually normalized (see Methods) using a repeated-measures ANOVA analysis taking into account the day of recording and the distinction between early (E) and late (L) light-phase SWS as factors. Results on the amplitudes of SW and spindles are presented in the following paragraphs; other characteristics of these sleep waves can be found in Figures S4 and S5, respectively.
Slow Waves
SW amplitudes increased in the F group throughout the protocol (+4% to +9% for E and L on D1 and D14, P < 0.001 for all, ANOVA, Figure 6C and 6F) and this was partly explained by deeper down deflections (−4% to −5% on D1 and D14, P < 0.05, ANOVA, Figure 6B and 6E). In addition, SW amplitudes retained an ultradian dependence of greater amplitudes during early sleep (P < 0.001 on D1 and D14, ANOVA, interaction > 0.05, Figure 6C and 6F). The slopes of the first and second segment were increased (+6% to +8% on D1 and D14, P < 0.001, ANOVA, Figure S4E and S4F) even when correcting for SW amplitudes (P < 0.05 for slope 1 and 2 on D1, P < 0.005 only for slope 1 on D14, ANOVA, Figure 6D and 6G). These results indicate that pressure for SW was elevated throughout the protocol suggesting increased synchrony between cortical networks generating SW.
Spindles
Spindles amplitude was increased throughout the protocol (+8% to +9% for E and L on D1 and D14, P < 0.001 for all, ANOVA, Figure 6H–6K) with a retained ultradian modulation being greater during the late phase of sleep (P < 0.005 on D1 and D14, interaction > 0.05 for both, ANOVA, Figure 6J and 6K). An increase in spindle amplitude was also present in the dark phase SWS with even greater magnitude (+18% on D14, P < 0.001, ANOVA). These results suggest higher synchrony in thalamocortical networks generating spindles.
Gene Expression and BDNF Levels
Three sets of genes known to be induced by acute sleep deprivation were selected to test whether SF would affect their regulation after 1 and 14 days. On D1, Hsp were increased with a pattern of induction specific to each brain region represented in Table S4 (supplemental material) for statistics. BiP (Hspa5) was induced in every region tested compared to QC and MC (increase ranged from + 11% to +37%, P < 0.001 to P < 0.03, ANOVA, Figure 7A). Among genes related to plasticity, we observed that Homer1a was induced in the hippocampus following 24 h of SF versus MC and QC (+ 19% to +26%, P < 0.01, ANOVA, Table S3), whereas FosB and Bdnf were decreased in the somatosensory cortex when F group was compared to MC and QC (respectively, −21%, P < 0.01, ANOVA and – 17%, P < 0.05, ANOVA, Dunn post hoc only significant test versus MC, Table S3). Values of cortical BDNF normalized by the protein content were in the range of previous reports in the literature.35 BDNF peptide assessed by enzyme-linked immunosorbent assay displayed no change in all brain structures (P > 0.05, Figure 8B). No variation of transcripts encoding proteins related to circadian clock was observed except for Npas2 that increased in the cerebellum (+17 to +20%, P < 0.05 ANOVA, versus MC and QC) and Clock that mildly increased in the somatosensory cortex in F compared to QC group only (+ 14%, P < 0.05, Dunn post hoc test) as well as in liver when F was compared to QC and MC groups (+25%, P < 0.05, Dunn post hoc test, Table S4). On D14, all transcripts coding for Hsp, clock-genes, and genes related to plasticity tested here were similar to control values (Figure 7B and Table S2). Both Bdnf messenger RNA (mRNA) and peptide were stable (Figure 8C and 8D).
Figure 7.
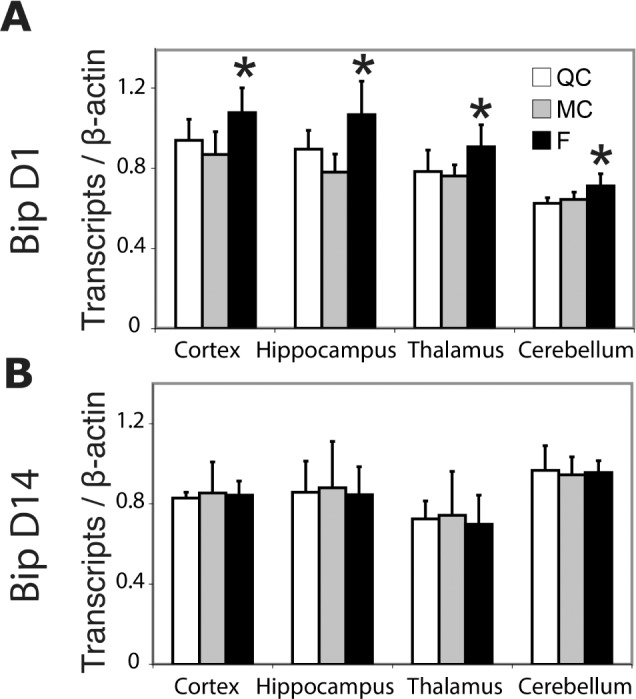
(A,B) Cortical messenger RNA (mRNA) levels of BiP. Transcript (mRNA) levels are normalized by β-actin (mean ± standard deviation). *P < 0.05 on Tukey test post-one–way analysis of variance for each structure. Complete experiments are reported in Table S3. Note consistent induction of BiP expression after 24 hours of sleep fragmentation (D1, n = 7) compared to 14 days (D14, n = 8). F, fragmented; MC, motor control; QC, quiet control.
Figure 8.
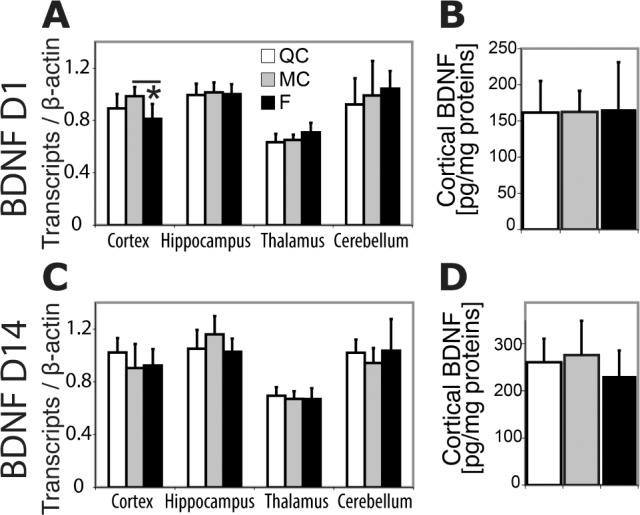
Cortical levels of BDNF messenger RNA (mRNA) and peptide. (A,C) Transcript (mRNA) levels normalized by β-actin (mean ± standard deviation). *With horizontal bar: P < 0.05 on Tukey test post-one–way analysis of variance in the cortex only significant compared to MC. (B,D) Peptide levels. Note the stability of BDNF on D1 and D14. F, fragmented; MC, motor control; QC, quiet control.
DISCUSSION
In the current study we used a new protocol of continuous automated SF and balanced the inherent effect of instrumental sleep disruption on locomotor activity by including the MC group. Thereby, we showed that it is possible to delineate the specific effects of SF in the absence of direct confounding factors, namely sleep loss, high stress levels, and disrupted circadian rhythm. As a matter of fact, SF shortened the duration of individual SWS bouts throughout the protocol but increased their number, so that the hourly cumulative SWS time was not different compared to MC beyond D1. This is consistent with previous studies where SF was induced for 2–4 w without any major effect on sleep duration12 or stress36 but increased the pressure for sleep as assessed by behavioral measures and spontaneous sleep latency. As an addition to this body of data, we established here that SWA homeostasis was engaged throughout the 2-w protocol and that chronic SF led to a subsequent rebound of SWA, SWS, and PS during recovery, which is viewed as the “gold standard” to evaluate sleep homeostasis. Interestingly, our protocol illustrates the differential and competitive homeostatic regulations of SWS and PS during chronic SF. PS pressure occasionally overrode SWS pressure during the short bouts of sleep but cumulative PS time remained slightly suppressed throughout the protocol until after a SWA rebound began during the recovery period. Our data show that the circadian modulation of the sleep-wake cycle persisted despite increased sleep pressure. Indeed, after SF cessation, mice maintained wakefulness during 6 h before entering a phase of sleep rebound, similar to the observed delay in sleep rebound after a 6-h sleep deprivation experiment ending during the active phase.25 This indicates that sleep pressure accumulated during chronic SF is likely insufficient to overrule the suprachiasmatic drive for wakefulness that is maximal during the peak of spontaneous activity at the beginning of the dark phase.37,38
Using spectral analysis and wave-triggered analysis, we showed that SWA buildup kinetics at sleep onset and increased amplitudes of SW are sensitive EEG markers of sleep homeostasis during chronic SF.39 In addition, by using longitudinal normalization methods (i.e., using a normalization parameter extrinsic to the data), we showed that there were global spectral changes (corresponding to global amplitude increase in the time domain) that could have been missed, had we used a normalization parameter intrinsic to a given day (e.g. the overall power). This method yielded an unanticipated global power increase in the 4–40 Hz range, suggesting that other sleep waves, including spindles, are also under homeostatic regulation during SF. Cortical SW (< 1 Hz) reflect slow oscillations of the neuronal cell membrane potential. Alternation of depolarization and hyperpolarization, also known as up (firing ON) and down states (firing OFF), is the key intracellular rhythm determining SW coalescence.40 The synchrony in large neuronal population recruitment into the slow oscillations and the duration of OFF periods are both under homeostatic regulation and respectively determine the slopes and amplitude of SW at the level of the EEG.16 Although we do not have intracellular recordings to confirm this, we may hypothesize that the shift toward higher SW amplitudes and steeper SW slopes observed in our study might be a reflection of homeostatic processes occurring at the cellular level during SF. Our observations therefore suggest that SF increases synchronization of cortical firing patterns. Accordingly, sleep pressure leads to an increase in distant synchronization of individual slow waves over frontal and parietal cortices.41 Similar effects on spindles suggest an additional synchronization effect of SF on the corticothalamic loops and are in agreement with human findings.42 The fact that SWA can never be fully expressed during SF might have a permissive effect on spindles expression (and maybe beta to gamma) because spindles and SW oscillations are mutually exclusive at the level of thalamocortical neurons.43 This suggests a convergent homeostatic regulation of these waves but different kinetics, spindles rising faster than SW after sleep onset.44 Recently, two human studies reported that short-term SF led to a 20% and 40% increase in power in 12–23 Hz and 1–25 Hz bands, respectively.45,46 These results point to an inter-species marker of SF consisting of an increase in ‘rapid’ sleep EEG frequencies during short bouts of SWS. Bearing in mind that in our model, SWS expresses itself at the expense of PS, a possible explanation is that the 4–40 Hz power increase reflects attempts at PS occurrence. Indeed, the range of the observed power increase is similar to transitional sleep observed just before PS, sometimes called “Pre-REM.”47,48 Contrary to SWS, PS did not undergo any qualitative changes during SF (its EEG spectrum remained the same) and its rebound was purely quantitative (its total duration increased) confirming the lack of PS EEG spectral marker of depth for PS pressure.49 In summary, widely distributed and sustained synchrony in slow oscillations might explain how constant sleep pressure during SF determines the coalescence of brain waves into a generalized increase in the EEG amplitude and consequently in the EEG power. Interestingly, spindles and SW have been linked to learning15,50 and synaptic plasticity51 at the molecular level.52 Changes in their dynamics, however, may interfere with their presumed function in sleep replays and could explain how SF affects memory in a spatial navigation task after training.36,53,54
A comparison with sleep deprivation and sleep restriction (SR) experiments further reveals how some aspects of sleep homeostasis pertain specifically to sleep quality. Five days of SR led to a rebound in SWA during daily 4-h periods of rest and to a final rebound during 2 recovery days in rodents39 and humans.55 Similarly, our SF protocol led to an increase in SWA build-up rate and amplitude of SW during the 40-sec periods without instrumental sleep disruption and to a final SWA rebound during 1 recovery day with subsequent normalization. Of note, SWA rebound observed here was approximately halved compared to a 6-h sleep deprivation experiment in our conditions14 (20% versus 50%), and compared to 5 days of sleep restriction in rodents39 and humans,55 with the caveat that SWA rebound after SF was shown to be dampened during the active period.56 Therefore, sleep disruption with preservation of its total duration seems to be sufficient to increase long-term sleep pressure, confirming that quality likely counts as much as quantity for sleep homeostasis. Consequently, the “sleep debt” may be sustained in the chronic setting of sleep disorders with SF. Importantly, we did not observe any intrusion of sleep waves during wakefulness, although our SF protocol shortened sustained awakenings. This shows that contrary to sleep deprivation and SR experiments,39 frequent partial sleep pressure relief allows for normal electrophysiological wakefulness41 but nevertheless culminate in long-term need for sleep consolidation. Accordingly, we observed an increase in theta frequency during D1 when sleep curtailment was greatest. This effect has been described as a marker for sleep propensity during enforced wakefulness39,57 and SF.12 Cumulative excess wakefulness may correlate best with brain processing speed as measured by reaction times,58 which we did not investigate in this study. In summary, the hallmark of sleep homeostasis is a sleep and SWA rebound during recovery. However, other EEG homeostatic markers such as SW amplitude and SWA kinetics might be more sensitive than averaged SWA in SF experiments because SWA expression is constrained in time. Shortened sleep latency and shortened consolidated wake time may be substitute markers for experiment not including a recovery phase.
In studying the expression of a set of genes expression at one circadian point on D1 and D14, we showed that important molecular pathways were transiently induced when sleep loss was maximal (D1) but normalized on D14 when sleep quantity returned to control levels, with the caveat that protein regulation was not broadly assessed in the current study. We opted to measure gene transcription at the end of the rest phase when effects of sleep disruption were presumably maximal. Moreover, although we showed elsewhere that peak corticosterone is increased toward the end of the protocol,13 we observed no difference in plasma corticosterone levels at that specific time of sacrifice, making a potential direct effect of corticosterone on gene expression less likely in our experiments.27
BiP (also known as Hspa5 or Grp78) was consistently induced by 1 day of SF in all studied brain structures along with other Hsps, possibly reflecting mild sleep loss on D1.59 Two Hsps were also induced in the liver after 24 h, confirming that sleep loss markers are not limited to the brain.18 However, Hsps expression normalized on D14, indicating that chronic sleep quality disruption is likely insufficient to cause a sustained cellular stress response or that this induction is normally not maintained over long periods of time. Along these lines, BiP mRNA levels were lower following 1 w of sleep deprivation than after 8 h of sleep deprivation.60 Also, markers of neocortical synaptic plasticity remained stable during SF contrary to sleep deprivation experiments.18 Homer1a increased in the hippocampus but remained stable in the cortex, and Bdnf transcripts decreased in the cortex whereas the corresponding peptide remained stable. Enforced wakefulness leads to locus coeruleus (LC) activation and extended exposure to noradrenergic transmission which is known to induce synaptic potentiation.61 In conserving total sleep time, SF might not activate LC and induce widespread forebrain plasticity. Moreover, recent evidence suggests that LC activity and excitability might be impaired during SF.36 Among clock-genes, only Clock mRNA levels increased in the somatosensory cortex on D1 when sleep loss was maximal contrary to results obtained in sleep deprivation experiments. Per1 and Per2 expression in the forebrain are under the regulation of Npas2 and Bmal1,26 which were both stable in the cortex. Different clock-genes may have different thresholds to sleep loss; for example, Per1 is induced after only 3 h of sleep deprivation, whereas Per2 is induced after 6 h.24 Our data suggest that sleep loss induced by 1 day of SF is insufficient to trigger the homeostatic response of clock-genes in the forebrain, with the caveat that we only studied their expression at one circadian point. However, recent data show that Per1 and Per2 expression can reflect sleep loss at any circadian time.26 Similar to other pathways studied here, the expression of clock-genes stabilized after 14 days of SF, paralleling normal circadian organization of sleep-wake cycle and locomotor activity toward the end of the protocol. EEG markers of sleep pressure measured here during chronic SF (decreased sleep latency, increased SWA) are insufficient to sustainably engage molecular markers of sleep loss. Our data suggest that cumulative excessive wakefulness and not altered sleep quality may be the proximate cause of transient induction of transcription observed on D1. Given that gene expression was measured only at one circadian time point, at the beginning of the dark phase, it remains unknown whether gene induction might occur concomitantly with the sleep rebound observed here when chronic SF is followed by 6 h of wake in the dark. Conceivably, chronic SF might lower the threshold for gene induction during excessive cumulative wakefulness.
In conclusion, by characterizing a rodent model of chronic SF in detail, we made observations that pertain to the fundamental understanding of sleep homeostasis. Although either the loss of quantity or quality of sleep tightly upregulates SWA, the current study suggests that circadian, Hsp, and plasticity related molecular pathways might not be engaged in the clinical setting of chronic sleep pathologies involving SF. Furthermore, by establishing that constant SF conveys specific sleep waves changes over weeks, this study brings new data to explore the mechanisms of cognitive dysfunctions observed in sleep disorders.
DISCLOSURE STATEMENT
This work was supported by a Swiss National Science Foundation grant (3100AO-108336/1) to Dr. Magistretti. Dr. Baud's work was directly supported by a Swiss National Science Foundation personal MD-PhD grant (323600-119351/1). The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank Joël Gyger for his excellent technical assistance.
SUPPLEMENTAL MATERIAL
Power spectral analysis performed by applying a fast Fourier transform to wake EEG during the sleep fragmentation (SF) protocol. Data were collapsed into 1-Hz bins and normalized by wake baseline recordings (n for QC = 6, MC = 8 and F = 7). Note that there is no slow wave activity intrusion during wakefulness on D1 (A) or D14 (B). Also note the slight increase in theta power in F and MC during D1 dark phase (A, upper panel, P > 0.05) and only in F during D1 light phase (A, lower panel, P < 0.05). F, fragmented; MC, motor control; QC, quiet control.
Analysis of the slow wave activity (SWA) dynamic. Exponential fit of SWA buildup after slow wave sleep onset during the light phase of D14 (A) and dark phase of R1 (B). Horizontal gray bars indicate significance (P < 0.05 on Tukey post-significant one-way analysis of variance). The best fits of individual data were obtained using a saturating exponential y = a(1−et/τ) + y0, (R2 > 0.9 for all) and were averaged (continuous line). Horizontal and vertical histograms plotted against corresponding axis represent mean ± standard deviation of the time constant (τ) and asymptote (a + y0), respectively. In panel A, note that in the F group, SWA build-up rate is significantly shorter on D14 (P < 0.001), In panel B, note that SWA asymptote is significantly higher on R1 (P < 0.001).
Analysis of the EEG amplitude during slow wave sleep. Envelope of the 10–35 Hz filtered EEG after sleep onset on D1 and D14 (black trace) normalized by the average amplitude on the reference day (gray trace). Note the increase in amplitude of the EEG signal in these frequencies. Horizontal gray bars represent paired t test (P < 0.05, D1 versus BSL and D14 versus R2).
Slow waves (SW) analysis. (A,B) Probability distribution histograms of SW on D1 and D14 showing a shift from low to high amplitude categories (P < 0.05, paired t test [(D1]) and P < 0.001, one-way analysis of variance [ANOVA, D14]). The x-axes represents the means of boundaries (in mV) set for individual mice to distribute amplitudes in percentiles (0.025, 0.1, 0.25, 0.5, 0.75, 0.9, 0.95). (C–F) Histograms of SW features in relative values for early (E, first third of light phase slow wave sleep) and late (L, third third) occurrence (100% = either BSL or R2). Note that incidence (number of SW per sec) does not change but that amplitude and slopes of SW increase on D1 and D14 compared to baseline and R2 respectively (P < 0.001, two-way ANOVA).
Spindles analysis. (A,B) Probability distribution histograms of spindles on D1 and D14 showing a shift from low to high amplitude (P < 0.05 on D1 and P < 0.001 on D14). At D1, spindles distribution was compared to baseline value while at D14, spindles distribution was compared to R1 and R2 values. (C–E) Histograms showing no difference in incidence but an increase in the amplitude, buildup, and decay rate of spindles on D1 and D14 compared to baseline and R2, respectively (P < 0.001, two-way analysis of variance [ANOVA] for all) with maintenance of circadian effects when considering late (L) versus early (E; P < 0.005, two-way ANOVA for all).
List of the Forward and Reverse primers used for gene expression assay.
Total time (24 h), 12 h of dark period (D), and 12 h of light period (L) values of wake (W), slow wave sleep (SWS), and paradoxical sleep (PS) during the sleep fragmentation protocol (D1, D7, and D14).
Total time (24 h), 12 h of dark period (D), and 12 h of light period (L) values of wake (W), slow wave sleep (SWS), and paradoxical sleep (PS) during the first and second day of the recovery period (R1 and R2).
Relative gene expression in four brain structures and in the liver following 1 day and 14 days of sleep fragmentation.
REFERENCES
- 1.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–5. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 2.Hossain JL, Shapiro CM. The prevalence, cost implications, and management of sleep disorders: an overview. Sleep Breath. 2002;6:85–102. doi: 10.1007/s11325-002-0085-1. [DOI] [PubMed] [Google Scholar]
- 3.Stepanski EJ. The effect of sleep fragmentation on daytime function. Sleep. 2002;25:268–76. doi: 10.1093/sleep/25.3.268. [DOI] [PubMed] [Google Scholar]
- 4.Aldrich MS. Automobile accidents in patients with sleep disorders. Sleep. 1989;12:487–94. doi: 10.1093/sleep/12.6.487. [DOI] [PubMed] [Google Scholar]
- 5.Spiegel K, Tasali E, Leproult R, Van Cauter E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol. 2009;5:253–61. doi: 10.1038/nrendo.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Dongen HPA, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–26. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- 7.Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 8.Bennett LS, Langford BA, Stradling JR, Davies RJ. Sleep fragmentation indices as predictors of daytime sleepiness and nCPAP response in obstructive sleep apnea. Am J Respir Crit Care Med. 1998;158:778–86. doi: 10.1164/ajrccm.158.3.9711033. [DOI] [PubMed] [Google Scholar]
- 9.Yue HJ, Bardwell W, Ancoli-Israel S, Loredo JS, Dimsdale JE. Arousal frequency is associated with increased fatigue in obstructive sleep apnea. Sleep Breath. 2009;13:331–9. doi: 10.1007/s11325-009-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GarciaBorreguero D, Larrosa O, Delallave Y, Josegranizo J, Allen R. Correlation between rating scales and sleep laboratory measurements in restless legs syndrome. Sleep Med. 2004;5:561–5. doi: 10.1016/j.sleep.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Jiménez-Correa U, Haro R, González RO, Velázquez-Moctezuma J. Correlations between subjective and objective features of nocturnal sleep and excessive diurnal sleepiness in patients with narcolepsy. Arq Neuropsiquiatr. 2009;67:995–1000. doi: 10.1590/s0004-282x2009000600006. [DOI] [PubMed] [Google Scholar]
- 12.Ramesh V, Nair D, Zhang SXL, et al. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-α pathway. J Neuroinflammation. 2012;9:91. doi: 10.1186/1742-2094-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baud MO, Magistretti PJ, Petit J-M. Sustained sleep fragmentation affects brain temperature, food intake and glucose tolerance in mice. J Sleep Res. 2012;22:3–12. doi: 10.1111/j.1365-2869.2012.01029.x. [DOI] [PubMed] [Google Scholar]
- 14.Petit J-M, Gyger J, Burlet-Godinot S, Fiumelli H, Martin J-L, Magistretti PJ. Genes involved in the astrocyte-neuron lactate shuttle (ANLS) are specifically regulated in cortical astrocytes following sleep deprivation in mice. Sleep. 2013;36:1445–58. doi: 10.5665/sleep.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber R, Felice Ghilardi M, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- 16.Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, et al. Cortical firing and sleep homeostasis. Neuron. 2009;63:865–78. doi: 10.1016/j.neuron.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull. 2003;62:143–50. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, et al. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–5. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terao A, Greco MA, Davis RW, Heller HC, Kilduff TS. Region-specific changes in immediate early gene expression in response to sleep deprivation and recovery sleep in the mouse brain. Neuroscience. 2003;120:1115–24. doi: 10.1016/s0306-4522(03)00395-6. [DOI] [PubMed] [Google Scholar]
- 20.Vyazovskiy VV, Riedner BA, Cirelli C, Tononi G. Sleep homeostasis and cortical synchronization: II. A local field potential study of sleep slow waves in the rat. Sleep. 2007;30:1631–42. doi: 10.1093/sleep/30.12.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28:4088–95. doi: 10.1523/JNEUROSCI.5510-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naidoo N. Cellular stress/the unfolded protein response: relevance to sleep and sleep disorders. Sleep Med Rev. 2009;13:195–204. doi: 10.1016/j.smrv.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naidoo N, Casiano V, Cater J, Zimmerman J, Pack AI. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep. 2007;30:557–65. doi: 10.1093/sleep/30.5.557. [DOI] [PubMed] [Google Scholar]
- 24.Wisor JP, Pasumarthi RK, Gerashchenko D, et al. Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J Neurosci. 2008;28:7193–201. doi: 10.1523/JNEUROSCI.1150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curie T, Mongrain V, Dorsaz S, Mang GM, Emmenegger Y, Franken P. Homeostatic and circadian contribution to EEG and molecular state variables of sleep regulation. Sleep. 2013;36:311–23. doi: 10.5665/sleep.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franken P. A role for clock genes in sleep homeostasis. Curr Opin Neurobiol. 2013;23:864–72. doi: 10.1016/j.conb.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Mongrain V, Hernandez SA, Pradervand S, et al. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep. 2010;33:1147–57. doi: 10.1093/sleep/33.9.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Refinetti R, Cornélissen G, Halberg F. Procedures for numerical analysis of circadian rhythms. Biol Rhythm Res. 2007;38:275–325. doi: 10.1080/09291010600903692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–79. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trachsel L, Tobler I, Borbély AA. Electroencephalogram analysis of non-rapid eye movement sleep in rats. Am J Physiol. 1988;255:R27–37. doi: 10.1152/ajpregu.1988.255.1.R27. [DOI] [PubMed] [Google Scholar]
- 31.Lee KS, Alvarenga TA, Guindalini C, Andersen ML, Castro RM, Tufik S. Validation of commonly used reference genes for sleep-related gene expression studies. BMC Mol Biol. 2009;10:45. doi: 10.1186/1471-2199-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szapacs ME, Mathews TA, Tessarollo L, Ernest Lyons W, Mamounas LA, Andrews AM. Exploring the relationship between serotonin and brain-derived neurotrophic factor: analysis of BDNF protein and extraneuronal 5-HT in mice with reduced serotonin transporter or BDNF expression. J Neurosci Methods. 2004;140:81–92. doi: 10.1016/j.jneumeth.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 33.Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat: an update of the 1989 paper. Sleep. 2002;25:18–24. doi: 10.1093/sleep/25.1.18. [DOI] [PubMed] [Google Scholar]
- 34.Schwierin B, Achermann P, Deboer T, Oleksenko A, Borbély AA, Tobler I. Regional differences in the dynamics of the cortical EEG in the rat after sleep deprivation. Clin Neurophysiol. 1999;110:869–75. doi: 10.1016/s1388-2457(99)00020-6. [DOI] [PubMed] [Google Scholar]
- 35.Klein AB, Williamson R, Santini MA, et al. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol. 2011;14:347–53. doi: 10.1017/S1461145710000738. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Panossian LA, Zhang J, et al. Effects of chronic sleep fragmentation on wake-active neurons and the hypercapnic arousal response. Sleep. 2014;37:51–64. doi: 10.5665/sleep.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kas MJ, Edgar DM. Circadian timed wakefulness at dawn opposes compensatory sleep responses after sleep deprivation in Octodon degus. Sleep. 1999;22:1045–53. doi: 10.1093/sleep/22.8.1045. [DOI] [PubMed] [Google Scholar]
- 38.Klerman EB, Boulos Z, Edgar DM, Mistlberger RE, Moore-Ede MC. Circadian and homeostatic influences on sleep in the squirrel monkey: sleep after sleep deprivation. Sleep. 1999;22:45–59. doi: 10.1093/sleep/22.1.45. [DOI] [PubMed] [Google Scholar]
- 39.Leemburg S, Vyazovskiy VV, Olcese U, Bassetti CL, Tononi G, Cirelli C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc Natl Acad Sci U S A. 2010;107:15939–44. doi: 10.1073/pnas.1002570107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 41.Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–7. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knoblauch V, Martens WLJ, Wirz-Justice A, Cajochen C. Human sleep spindle characteristics after sleep deprivation. Clin Neurophysiol. 2003;114:2258–67. doi: 10.1016/s1388-2457(03)00238-4. [DOI] [PubMed] [Google Scholar]
- 43.Amzica F, Steriade M. Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol. 1998;107:69–83. doi: 10.1016/s0013-4694(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 44.Dijk DJ. EEG slow waves and sleep spindles: windows on the sleeping brain. Behav Brain Res. 1995;69:109–16. doi: 10.1016/0166-4328(95)00007-g. [DOI] [PubMed] [Google Scholar]
- 45.Mongrain V, Dumont M. Increased homeostatic response to behavioral sleep fragmentation in morning types compared to evening types. Sleep. 2007;30:773–80. doi: 10.1093/sleep/30.6.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferri R, Drago V, Aricò D, et al. The effects of experimental sleep fragmentation on cognitive processing. Sleep Med. 2010;11:378–85. doi: 10.1016/j.sleep.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franken P. Long-term vs. short-term processes regulating REM sleep. J Sleep Res. 2002;11:17–28. doi: 10.1046/j.1365-2869.2002.00275.x. [DOI] [PubMed] [Google Scholar]
- 48.Benington JH, Kodali SK, Heller HC. Scoring transitions to REM sleep in rats based on the EEG phenomena of pre-REM sleep: an improved analysis of sleep structure. Sleep. 1994;17:28–36. doi: 10.1093/sleep/17.1.28. [DOI] [PubMed] [Google Scholar]
- 49.Beersma DG, Dijk DJ, Blok CG, Everhardus I. REM sleep deprivation during 5 hours leads to an immediate REM sleep rebound and to suppression of non-REM sleep intensity. Electroencephalogr Clin Neurophysiol. 1990;76:114–22. doi: 10.1016/0013-4694(90)90209-3. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt C. Encoding difficulty promotes postlearning changes in sleep spindle activity during napping. J Neurosci. 2006;26:8976–82. doi: 10.1523/JNEUROSCI.2464-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- 52.Hanlon EC, Faraguna U, Vyazovskiy VV, Tononi G, Cirelli C. Effects of skilled training on sleep slow wave activity and cortical gene expression in the rat. Sleep. 2009;32:719–29. doi: 10.1093/sleep/32.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward CP, McCarley RW, Strecker RE. Experimental sleep fragmentation impairs spatial reference but not working memory in Fischer/Brown Norway rats. J Sleep Res. 2009;18:238–44. doi: 10.1111/j.1365-2869.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward CP, McCoy JG, McKenna JT, Connolly NP, McCarley RW, Strecker RE. Spatial learning and memory deficits following exposure to 24 h of sleep fragmentation or intermittent hypoxia in a rat model of obstructive sleep apnea. Brain Res. 2009;1294:128–37. doi: 10.1016/j.brainres.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akerstedt T, Kecklund G, Ingre M, Lekander M, Axelsson J. Sleep homeostasis during repeated sleep restriction and recovery: support from EEG dynamics. Sleep. 2009;32:217–22. doi: 10.1093/sleep/32.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna JT, Tartar JL, Ward CP, et al. Sleep fragmentation elevates behavioral, electrographic and neurochemical measures of sleepiness. Neuroscience. 2007;146:1462–73. doi: 10.1016/j.neuroscience.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vyazovskiy VV, Tobler I. Theta activity in the waking EEG is a marker of sleep propensity in the rat. Brain Res. 2005;1050:64–71. doi: 10.1016/j.brainres.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 58.Van Dongen HPA, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–26. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- 59.Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- 60.Cirelli C, Faraguna U, Tononi G. Changes in brain gene expression after long-term sleep deprivation. J Neurochem. 2006;98:1632–45. doi: 10.1111/j.1471-4159.2006.04058.x. [DOI] [PubMed] [Google Scholar]
- 61.Cirelli C, Tononi G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci. 2000;20:9187–94. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Power spectral analysis performed by applying a fast Fourier transform to wake EEG during the sleep fragmentation (SF) protocol. Data were collapsed into 1-Hz bins and normalized by wake baseline recordings (n for QC = 6, MC = 8 and F = 7). Note that there is no slow wave activity intrusion during wakefulness on D1 (A) or D14 (B). Also note the slight increase in theta power in F and MC during D1 dark phase (A, upper panel, P > 0.05) and only in F during D1 light phase (A, lower panel, P < 0.05). F, fragmented; MC, motor control; QC, quiet control.
Analysis of the slow wave activity (SWA) dynamic. Exponential fit of SWA buildup after slow wave sleep onset during the light phase of D14 (A) and dark phase of R1 (B). Horizontal gray bars indicate significance (P < 0.05 on Tukey post-significant one-way analysis of variance). The best fits of individual data were obtained using a saturating exponential y = a(1−et/τ) + y0, (R2 > 0.9 for all) and were averaged (continuous line). Horizontal and vertical histograms plotted against corresponding axis represent mean ± standard deviation of the time constant (τ) and asymptote (a + y0), respectively. In panel A, note that in the F group, SWA build-up rate is significantly shorter on D14 (P < 0.001), In panel B, note that SWA asymptote is significantly higher on R1 (P < 0.001).
Analysis of the EEG amplitude during slow wave sleep. Envelope of the 10–35 Hz filtered EEG after sleep onset on D1 and D14 (black trace) normalized by the average amplitude on the reference day (gray trace). Note the increase in amplitude of the EEG signal in these frequencies. Horizontal gray bars represent paired t test (P < 0.05, D1 versus BSL and D14 versus R2).
Slow waves (SW) analysis. (A,B) Probability distribution histograms of SW on D1 and D14 showing a shift from low to high amplitude categories (P < 0.05, paired t test [(D1]) and P < 0.001, one-way analysis of variance [ANOVA, D14]). The x-axes represents the means of boundaries (in mV) set for individual mice to distribute amplitudes in percentiles (0.025, 0.1, 0.25, 0.5, 0.75, 0.9, 0.95). (C–F) Histograms of SW features in relative values for early (E, first third of light phase slow wave sleep) and late (L, third third) occurrence (100% = either BSL or R2). Note that incidence (number of SW per sec) does not change but that amplitude and slopes of SW increase on D1 and D14 compared to baseline and R2 respectively (P < 0.001, two-way ANOVA).
Spindles analysis. (A,B) Probability distribution histograms of spindles on D1 and D14 showing a shift from low to high amplitude (P < 0.05 on D1 and P < 0.001 on D14). At D1, spindles distribution was compared to baseline value while at D14, spindles distribution was compared to R1 and R2 values. (C–E) Histograms showing no difference in incidence but an increase in the amplitude, buildup, and decay rate of spindles on D1 and D14 compared to baseline and R2, respectively (P < 0.001, two-way analysis of variance [ANOVA] for all) with maintenance of circadian effects when considering late (L) versus early (E; P < 0.005, two-way ANOVA for all).
List of the Forward and Reverse primers used for gene expression assay.
Total time (24 h), 12 h of dark period (D), and 12 h of light period (L) values of wake (W), slow wave sleep (SWS), and paradoxical sleep (PS) during the sleep fragmentation protocol (D1, D7, and D14).
Total time (24 h), 12 h of dark period (D), and 12 h of light period (L) values of wake (W), slow wave sleep (SWS), and paradoxical sleep (PS) during the first and second day of the recovery period (R1 and R2).
Relative gene expression in four brain structures and in the liver following 1 day and 14 days of sleep fragmentation.