

Abstract
The major neuronal gap junction protein Connexin 36 (Cx36) exhibits the remarkable property of “run-up”, in which junctional conductance typically increases by ten-fold or more within 5–10 min following cell break-in with patch pipettes. Such conductance “run-up” is a unique property of Cx36, as it has not been seen in cell pairs expressing other connexins. Because of the recent observation describing CaMKII binding and phosphorylation sites in Cx36 and evidence that calmodulin dependent protein kinase II (CaMKII) may potentiate electrical coupling in neurons of teleosts, we have explored whether CaMKII activates mammalian Cx36. Consistent with this hypothesis, certain Cx36 mutants lacking the CaMKII binding and phosphorylation sites or wild type Cx36 treated with certain cognate peptides corresponding to binding or phosphorylation sites blocked or strongly attenuated run-up of junctional conductance. Likewise, KN-93, an inhibitor of CaMKII, blocked run-up, as did a membrane permeable peptide corresponding to the CaMKII autoinhibitory domain. Furthermore, run-up was blocked by phosphatase delivered within the pipette and not affected by treatment with the phosphatase inhibitor okadaic acid. These results imply that phosphorylation by CaMKII strengthens junctional currents of Cx36 channels, thereby conferring functional plasticity on electrical synapses formed of this protein.
Keywords: electrical synapse, synaptic plasticity, CaMKII
INTRODUCTION
Connexins (Cx) comprise a family of proteins containing four membrane-spanning domains that form hexameric connexons (or hemichannels) in the plasma membrane of mammalian cells. When hemichannels of neighboring cells dock with one another, a gap junction channel can be formed that allows electrical and metabolic communication between neighboring cells. Gap junctions between neurons function as electrical synapses (Bennett, 1977; Connors and Long, 2004; Spray, 2004). The most prominent neuronal gap junction protein is Cx36 in mammals, which was first identified in screens for brain connexins homologous to the fish Cx35 (Condorelli et al., 1998; O’Brien et al., 1996; Söhl et al., 1998). More recently Cx45, Cx50, Cx57, and Cx30.2 have also been reported to be expressed in certain mammalian neurons (Hombach et al., 2004; Kreuzberg et al., 2008; Maxeiner et al., 2003; O’Brien et al, 2006).
Whereas chemical synapses exhibit use-dependent plasticity and are therefore thought to underlie learning and memory storage, electrical synapses formed by gap junctions have been widely believed to be passive intercellular conduits with little or no dynamic control. However, electrical synapses in both fish and mammalian brain have been shown to undergo long-term changes in strength (Landisman and Connors, 2005; Yang et al., 1990). Recent evidence implicates two protein kinases [calcium/calmodulin-dependent kinase (CaMKII) and cAMP dependent protein kinase (PKA)] in long term functional plasticity of gap junctions in goldfish Mauthner cell (Pereda et al., 2003; Pereda et al., 1998; Cachope et al., 2007) and in both teleost and rodent retina (Kothmann et al., 2009; Li et al., 2009). We recently reported that CaMKII binds Cx36 in a manner analogous to binding of this kinase to the NR2B subunit of the NMDA receptor and phosphorylates the gap junction protein both in situ and in vitro (Alev et al., 2008).
Early studies on exogenously expressed green fluorescent protein (GFP) – tagged and unmodified Cx36 revealed that junctional conductance progressively increased over time after establishment of the whole cell recording configuration with patch clamp electrodes (Zoidl et al., 2002). We now report that certain peptides corresponding to sites where we have shown CaMKII binds and phosphorylates Cx36 (Alev et al, 2008)and deletion of corresponding CaMKII binding and phosphorylation sitesleads to a significant loss of this “run up” activity. These results indicate that Cx36 interaction with CaMKII endows electrical synapses with a high degree of functional plasticity, implying that activity dependent modulation is a characteristic of electrical as well as chemical synapses.
RESULTS
Neuroblastoma cells (N2A) were transiently transfected with the cDNA encoding rat connexin36 (rCx36) tagged with green fluorescence protein (Cx36-EGFP) or double transfected with the full length coding regions of rCx36 and EGFP in separate plasmids. Electrical coupling was assessed in transiently transfected rCx36 cell pairs using dual whole cell patch clamp technique within 18 to 60 hours after transfection. A combined brightfield and immunoflourescence micrograph of a cell pair is shown in Figure 1A; note the abundant Cx36-EGFP distribution of Cx36 in membrane domains, with only minor amounts in cytoplasmic or intracellular membrane compartments. We have previously shown the phenomenon of “run-up” in Cx36 transfected cells and concluded that it is not appreciably affected by the presence of a fluorophore on the carboxyl terminus (Zoidl et al., 2002). A movie illustrating the accrual of junctional current as a function of time after dual whole cell recording is shown in Supplemental Figure S1. Note that at the beginning of the recording illustrated in this film clip, electrical coupling measured as junctional current (Ij) and junctional conductance (Gj) is very low, but in subsequent recordings “run-up” occurs in which junctional current and conductance gradually increased about 10-fold.
Figure 1.

Time course of “run-up” in Cx36-GFP transfected Neuro2A cells A. Phase (upper) and fluorescence (lower) micrograph of a pair of Cx36-GFP transfected Neuro2A cells. Arrows indicate junctional plaques between the cells. B. Normalized “run-up” from 18 cell pairs in which long term recording was achieved. Continuous black line represents data fitting with Boltzmann equation with half time of 6.35 min and maximal slope of 0.45 fold/min.
In order to measure the time course of this response with more precision, we quantified normalized junctional currents in eighteen cell pairs during 14 min recordings of responses to voltage ramps (mean values and SEs shown in Fig. 1B). When the resulting composite curve was fit with a Boltzmann relation using Graphpad Prism, halftime was about 6.35 min and maximal slope was 0.45 fold/min. The unitary conductance of Cx36 channels is about 15 pS (Srinivas et al., 1999) and the initial conductance averaged 160±10 pS, so that this slope corresponds to the accrual of about thirty channels/min during the period of the experiment before apparent saturation occurred.
The “run-up” phenomenon might be due to the loss, redistribution or rearrangement of intracellular proteins during cell dialysis induced by the patch clamp procedure. To test this hypothesis we used the perforated patch method (100 μg/ml amphotericin B) to obtain electrical access to the cell interior with limited dialysis. In all cases, electrical access was identified by the larger and slower capacitive transient current response to a 10 mV voltage step after 5 to 20 minutes in both cells from the pair. Figure 2A shows averaged results for increase in Gj observed in five cell pairs that were first recorded in perforated patch conditions (open circles) and after the breakage of the previously perforated membrane patch, which allows dialysis of the cell interior by pipette solution (closed circles). Figure 2B shows a similar perforated patch experiment in which cells were exposed to the calcium ionophore ionomycin after amphotericin access had been achieved. Note that the ionophore application significantly increased the extent of run-up (quantified in Fig. 2B; values given in Fig. 2D). This result would be consistent with an acute elevation of Ca2+ upon break in triggering run-up. In order to further test this hypothesis we exposed cell pairs to the membrane permeant Ca2+ buffer BAPTA AM (25 uM) for 15 min to two hours. The average normalized results from four experiments are shown in Fig. 2C, where run-up under these conditions is shown to be minimal, significantly less than with normal intracellular solution.
Figure 2.
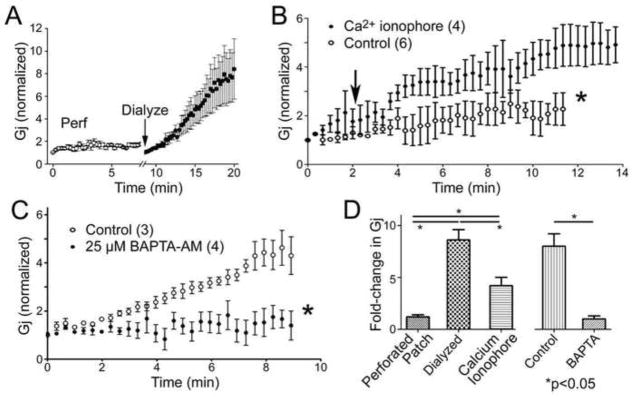
Perforated patch clamp recordings of “run-up”. A. Using amphotericin B, cells were held for 10 min at which time electrical access to cell interior was achieved through insertion of monovalent cation channels. The degree of run-up during the subsequent 10 min period was minimal, whereas run-up was quickly obtained when access to cell interior was obtained with strong suction “Dialyze”, arrow. N=3 cell pairs. B. Run-up was obtained in perforated patch recordings in the presence of calcium ionophore (ionomycin, 1 μM, added at arrow). C. Run-up was largely prevented in dialyzed cells by the Ca buffer BAPTA (25 μM) included in the patch pipette. D. Quantitative comparison of the fold changes during run-up in multiple experiments under perforated patch, dialyzed and perforated patch and Ca ionophore conditions.
These studies indicate that elevated intracellular Ca2+ likely contributes to the run-up phenomenon, which could ultimately lead to increased trafficking and insertion of Cx36 channels or to altered function of channels already present in the junctional membrane. One way in which gap junction channel function has been shown to modulated is through phosphorylation of the connexin proteins, and we have recently shown that CaMKII binds to and phosphorylates Cx36 (Alev et al., 2008). Moreover, CaMKII inhibition is reported to interfere with potentiation of electrotonic PSPs in Mauthner cell (Pereda et al., 1998). Therefore, we tested whether inhibition of CaMKII would interfere with the run-up phenomenon, as illustrated in Figure 3. When cells were exposed to the CaMKII inhibitor KN-93 (50μM) during the “run up” process, the conductance increase was blocked until the drug was removed (Fig. 3A); moreover, pretreatment of cell pairs with the same CaMKII inhibitor (100 μM) largely inhibited the “run-up” phenomenon (Fig. 3B). In addition, we tested for effects on run-up of a membrane permeable inhibitor of CaMKII (TAT-CN21a). As illustrated in Fig. 3C, exposure of Cx36 transfected cell pairs to 50uM TAT peptide largely reduced the extent of run-up seen under dual whole cell recording conditions. Together, these data support the hypothesis that CaMKII activation may play an essential role in the plasticity of Cx36 junctional conductance in Cx36 transfected cells. Moreover, it leads to the new hypothesis that this modulation could involve binding and phosphorylation sites in Cx36 cytoplasmic domains.
Figure 3.

Calmodulin inhibitors arrest and block run-up in Cx36 transfectants. A. Application of KN-93 (50 μM) during run-up reversibly blocked the increase in conductance, which resumed after the drug is washed away. B. Pre-treatment (20 min) with 100 μM KN-93 substantially reduced the extent of run-up. C. Treatment with the membrane permeable TAT-CN21 at the beginning of the experiment substantially reduced final junctional conductance. D. Quantitation of effects of KN-93 and the peptide CaMKII inhibitor on runup. *P<0.05
We previously identified distinct cytoplasmic domains of Cx36 that are bound and phosphorylated by CaMKII (Alev et al., 2008); see Fig 4A). In order to evaluate the role of these individual domains in conductance run-up, we introduced peptides corresponding to these domains that we had previously used for the biochemistry experiments through patch pipettes and also tested Cx36 mutants lacking these domains. As illustrated in Fig. 4 peptide corresponding to the sequence of the cytoplasmic loop phosphorylated by CaMKII (CLP) had minimal effect on run-up (Fig 4B), whereas peptides corresponding to the CT phosphorylation site (CTP) and to binding sites at the cytoplasmic loop (CLB) and carboxy-terminal domain (CTB) substantially decreased the “run-up” (Fig. C,D,E). This finding indicates that CaMKII binding sites and the run-up phenomenon are linked. Together, these data indicate that CaMKII seems to bind either to CLB and CTB sites of Cx36 and that phosphorylation of CTP is critical to trigger the “run-up” phenomenon.
Figure 4.

Effects of intracellular peptides on rate and extent of run-up. A. Peptide sequences corresponding to binding and phosphorylation domains of Cx36 cytoplasmic regions. In each panel, control and peptide experiments were done on the same days for comparison. B. CLP peptide had no effect on run-up. C. CLB peptide blocked run-up virtually completely. D. CTB peptide strongly inhibited run-up. E. CTP (CT2) peptide also reduced extent of run-up. Asterisks in C-E indicate p<0.5 compared to control.
Since the peptide application procedure itself might obscure essential protein interactions involved in the observed phenomenon, we evaluated mutants in which site directed mutagenesis selectively deleted Cx36 binding and phosphorylation sites. Consistent with the result shown above (Fig. 4) deletion of the CTB region led to a substantial decrease of the run-up, as did the construct representing double deletion of CTB and CLB (compare Fig. 5A,D). Although run-up was not totally eliminated by the CLB deletion (Fig 5B), plotting normalized data on the same axis reveals substantial blockade by this mutant as well. In contrast, the deletion of the CLP region was without effect (not shown). This suggests that the both CaMKII binding regions and the CT phosphorylation region are essential for the run-up.
Figure 5.

Deletions of specific regions of Cx36 reduce the extent of run-up. A. Combined controls for all experiments, performed on the same days as the mutant experiments. Note approximately >5 fold run-up at 13 min. N=9 cell pairs. B. Deletion of region CLB decreased but did not prevent run-up. N=7–9 cell pairs/time point. C. Deletion of region CTB (N=4) or both CTB and CLB (N=6) (D) strongly reduced run-up. E. Normalized plots of each of these data sets illustrating that each mutant (n=4–6 for each) showed strong differences in extent of run-up. Although not graphed, ΔCTB was also significantly different from control. Asterisks indicate p<0.05.
In order to determine whether phosphorylation/dephosphosphorylation was necessary for the run-up phenomenon, we tested effects of alkaline phosphatase and a phosphatase inhibitor (okadaic acid). When added to the pipette solution alkaline phosphatase (1 U/ml) led to decreased junctional conductance during the course of the experiment (Fig. 6A,B), whereas run-up still occurred in the presence of okadaic acid (6 μM, 30–75 min pretreatment at 37° C: Fig 6C,D).
Figure 6.

Runup was inhibited by phosphatase treatment and was preserved when cells are pretreated with a phosphatase inhibitor. A. An example of a recording from a cell pair in which each pipette contained alkaline phosphatase (1U/ml). In three experiments of this type, junctional conductance was reduced, although time course of uncoupling varied, as illustrated in B. C. An example of a recording of runup following incubation in the presence of okadaic acid (6 μM for 30–75 min). D. Means and SE of runup in three experiments in the presence of okadaic acid.
DISCUSSION
In this article we provide evidence that CaMKII can directly modulate the junctional currents of Cx36 channels to induce a conductance “run-up” phenomenon. This property is unique to Cx36 and has not been seen in cell pairs expressing other connexins, although it may be similar in some respects to the long term increase in coupling (LINC) that occurs in astrocytes following elevation of intracellular Ca2+ (De Pina-Benabou et al., 2001). Our in vitro observation is supported by a similar phenomenon recently described by Veruki and colleagues (Hartveit and Veruki, 2012; Veruki et al., 2008). In the retina AII amacrine cells express Cx36 and form a network of electrically coupled interneurons (Feigenspan et al., 2001). Targeted disruption of Cx36 is known to cause visual transmission deficits in mice (Guldenagel et al., 2001) underlining the importance of Cx36 mediated gap junction coupling. Cx36 in AII amacrine cells undergoes a very similar physiological modulation of Gj, appearing as a time-dependent increase from about 500 pS to a maximum of about 3,000 pS after 30–90 min of patch clamp recording (Veruki et al., 2008).
The increased junctional conductance over time could be due to increased unitary conductance, increased open probability of existing channels or addition of new channels to the junctional plaque. In our original publication describing Cx36 channels (Srinivas et al, 1999), we showed that unitary conductance was very similar in poorly coupled cells with only a few channels open and after acutely uncoupling cell pairs with halothane (Fig. 3). Thus, it is highly unlikely that altered channel unitary conductance accounts for the difference observed in the runup phenomenon.
The most likely explanation for the time-dependent run-up of Gj is increased open probability following intracellular washout of an intrinsic regulatory factor and/or altered phosphorylation of Cx36. However, alternative possibilities have to be considered. For example, it is possible that longer-lasting whole cell recordings lead to a transport dependent increase in the total number of channels in the gap-junction contacts between coupled cells. Assuming a continuous turnover of Cx36, an increase in the total number of channels could be equally well caused by an increase in the number of inserted channels, a decrease in the number of internalized channels, or both (for review, see (Laird, 2005).
We believe that the experimental evidence presented here favors a role of CaMKII increasing channel open time rather than affecting channel insertion or internalization. The results showing pharmacological blockade of CaMKII using KN-93 and with cognate peptides applied intracellularly, together with site directed mutagenesis of previously described CaMKII binding and phosphorylation sites of Cx36 connect the previously reported direct interaction of CaMKII with its binding sites CLB and CTB as well as the CaMKII phosphorylation site at S315 of Cx36 (Alev et al., 2008) to open probability of neuronal gap junctions. It is very likely that the modulatory control of Gj between electrically coupled cells is preferentially mediated by changing the open probability of Cx36 connexons through interaction with CaMKII.
Electrical synapses in both fish and mammalian brain show long-term changes in strength (Hass and Landisman, 2012; Pereda et al., 2004). More recent studies implicate two protein kinases [calcium/calmodulin-dependent kinase (CaMKII) and cAMP dependent protein kinase (PKA)] and postsynaptic elevation of Ca2+ in long term functional plasticity of gap junctions in goldfish Mauthner cell (Pereda et al., 2003; Pereda et al., 1998; Smith and Pereda, 2003). The calcium/calmodulin-dependent kinase II (CaMKII) appears to play a critical role in this phenomenon, but the underlying mechanisms of how CaMKII affects the neuronal gap junction protein connexin36 (Cx36) were essentially unknown until we demonstrated effective binding of CaMKII to two juxtamembrane cytoplasmic domains of Cx36 and in vitro phosphorylation of this protein by the kinase. Since both domains reveal striking similarities with segments of the regulatory subunit of CaMKII including the pseudosubstrate and pseudotarget sites of the kinase, we proposed at that time that Cx36, in analogy to NR2B interaction with CaMKII, provides a mechanistic framework for CaMKII and Cx36 interaction at electrical synapses. The functional studies presented here indicate that, like chemical synapses, the gap junction type that forms electrical synapses possesses plasticity of strength of intercellular interaction that depends on CaMKII activation.
An additional prominent feature of NMDA receptors is magnesium dependent blockade. There have been numerous reports of gap junction channel closure following exposure of the channel lumen to moderate Mg2+ concentrations (see Matsuda et al., 2010), and recent experiments on Cx36 channels indicate such effects (personal communication, N. Palacios- Prado, MVL Bennett and F. Bukauskas). Whether the underlying cause of the Mg2+ effect on gap junction channels is channel blockade, phosphatase inhibition or another mechanism remains to be determined, but may provide additional evidence of similarities of mechanisms for plasticity in electrotonic and chemical (glutamatergic) synapse, respectively.
The studies reported here were carried out entirely on transfected cells in tissue culture. Although we believe that the CaMKII-mediated change in juncitonal conductance likely underlies the long term functional plasticity of intercellular coupling in goldfish Mauthner cell (Pereda et al., 2003; Pereda et al., 1998; Cachope et al., 2007) and in teleost and rodent retinas (Kothmann et al., 2009; Li et al., 2009), it is also possible that it is responsible for such phenomena as the increasing frequency of oscillatory pre-ictal activity (“glissandi”) that the authors modeled as attributable to increased coupling among cortical pyramidal cells (Cunningham et al, 2012). Based on our experiments, we expect that such runup should occur in any cell type expressing Cx36 under conditions in which CaMKII is activated, which might include not only highly active neurons, but also β cells of pancreatic islets.
MATERIAL AND METHODS
Cx36 Peptides and Cx36-EYFP expression plasmids
Peptides corresponding to cytoplasmic domains of Cx36 were synthesized at >98% purity (HPLC) at the Einstein Laboratory of Molecular Analysis and Proteomics (LMAP). Those used in this study were those that we previously used for biochemical identification of CaMKII binding and phosphorylation sites (Alev et al, 2008) and for which we have published structural features (Fort and Spray, 2009). They corresponded to the predicted CaMKII phosphorylation sites in the cytoplasmic loop (aa99–119, termed CLP here and in (Alev et al., 2008), and termed Cx36CL1 in (Fort and Spray, 2009) and carboxyl terminus (aa 300–321, CTP or Cx36CT2) and the predicted CaMKII binding regions in cytoplasmic loop (aa 175–195, CLB or Cx36CL3) and carboxyl terminus (aa 272–292, CTB or Cx36CT1). These peptides were diluted in the pipette solution (final concentration: 200 μM). Membrane permeable TAT-CN21a, a peptide that corresponds to the autoinhibitory domain of CaMKII and totally blocks CaMKIIα and β activity (Vest et al., 2007) was a kind gift of Dr. Ulrich Bayer (University of Colorado School of Medicine, Boulder, CO).
Full-length rat Cx36 cDNA was a gift from Dr. D. Condorelli (University of Catania, Italy). The EYFP expression vector pEYFP-N1 was purchased from Clontech (Palo Alto, CA). Isolation and subcloning of Cx36 has been described previously (Zoidl et al., 2002). The plasmids pEYFP-Cx36wt and the Cx36 mutants Cx36CLB-Del (Δ175–185) or Cx36CTB-Del (Δ272–292) and Cx36 double site mutant (Cx36CLB and CTB-Del) were generated using the Transformer TM Site-Directed Mutagenesis Kit (Clontech) based on the protocol of Deng and Nickoloff (Deng and Nickoloff, 1992) as described previously (Zoidl et al., 2002).
Cells and Electrophysiology
The data shown here were entirely obtained from mouse neuroblastoma (Neuro2A) cells that were either transiently co-transfected with rat (r) Cx36 and enhanced green fluorescent protein (EGFP) cDNAs in separate vectors, transiently transfected with c-terminally tagged rCx36-EGFP (see Fig. 1A) or stably co-transfected with rCx36- and EGFP- Puromycin after antibiotic selection [as described: (Alev et al., 2008)]. Although not quantified, similar responses were obtained from rat insulinoma cells transfected with the same vectors (data not shown). Cells were kept overnight in transfection media and the next morning plated with DMEM on round sterile glass coverslips maintained within 35 mm culture dishes. For electrophysiological recordings, coverslips at 24–48 hr after transfection were placed in a recording chamber on an inverted phase-contrast microscope (Nikon Diaphot equipped for epifluorescence). The bathing solution was composed of (in mM): NaCl 140, KCl 5, CsCl 2, CaCl2 2, MgCl2 1, Hepes 5, D-glucose 5, pyruvate 2 and BaCl2 1, pH 7.4, adjusted with NaOH. Patch pipettes pulled with a P97 Flaming/Brown micropipette puller (Sutter Instrument Co., Novato, CA, USA) had resistances of 3 – 5 MΩ when filled with internal solution containing (in mM): 130 CsCl, 10 EGTA, 0.5 CaCl2, and 10 Hepes, pH titrated to 7.3 with CsOH. Cells were intermittently or constantly perfused with bathing solution through a gravity-fed perfusion system with a solution exchange time of about 25 seconds and flow of 1.5 ml/min.
Junctional current was measured between cell pairs [described in (del Corsso et al., 2006)] using the dual whole cell voltage clamp technique with Axopatch 1D patch clamp amplifiers (Axon Instruments Inc., Union City, CA, USA) at room temperature (22 to 25°C). Data were acquired using pClamp6 or later software (Axon Instruments Inc.) and digitized at 1 – 2 kHz sampling rate. Each cell of a pair was initially held at a holding potential (Hp) of 0 mV. To evaluate junctional coupling, 300 msec hyperpolarizing pulses from a Hp of 0 mV to −10 mV were applied to one cell to establish a transjunctional voltage gradient (Vj) and the junctional current was measured in the second cell. Macroscopic current (Ij) recordings were filtered at 0.2 – 0.5 kHz. Macroscopic junctional conductances (Gj) were calculated as Gj = Ij / Vj, where Ij is the measured junctional current and Vj is transjunctional voltage (Srinivas et al., 1999). Time course of Gj runup was obtained by applying slow voltage ramps (from −100 mV to +100 mV, 6 seconds duration) every 20 seconds for 10 to 15 minutes, where the voltage was applied to one cell of the pair and the junctional current response was recorded from the neighboring cell. Data are presented as means ± SEM.
Perforated patch recordings
In order to minimize dialysis of internal molecules, we used the perforated patch method with Amphotericin B (100μg/ml) included in the internal solution. After seal formation, permeabilization was evidenced by stable time constant of the capacitive current in response to −10 mV steps until time constant of the capacitive current was stable (5–20 min). see (del Corsso et al., 2006). Recordings were generally begun at 10 min after seal formation, at which time the capacitive transient was generally stable.
Ca2+ buffering, inhibition of CaMKII activation and manipulation of phosphorylation
In order to block the rise in intracellular Ca2+ that accompanies “break-in” during whole cell recordings, we exposed cells to 25 uM BAPTA-AM (Invitrogen) for 30 min to two hours and then performed recordings. To inhibit activation of CaMKII, we applied the drug KN-93 at 50–100 μM; in addition, we applied a membrane permeant inhibitor of CaMKII at the start of the recording [50 μM TAT-CN21 (Vest et al., 2007)]. In order to dephosphorylate Cx36, we added alkaline phosphate to the pipette solution (1U/ml; bovine intestinal mucosal alkaline phosphatase, Sigma Aldrich); to inhibit phosphatases, we incubated cells for 30–75 min with 6 μM okadaic acid, an inhibitor of phosphatases 1 and 2A.
Membrane permeable drugs (KN-93, inomycin, okadaic acid, BAPTA-AM and TAT-CN21) were added to extracellular solution; nonpermeant compounds (peptides, alkaline phosphatase) were added to the solution in both patch pipettes, as was Amphotericin B.
Data were analyzed from at least three independent sets of experiments using GraphPad Prism 5 software (San Diego, CA, USA) or Origin (OriginLab Corporation; Northampton, MA.. Statistical differences were determined by t-tests and one way ANOVA followed by Tukey’s multiple comparison test. P < 0.05 was considered statistically significant.
All compounds were from Sigma Aldrich unless otherwise noted.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH RO1 NS041282 to David C. Spray) and a grant from the German Research Council (DFG De 292/11) to R. D. and G.Z. We are grateful to Ms. Marcia Urban-Maldonado (NY) and Ms. Christiane Zoidl (Germany) for the excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alev C, et al. The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc Natl Acad Sci U S A. 2008;105:20964–9. doi: 10.1073/pnas.0805408105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MVL. Electrical transmission; a functional analysis and comparison to chemical transmission, Vol. American Physiological Society; Washington: 1977. [Google Scholar]
- Cachope R, et al. Potentiation of electrical and chemical synaptic transmission mediated by endocannabinoids. Neuron. 2007;56:1034–47. doi: 10.1016/j.neuron.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli DF, et al. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. European Journal of Neuroscience. 1998;10:1202–1208. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- Connors BW, Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- Cunningham MO, Roopun A, Schofield IS, Whittaker RG, Duncan R, Russell A, Jenkins A, Nicholson C, Whittington MA, Traub RD. Glissandi: transient fast electrocorticographic oscillations of steadily increasing frequency, explained by temporally increasing gap junction conductance. Epilepsia. 2012 doi: 10.1111/j.1528-1167.2012.03530.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pina-Benabou MH, et al. Calmodulin kinase pathway mediates the K+-induced increase in Gap junctional communication between mouse spinal cord astrocytes. J Neurosci. 2001;21:6635–43. doi: 10.1523/JNEUROSCI.21-17-06635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Corsso C, et al. Transfection of mammalian cells with connexins and measurement of voltage sensitivity of their gap junctions. Nat Protoc. 2006;1:1799–809. doi: 10.1038/nprot.2006.266. [DOI] [PubMed] [Google Scholar]
- Deng WP, Nickoloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal Biochem. 1992;200:81–8. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, et al. Expression of neuronal connexin36 in AII amacrine cells of the mammalian retina. J Neurosci. 2001;21:230–9. doi: 10.1523/JNEUROSCI.21-01-00230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort AG, Spray DC. Trifluoroethanol reveals helical propensity at analogous positions in cytoplasmic domains of three connexins. Biopolymers. 2009;92:173–82. doi: 10.1002/bip.21166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldenagel M, et al. Visual transmission deficits in mice with targeted disruption of the gap junction gene connexin36. J Neurosci. 2001;21:6036–44. doi: 10.1523/JNEUROSCI.21-16-06036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E, Veruki MI. Electrical synapses between AII amacrine cells in the retina. Function and modulation. Brain Research. 2012 doi: 10.1016/j.brainres.2012.05.060. in press. [DOI] [PubMed] [Google Scholar]
- Hass J, Landisman CE. Bursts modify electrical synaptic strength. Brain Research. 2012 doi: 10.1016/j.brainres.2012.05.061. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach S, et al. Functional expression of connexin57 in horizontal cells of the mouse retina. Eur J Neurosci. 2004;19:2633–40. doi: 10.1111/j.0953-816X.2004.03360.x. [DOI] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC, O’Brien J. Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci. 2009;29:14903–11. doi: 10.1523/JNEUROSCI.3436-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzberg MM, et al. Expression of connexin30.2 in interneurons of the central nervous system in the mouse. Mol Cell Neurosci. 2008;37:119–34. doi: 10.1016/j.mcn.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–82. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Connors BW. Long-term modulation of electrical synapses in the mammalian thalamus. Science. 2005;310:1809–13. doi: 10.1126/science.1114655. [DOI] [PubMed] [Google Scholar]
- Li H, Chuang AZ, O’Brien J. Photoreceptor coupling is controlled by connexin 35 phosphorylation in zebrafish retina. J Neurosci. 2009;29:15178–86. doi: 10.1523/JNEUROSCI.3517-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, et al. Magnesium gating of cardiac gap junction channels. Prog Biophys Mol Biol. 2010;103:102–10. doi: 10.1016/j.pbiomolbio.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Maxeiner S, et al. Spatiotemporal transcription of connexin45 during brain development results in neuronal expression in adult mice. Neuroscience. 2003;119:689–700. doi: 10.1016/s0306-4522(03)00077-0. [DOI] [PubMed] [Google Scholar]
- O’Brien J, al-Ubaidi MR, Ripps H. Connexin 35: a gap-junctional protein expressed preferentially in the skate retina. Mol Biol Cell. 1996;7:233–43. doi: 10.1091/mbc.7.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JJ, Li W, Pan F, Keung J, O’Brien J, Massey SC. Coupling between A-type horizontal cells is mediated by connexin 50 gap junctions in the rabbit retina. J Neurosci. 2006;26:11624–36. doi: 10.1523/JNEUROSCI.2296-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A, et al. Connexin35 mediates electrical transmission at mixed synapses on Mauthner cells. J Neurosci. 2003;23:7489–503. doi: 10.1523/JNEUROSCI.23-20-07489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, et al. Ca2+/calmodulin-dependent kinase II mediates simultaneous enhancement of gap-junctional conductance and glutamatergic transmission. Proc Natl Acad Sci U S A. 1998;95:13272–7. doi: 10.1073/pnas.95.22.13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE, et al. Dynamics of electrical transmission at club endings on the Mauthner cells. Brain Res Brain Res Rev. 2004;47:227–44. doi: 10.1016/j.brainresrev.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Smith M, Pereda AE. Chemical synaptic activity modulates nearby electrical synapses. Proc Natl Acad Sci U S A. 2003;100:4849–54. doi: 10.1073/pnas.0734299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl G, et al. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Letters. 1998;428:27–31. doi: 10.1016/s0014-5793(98)00479-7. [DOI] [PubMed] [Google Scholar]
- Spray DC, Rozental R, Dermietzel R, Scemes E. cell-Cell Communcation: an overview emphasizing gap junctions. Elsevier; New York: 2004. [Google Scholar]
- Srinivas M, et al. Functional properties of channels formed by the neuronal gap junction protein connexin36. J Neurosci. 1999;19:9848–55. doi: 10.1523/JNEUROSCI.19-22-09848.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Oltedal L, Hartveit E. Electrical synapses between AII amacrine cells: dynamic range and functional consequences of variation in junctional conductance. J Neurophysiol. 2008;100:3305–22. doi: 10.1152/jn.90957.2008. [DOI] [PubMed] [Google Scholar]
- Vest RS, et al. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–33. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XD, Korn H, Faber DS. Long-term potentiation of electrotonic coupling at mixed synapses. Nature. 1990;348:542–5. doi: 10.1038/348542a0. [DOI] [PubMed] [Google Scholar]
- Zoidl G, et al. Evidence for a role of the N-terminal domain in subcellular localization of the neuronal connexin36 (Cx36) J Neurosci Res. 2002;69:448–65. doi: 10.1002/jnr.10284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.