

Abstract
Glioblastoma multiforme (GBM) is an intractable brain tumor, associated with poor prognosis and low survival rate. Combination therapy such as surgery, radiotherapy and temozolomide is considered standard in overcoming this aggressive cancer, despite poor prognosis. There is a need to identify potential agents which may augment the chemotherapeutic effects of standard drugs such as temozolomide. In this project, we evaluated the effects of silibinin, a natural plant component of milk thistle seeds, to potentiate toxic effects of chemotherapy drugs such as temozolomide, etoposide and irinotecan on LN229, U87 and A172 (P53 and phosphatase and tensin homolog (PTEN) -tumor suppressormutated) glioma cell lines. The data from this work suggest that silibinin was effective in potentiating the cytotoxic efficacy of temozolomide in LN229, U87 and A172 cells. While silibinin reduced survivin protein expression only in LN229 cells, its ability to potentiate cytotoxicity of chemotherapy drugs occurred irrespective of survivin protein level. The data also demonstrated that silibinin potentiated the effect of etoposide and but not irinotecan in LN229 cells. Future research will be required to evaluate the in vivo efficacy of silibinin and to delineate its mechanism of action and its ability to cross the blood-brain barrier.
Keywords: Glioblastoma, milk thistle, silybinin, temozolomide, natural compounds
Primary brain tumors are a heterogeneous group of diseases arising from different cells of origin and with characteristic age distributions (1). Brain tumors almost always present a therapeutic challenge because of their location, ag¬gressive biological behavior, diffuse infiltrative growth and limited surgical option. Almost half of the 18,000 new cases of brain tumors diagnosed in the United States each year are categorized as glioblastoma multiforme (GBM) (2). GBM / World Health Organization (WHO) grade IV gliomas occur in 50- to 70-year old patients, with a median survival time of approximately 10-15 months or up to 30-50 months for anaplastic / WHO grade III astrocytomas, respectively (3). The majority of these patients will die within a year of diagnosis.
While combination therapies include surgery, radiotherapy (RT), and chemotherapy agents such as temozolomide [an alkylating agent with the capacity to cross the blood-brain barrier (BBB)], the prognosis remains poor (2, 4). Although temozolomide is somewhat effective, response duration is short and overall survival is typically one year (5). Unfortunately, effective treatment for fatal and aggressive GBM is hard to achieve due to the aggressive metastatic and infiltrative nature of the disease, combined with limited drug transport across the blood brain barrier (BBB) compounded by further limitations on surgical resection. Thus, achieving therapeutic concentrations in distal, seemingly intact areas contain infiltrating tumor cells remains an enormous challenge. Lastly, is the inability to identify tumor cells that are resistant to chemotherapy agents or radiation to target (6). While strides are being made in nano-targeted drug delivery systems to treat gliomas (7), there is still a need to develop effective therapeutic combination strategies of existing chemotherapy agents (8).
In the current study, we evaluate the potentiating effects of a natural compound, silibinin [a constituent of milk thistle seeds], to augment tumoricidal effects of temozolomide, irinotecan and etoposide in diverse human glioma cell lines. Previous studies have shown that silibinin alone has the capacity to halt proliferation of tumor cells by attenuating the early phase of the cell cycle, G1, by inhibiting various kinases (9) and the mitogen-activated protein kinase (MAPK) pathway (10). In glioblastoma cells, silibinin can also induce apoptosis through activation of calpain and protein kinase C-δ (PKC δ) (11) and attenuate metastatic processes by suppression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB) and downstream effects on the stimulation of matrix metalloproteinase-9 (12). In contrast to standard chemotherapy agents, silibinin is not toxic, as demonstrated in animal studies (13) and in human patients (14, 15), it has a hepatoprotective, cardioprotective and nephroprotective effect against various toxicological injurries (13, 14, 16, 17), making this a potential component to augment traditional chemotherapy for aggressive gliomas.
In this investigation, we used a number of diverse glioma cell lines carrying genetic mutations of tumor suppressor genes [p53, phosphatase and tensin homolog (PTEN)], which would make them otherwise resistant to chemotherapy. The U87 and A172 cell lines are mutated at PTEN (18) which is associated with up-regulation of p27/KIP1 and dephosphorylation of focal adhesion kinases which are integral to uncontrolled cell growth and tumor formation (19). Likewise, the LN229 and A172 cell lines are mutated at p53 (tumor-suppressor gene) (20) which leads to hampered regulatory controls over cell cycle, senescence, apoptosis (21) and chemo-resistance (22, 23).
Materials and Methods
Growth and experimental culture media were purchased from Mediatech Inc. (Manassas, VA and Life Technologies, Carlsbad, CA, USA), penicillin/streptomycin from Life Technologies; (Carlsbad, CA, USA) and fetal calf serum (FCS) from Omega Scientific (Tarzana, CA, USA). Experimental compounds, including temozolomide and silibinin, were purchased from Sigma-Aldrich (St. Louis, MO, USA), etoposide from Calbiochem (San Diego, CA, USA) and irinotecan from Pfizer Pharmaceutical Group (NY, USA). Other scientific/blotting supplies such as phenylmethanesulfonyl fluoride (PMSF) and protease inhibitors, dimethyl sulfoxide (DMSO) and 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) were purchased from Sigma-Aldrich and Thermo Scientific (Waltham, MA, USA). Antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and cell lines from American Type Culture Collection (Manassas, VA, USA).
Cell culture
Primary brain tumors, GBM (glioma tumor cell lines, LN229, U87, and A172) were obtained in accordance with the University of Southern California Institutional Review Board guidelines. The temozolomide-resistant LN229 cell line was developed in our laboratory by subjecting LN229 cell line to a gradual increase in temozolomide, rendering them gradually resistant (TR-LN229). The LN229 cell line and TR-LN229 cell lines were cultured in RPMI-1640 containing L-glutamine, supplemented with 1% penicillin/streptomycin and 10% FCS. Other cell lines (U87, A172) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with L-glutamine, supplemented with 1% penicillin/streptomycin and 10% FCS. Astrocytes were maintained in specific astrocyte medium (ScienceCell Research Laboratory, Carlsbad, CA, USA) and all cell lines were maintained in culture at 37°C in a 5% CO2 atmosphere in humidified cell culture incubator. All experiments were carried out on sub-confluent (60-80%) cultures.
Cell viability
MTT assay: The different cell lines were plated in triplicate (5×103 per well; 100 μl/well) in 96-well plates, using either RPMI/10%FCS or DMEM/10%FCS. Cells were treated with the designated drugs at different concentrations (etoposide, silibinin, or irinotecan) and incubated at 37°C at 5% CO2/air for two days. MTT reagent (5 mg/ml in phosphate buffered saline (PBS) was prepared and was added at 20 μl/well. Plates were then incubated again for 4 hours at 37°C. The supernatants were removed and 150 μl/well of DMSO was added and plates left to shake in shaker for 30 minutes. Absorbance was measured at 490 nm, and the percentage cell viability was calculated relative to that of untreated controls. Experiments were repeated three times.
Trypan blue exclusion
The viability of cell lines was determined using Trypan blue exclusion analysis. Cells were seeded at density of 5×104 cells/well with 1 ml cell suspensions being added into 6-well plates. After treatment with the designated drugs, cells were incubated at 37°C in a 5% CO2 atmosphere in humidified cell culture incubator for the desired time. Cell suspension (0.1 ml) was treated with temozolomide at different concentrations and transferred to test tubes. Then 10 μl from each cell group were added to 10 μl Trypan blue dye in another Eppendorf tube. Cell survival was examined under an inverted microscope using the hemocytometer method. The analysis was evaluated in two independent studies, each conducted in quadruplicate [% cell viability=(live cell count×2×104) /total cell count)×100].
Western blot
Cells were evaluated for survivin after the following treatments: 50 μM silibinin; 25 μM TMZ; 50 μM silibinin and 25 μM TMZ; and 100 μM silibinin. Cells were lysed in buffer containing [20 mmol/L Tris-base, 300 mmol/ l NaCl, 5 mmol/l EDTA, 0.1% sodium dodecyl sulfate (SDS), 1% deoxycholate, 1% Triton X-100], phenylmethanesulfonylfluoride (PMSF) solution at 1:100 dilution, and protease inhibitor cocktail at 1:100 dilution. About 50 to 100 μg of protein lysate were subjected and electro-phoresed on 15% SDS-PAGE gel and then electrotransferred onto nitrocellulose membranes. The blotted membranes were blocked for 2 hours at room temperature using blocking buffer containing Sea Block Blocking Buffer (Thermo Scientific) 50%, PBS 50%, and 0.1% Tween-20. Membranes were then incubated in a cold room overnight with primary antibodies. The primary antibodies used for western blots were mouse monoclonal anti-survivin (1:500), and polyclonal rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:5,000). Membranes were then incubated with secondary antibodies for 45-60 minutes. The secondary antibodies used for western blots were goat anti-mouse IgG (H+L), Dylight 800 conjugated for survivin (1:5,000), and goat anti-rabbit IgG (H+L), Dylight 680 conjugated for GAPDH (1:15.000), all purchased from Pierce (Rockford, IL, USA). Protein bands were detected by Odyssey infrared imaging (LI-COR Biosciences, Lincoln, NE, USA) and densitometry conduced using Scion Image software. (Scion Corp., Frederick, MD., USA)
Data analysis and statistics
Statistical analysis was performed using Graph Pad Prism (version 3.0; Graph Pad Software Inc. San Diego, CA, USA) and significance of difference between multiple groups evaluated by a one-way ANOVA, followed by Bonferroni's multiple comparison test or a Students t-test. IC50s were determined by regression analysis.
Results
The objective of this study was to determine if silibinin attenuated survivin levels thereby rendering greater sensitivity of glioma cells to chemotherapeutic agents. The basic toxicity of silibinin on diverse cell lines was first analyzed (Figure 1), where the A172 cell line was found to be most sensitive, and the LN229 most resistant. In the LN-229 and TR-LN229 cell line, the cytotoxic effects of temozolomide (0-50 μM) with and without 50 μM of silibinin were evaluated (Figure 2). In the LN229 cells, 50 μM of silibinin potentiated the toxicity of temozolomide where the IC50 was reduced from [16 μM to 8 μM], and in the TR-LN229 cells from [77 μM to 18 μM]. The data show the additive effects of 50μM of silibinin on the toxicity of temozolomide (0-50μM) in U87 cells (Figure 3), and on etoposide (0-50μM) in LN229 cells (Figure 4A) which were unaffected by a combination of silbinin and irinotecan (Figure 4B).
Figure 1.
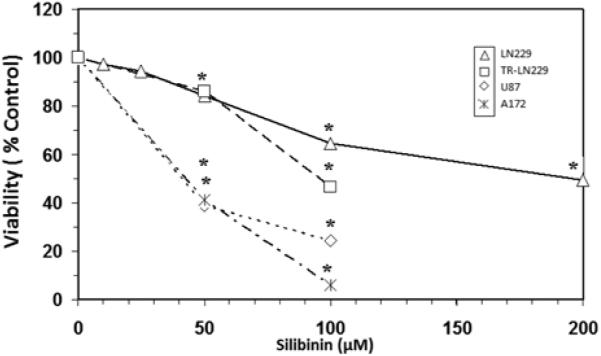
Toxicity of silibinin on A172, U87, LN229 and temozolomide-resistant LN229 (TR-LN229) cell lines. The data is presented as viability % control, and represents the Mean±S.E.M, n=3. Significant differences from the controls were evaluated by a one-way ANOVA, followed by Bonferroni's multiple comparison test, *=p<.05. The IC50s were established by regression analysis corresponding to: A172=40 μM, U87=45 μM, LN229=200 μM and TR-LN229=95 μM.
Figure 2.
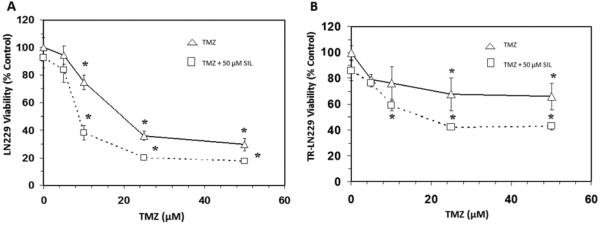
Effects of silibinin (SIL) on temozolomide (TMZ) cytotoxicity in LN229 cells (A) and TR-LN229 cells (B). The data is presented as viability % control, and represents the Mean±S.E.M, n=3. Significant differences from the controls were evaluated by a one-way ANOVA, followed by Bonferroni's multiple comparison test, *=p<.05.
Figure 3.
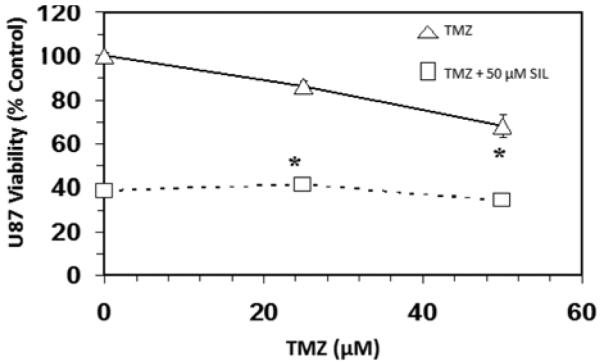
Effects of silibinin (SIL) on temozolomide (TMZ) on temozolomide cytotoxicity in U87 cells. The data is presented as viability % control, and represents the Mean±S.E.M, n=3. Significant differences from the controls were evaluated by a one-way ANOVA, followed by Bonferroni's multiple comparison test, *=p<.05.
Figure 4.
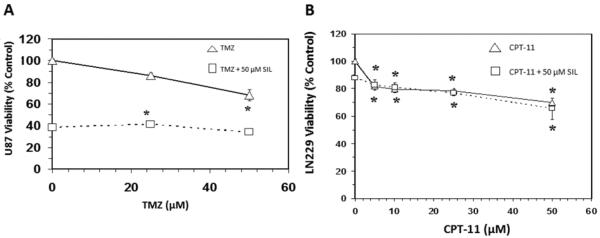
Effects of silibinin on etoposide (A) and irinotecan (CPT-11) (B) cytotoxicity in LN229 cells. The data is presented as viability % control, and represents the Mean±S.E.M, n=3. Significant differences from the controls were evaluated by a one-way ANOVA, followed by Bonferroni's multiple comparison test, *=p<.05.
In the next experiment, protein expression of survivin was quantified to determine if its presence plays a role in the potentiation of the cytotoxic effects of chemotherapy drugs. The results show that 50 μM silibinin down-regulated survivin expression in LN229 cells to 63.5±1.6% of the control value but did not have significant effects in either A172 or U87 cell lines (Figure 5-7, respectively). Increased concentrations of silibinin (100 μM) resulted in the loss of survivin in A172 cells (Figure 6) but not U87 cells (Figure 7), and these effects were likely a result of cell death (Figure 1) rather than on effects of protein expression. The data in Figure 1 suggest that A172 and U87 cells were more sensitive to the tumoricidal effects of silibinin, despite lack of attenuating effects on survivin expression as observed in the LN229 cells. In contrast, these data show that the only cell line in which survivin was reduced, namely LN229, was also the most resistant to the effects of silibinin itself.
Figure 5.
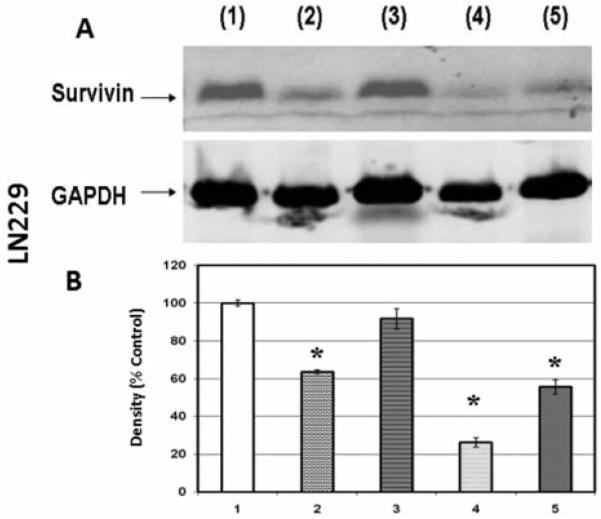
Western blot (A) and densitometry (n=3) (B) for survivin expression in LN229 cell line where 1: control; 2: 50 μM silibinin; 3: 25 μM temozolomide; 4: 50 μ M silibinin and 25 μM temozolomide; 5: 100 μM silibinin. Significance of difference between control and treatments were evaluated by a Students t-test. *=p<.05
Figure 7.
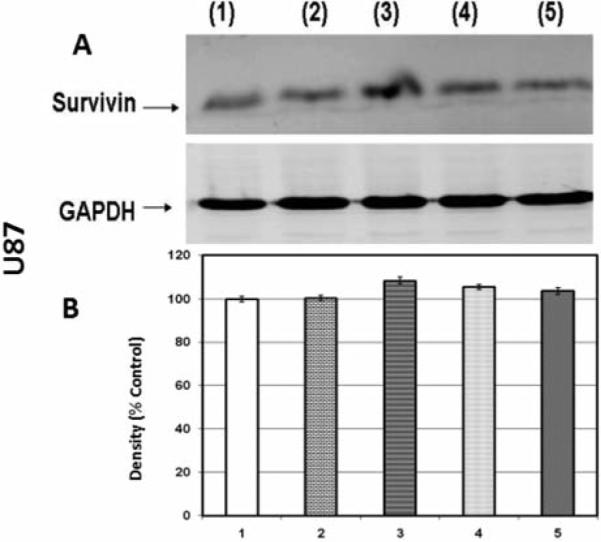
Western blot (A) and densitometry (n=3) (B) for survivin expression in U87 cell line where 1: control; 2: 50 μM silibinin; 3: 25 μM temozolomide; 4: 50 μM silibinin and 25 μM temozolomide; 5: 100 μM silibinin.. Significance of difference between control and treatments were evaluated by a Students t-test. *=p<.05
Figure 6.
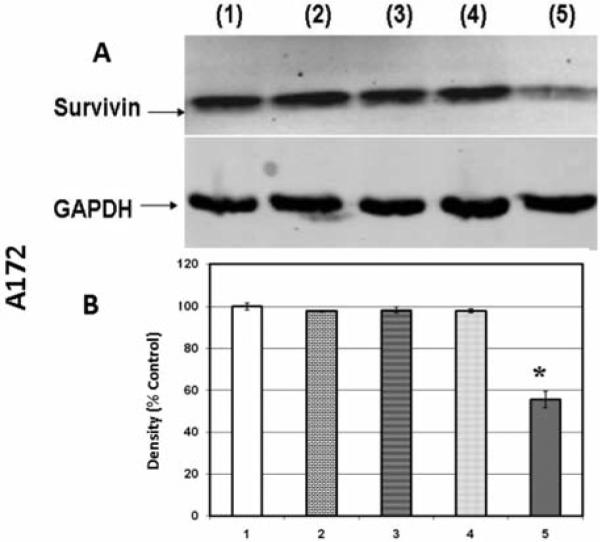
Western blot (A) and densitometry (n=3) (B) for survivin expression in A172 cell line where 1: control; 2: 50 μM silibinin; 3: 25 μM temozolomide; 4: 50 μM silibinin and 25 μM temozolomide; 5: 100 μM silibinin. Significance of difference between control and treatments were evaluated by a Students t-test. *=p<.05
Discussion
GBM is the most common and the most deadly malignant primary brain tumor in adults (2). Combination therapy targeting the disease from different mechanisms remains the hope to treat this aggressive cancer. Temozolomide emerged as one of the most significant advances in the treatment of this disease as a new chemotherapeutic agent given in combination with radiation therapy (2). Many glioblastoma cases continue to be refractory to temozolomide, making it critical to continue the search for novel adjunctive chemotherapeutic regimens to augment its efficacy.
Several molecular epidemiological studies have concluded that there is a strong association between increased survivin and progression of human cancers (24-27). This is also the case for GBM to which development of drugs are now including small molecule survivin antagonists (28) for treatment of non-responsive tumors (24-27, 29). Survivin is the smallest (16.5 kDa) member of the mammalian inhibitor of apoptosis (IAP) family, which is located on chromosome 17q25 and encodes mRNA that is divided into three introns and four exons (30-32). Survivin regulates the anti-apoptotic activity of the protooncogene (v-REL) and NFKB transcription factor family (30, 33), also playing a role in cancer cell survival by inhibiting caspase-7 and caspase-3 activation (30, 33).
The objective of the current study was to evaluate influential loss of survivin, if any, by silibinin and quantify its efficacy of potentiating toxicity with drugs such as temozolomide. The three cell lines, selected for this study, LN229, A172, and U87, differ in their genetic mutations: LN229 carries a mutated heterozygous mutation at p53 (20), A172 at p53 and PTEN (22, 23), and U87 at PTEN only (18) making these cell lines suitable to study aggressive glioma, largely due to defect in cell cycle regulation and senescence (34-37). We also used a temozolomide-resistant strain of LN229 that was developed in our laboratory by subjecting LN229 cell line to a gradual increase in temozolomide concentrations, rendering them resistant to temozolomide. The data from this study show that temozolomide and etoposide demonstrated additive effects in the presence of silibinin at 50 μM in the LN229 cell line, possibly linked to the hampered protein expression of survivin, and in two other cell lines A172, and U87, to which silibinin exerted no influence on survivin. The results are inconclusive as to the effects of silibinin on survivin protein expression, but demonstrate that its toxic effects, while not synergistic with chemotherapy agents-could provide an additive effect.
The independent toxic effects of silibinin directly on glioma cells have been reported in the literature, and are known to involve generation of reactive oxygen species and subsequent activation of extracellular signal-regulated kinase, p38 kinase, and c-Jun N-terminal kinase, protein kinase C in addition to initiating apoptosis. (11, 38) In other types of cancers, reports consistently provide evidence to support diverse therapeutic values of silibinin against oncogenic processes including, ability to initiate cell cycle arrest at G2/M by down-regulating cyclinB1 (39), inhibit migration through impairing chemokine signaling (40) protect against UV induced DNA damage (41) and attenuate migration of radiotherapy pro-survival tumor signaling. (42) The use of silibinin in augmenting the effects of temozolomide for GBM could be beneficial because it is also a non-toxic compound widely consumed as a component in ‘milk thistle extract’ known for its health promoting and anti-hepatotoxic effects (14, 15). At the same time, silibinin can impede metastatic processes and initiate apoptotic cell death in glioma. (12)
Future research will be required to evaluate similar natural products for efficacy and synergistic effects with standard chemotherapy agents such as etoposide, irinotecan and temozolomide.
Acknowledgements
This research was supported in part by the National Institute on Minority Health and Health Disparities of the National Institutes of Health through Grants Number 8 G12MD007582-28 and Number 1P20 MD006738.
References
- 1.Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. doi: 10.1007/s00401-005-0991-y. [DOI] [PubMed] [Google Scholar]
- 2.Robins HI, Chang S, Butowski N, Mehta M. Therapeutic advances for glioblastoma multiforme: current status and future prospects. Curr Oncol Rep. 2007;9:66–70. doi: 10.1007/BF02951428. [DOI] [PubMed] [Google Scholar]
- 3.Nieder C, Grosu AL, Mehta MP, Andratschke N, Molls M. Treatment of malignant gliomas: radiotherapy, chemotherapy and integration of new targeted agents. Expert Rev Neurother. 2004;4:691–703. doi: 10.1586/14737175.4.4.691. [DOI] [PubMed] [Google Scholar]
- 4.Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging Therapies for Glioblastoma. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.1701. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert MR, Friedman HS, Kuttesch JF, Prados MD, Olson JJ, Reaman GH, Zaknoen SL. A phase II study of temozolomide in patients with newly diagnosed supratentorial malignant glioma before radiation therapy. Neuro Oncol. 2002;4:261–267. doi: 10.1093/neuonc/4.4.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parvez T. Present trend in the primary treatment of aggressive malignant glioma: glioblastoma multiforme. Technol Cancer Res Treat. 2008;7:241–248. doi: 10.1177/153303460800700310. [DOI] [PubMed] [Google Scholar]
- 7.Li XT, Ju RJ, Li XY, Zeng F, Shi JF, Liu L, Zhang CX, Sun MG, Lou JN, Lu WL. Multifunctional targeting daunorubicin plus quinacrine liposomes, modified by wheat germ agglutinin and tamoxifen, for treating brain glioma and glioma stem cells. Oncotarget. 2014;5:6497–6511. doi: 10.18632/oncotarget.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol. 2014;71:1437–1444. doi: 10.1001/jamaneurol.2014.1701. [DOI] [PubMed] [Google Scholar]
- 9.Hoh C, Boocock D, Marczylo T, Singh R, Berry DP, Dennison AR, Hemingway D, Miller A, West K, Euden S, Garcea G, Farmer PB, Steward WP, Gescher AJ. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharma-codynamic consequences. Clin Cancer Res. 2006;12:2944–2950. doi: 10.1158/1078-0432.CCR-05-2724. [DOI] [PubMed] [Google Scholar]
- 10.Son YG, Kim EH, Kim JY, Kim SU, Kwon TK, Yoon AR, Yun CO, Choi KS. Silibinin sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5 up-regulation and down-regulation of c-FLIP and survivin. Cancer Res. 2007;67:8274–8284. doi: 10.1158/0008-5472.CAN-07-0407. [DOI] [PubMed] [Google Scholar]
- 11.Jeong JC, Shin WY, Kim TH, Kwon CH, Kim JH, Kim YK, Kim KH. Silibinin induces apoptosis via calpain-dependent AIF nuclear translocation in U87MG human glioma cell death. J Exp Clin Cancer Res. 2011;30:44. doi: 10.1186/1756-9966-30-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Momeny M, Malehmir M, Zakidizaji M, Ghasemi R, Ghadimi H, Shokrgozar MA, Emami AH, Nafissi S, Ghavamzadeh A, Ghaffari SH. Silibinin inhibits invasive properties of human glioblastoma U87MG cells through suppression of cathepsin B and nuclear factor kappa B-mediated induction of matrix metalloproteinase 9. Anticancer Drugs. 2010;21:252–260. doi: 10.1097/cad.0b013e3283340cd7. [DOI] [PubMed] [Google Scholar]
- 13.Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene. 2005;24:1188–1202. doi: 10.1038/sj.onc.1208276. [DOI] [PubMed] [Google Scholar]
- 14.Singh RP, Agarwal R. Flavonoid antioxidant silymarin and skin cancer. Antioxid Redox Signal. 2002;4:655–663. doi: 10.1089/15230860260220166. [DOI] [PubMed] [Google Scholar]
- 15.Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002;62:3063–3069. [PubMed] [Google Scholar]
- 16.Wellington K, Jarvis B. Silymarin: a review of its clinical properties in the management of hepatic disorders. BioDrugs. 2001;15:465–489. doi: 10.2165/00063030-200115070-00005. [DOI] [PubMed] [Google Scholar]
- 17.Singh RP, Agarwal R. Tumor angiogenesis: a potential target in cancer control by phytochemicals. Curr Cancer Drug Targets. 2003;3:205–217. doi: 10.2174/1568009033481985. [DOI] [PubMed] [Google Scholar]
- 18.Pore N, Liu S, Haas-Kogan DA, O'Rourke DM, Maity A. PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by trans-activating the proximal VEGF promoter. Cancer Res. 2003;63:236–241. [PubMed] [Google Scholar]
- 19.Zhou XP, Li YJ, Hoang-Xuan K, Laurent-Puig P, Mokhtari K, Longy M, Sanson M, Delattre JY, Thomas G, Hamelin R. Mutational analysis of the PTEN gene in gliomas: molecular and pathological correlations. Int J Cancer. 1999;84:150–154. doi: 10.1002/(sici)1097-0215(19990420)84:2<150::aid-ijc10>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Combs SE, Bohl J, Elsasser T, Weber KJ, Schulz-Ertner D, Debus J, Weyrather WK. Radiobiological evaluation and correlation with the local effect model (LEM) of carbon ion radiation therapy and temozolomide in glioblastoma cell lines. Int J Radiat Biol. 2009;85:126–137. doi: 10.1080/09553000802641151. [DOI] [PubMed] [Google Scholar]
- 21.Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 2004;61:822–842. doi: 10.1007/s00018-003-3304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan X, Aalto Y, Sanko SG, Knuutila S, Klatzmann D, Castresana JS. Genetic profile, PTEN mutation and therapeutic role of PTEN in glioblastomas. Int J Oncol. 2002;21:1141–1150. [PubMed] [Google Scholar]
- 23.Kataoka Y, Murley JS, Patel R, Grdina DJ. Cytoprotection by WR-1065, the active form of amifostine, is independent of p53 status in human malignant glioma cell lines. Int J Radiat Biol. 2000;76:633–639. doi: 10.1080/095530000138295. [DOI] [PubMed] [Google Scholar]
- 24.Patton SE, Hall MC, Ozen H. Bladder cancer. Curr Opin Oncol. 2002;14:265–272. doi: 10.1097/00001622-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Gazzaniga P, Gradilone A, Giuliani L, Gandini O, Silvestri I, Nofroni I, Saccani G, Frati L, Agliano AM. Expression and prognostic significance of LIVIN, SURVIVIN and other apoptosis-related genes in the progression of superficial bladder cancer. Ann Oncol. 2003;14:85–90. doi: 10.1093/annonc/mdg002. [DOI] [PubMed] [Google Scholar]
- 26.Sharp JD, Hausladen DA, Maher MG, Wheeler MA, Altieri DC, Weiss RM. Bladder cancer detection with urinary survivin, an inhibitor of apoptosis. Front Biosci. 2002;7:e36–41. doi: 10.2741/sharp. [DOI] [PubMed] [Google Scholar]
- 27.Swana HS, Grossman D, Anthony JN, Weiss RM, Altieri DC. Tumor content of the antiapoptosis molecule survivin and recurrence of bladder cancer. N Engl J Med. 1999;341:452–453. doi: 10.1056/NEJM199908053410614. [DOI] [PubMed] [Google Scholar]
- 28.Guvenc H, Pavlyukov MS, Joshi K, Kurt H, Banasavadi-Siddegowda YK, Mao P, Hong C, Yamada R, Kwon CH, Bhasin D, Chettiar S, Kitange G, Park IH, Sarkaria JN, Li C, Shakhparonov MI, Nakano I. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013;19:631–642. doi: 10.1158/1078-0432.CCR-12-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith SD, Wheeler MA, Plescia J, Colberg JW, Weiss RM, Altieri DC. Urine detection of survivin and diagnosis of bladder cancer. JAMA. 2001;285:324–328. doi: 10.1001/jama.285.3.324. [DOI] [PubMed] [Google Scholar]
- 30.Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 31.Giodini A, Kallio MJ, Wall NR, Gorbsky GJ, Tognin S, Marchisio PC, Symons M, Altieri DC. Regulation of micro-tubule stability and mitotic progression by survivin. Cancer Res. 2002;62:2462–2467. [PubMed] [Google Scholar]
- 32.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle check-point by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 33.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17:3247–3259. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 34.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 35.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 36.Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- 37.Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–181. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Kim KW, Choi CH, Kim TH, Kwon CH, Woo JS, Kim YK. Silibinin inhibits glioma cell proliferation via Ca2+/ROS/ MAPK-dependent mechanism in vitro and glioma tumor growth in vivo. Neurochem Res. 2009;34:1479–1490. doi: 10.1007/s11064-009-9935-6. [DOI] [PubMed] [Google Scholar]
- 39.Wang YX, Cai H, Jiang G, Zhou TB, Wu H. Silibinin inhibits proliferation, induces apoptosis and causes cell cycle arrest in human gastric cancer MGC803 cells via STAT3 pathway inhibition. Asian Pac J Cancer Prev. 2014;15:6791–6798. doi: 10.7314/apjcp.2014.15.16.6791. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Liang WC, Pan WL, Law WK, Hu JS, Ip DT, Waye MM, Ng TB, Wan DC. Silibinin, a novel chemokine receptor type 4 antagonist, inhibits chemokine ligand 12-induced migration in breast cancer cells. Phytomedicine. 2014;21:1310–1317. doi: 10.1016/j.phymed.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 41.Narayanapillai S, Agarwal C, Deep G, Agarwal R. Silibinin inhibits ultraviolet B radiation-induced DNA-damage and apoptosis by enhancing interleukin-12 expression in JB6 cells and SKH-1 hairless mouse skin. Mol Carcinog. 2014;53:471–479. doi: 10.1002/mc.22000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nambiar DK, Rajamani P, Singh RP. Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem Biophys Res Commun. 2014 doi: 10.1016/j.bbrc.2014.11.069. [DOI] [PubMed] [Google Scholar]