

Abstract
Purpose
To determine the efficacy and safety of repeated intravitreal and subconjunctival administrations of sirolimus in patients with noninfectious uveitis at 1 year in the Sirolimus as a Therapeutic Approach UVEitis (SAVE) Study.
Methods
Open-label, prospective, and randomized interventional clinical trial in which 30 patients with noninfectious intermediate, posterior, or panuveitis were randomized 1:1 to receive sirolimus 352-μg intravitreal or 1320-μg subconjunctival. Sirolimus was administered at days 0, 60, and 120. At month 6, all subjects were allowed to receive sirolimus at intervals greater than or equal to 2 months and until month 12. Changes in vitreous haze (VH), visual acuity (VA), and retinal thickness at month 12 were compared with baseline.
Results
Of patients with active uveitis at baseline (n = 20), 70% showed greater than or equal to 2 steps reduction of VH at month 12 (P < 0.05), 88% (n = 7) of patients with inactive uveitis at baseline showed either no change or reduction of VH to no haze, 36% (n = 10) of all patients (n = 28) gained greater than or equal to one line of VA, 21% (n = 6) lost greater than or equal to 1 line, and 43% (n = 12) showed no change. At the end of 1 year, no statistical differences in efficacy were found between intravitreal and subconjunctival groups. No serious adverse events were determined to be secondary to sirolimus.
Conclusions
Repeated subconjunctival/intravitreal injections of sirolimus appear to be tolerated by patients with noninfectious uveitis over 12 months. Results from the index study suggest that sirolimus may provide benefits to patients with uveitis. Both intravitreal and subconjunctival routes demonstrate similar bioactivity/efficacy. The intravitreal route, however, was better tolerated.
Translational Relevance
The SAVE Study illustrates for the first time the application of local formulations of sirolimus in non-infectious intermediate, posterior, and pan-uveitis. Subconjunctival/Intravitreal sirolimus may help to control inflammation while offering better tolerability/safety profiles than systemic therapies, including immunosuppressants and corticosteroids.
Keywords: Sirolimus, mTOR, uveitis, intravitreal, subconjunctival
Introduction
Sirolimus, also known as rapamycin, is an immunomodulatory therapeutic (IMT) agent that suppresses T-cell proliferation through inhibiting the expression of the interleukins 2, 4, and 15. Such inhibition is mediated though binding to the immunophilin FK12 binding protein (FKBP-12) and therefore, preventing it from binding and activating the mammalian target of rapamycin (mTOR). A systemic formulation of sirolimus is US Food and Drug Administration approved for use to prevent kidney transplant rejection and a sirolimus eluting coronary stent is approved for the use in patients with ischemic heart disease to enhance coronary luminal1,2 diameter. Systemic use of sirolimus, however, was associated with a number of cytotoxic, especially hematological, adverse effects that limit its use in the management of uveitis.3–10 A local ocular formulation of sirolimus has been developed and proved, based on animal studies, to be suitable for both subconjunctival (SCJ) and intravitreal (IVT) injection.11
Based on such knowledge of sirolimus and its potential anti-inflammatory effect, the Sirolimus as a Therapeutic Approach UVEitis (SAVE) Study has been conducted. In the SAVE Study, patients with noninfectious intermediate, posterior, and panuveitis were randomized to receive either intravitreal or subconjunctival sirolimus.12 At the primary endpoint at month 6, 80% showed significant improvement of the inflammatory indices (P < 0.05) in both study groups.12 After the primary end point, if retreatment criteria were met, patients in either group were treated with sirolimus injections in the same dose and route of injection they were initially assigned to during the active phase of the study and in intervals no more frequent than every 2 months.12 The follow-up period continued until month 12. The index manuscript herein reports the 1-year outcomes of the SAVE Study.
Methods
Sirolimus as a therapeutic Approach for uVEitis (SAVE) is a pilot, open-label, randomized clinical study conducted at the Wilmer Eye Institute, Johns Hopkins University School of Medicine (Baltimore, Maryland), to assess the safety, tolerability, and bioactivity of intravitreal and subconjunctival injections of sirolimus in patients with noninfectious uveitis. The study was approved by the Johns Hopkins University institutional review board and was conducted in compliance with the declaration of Helsinki, US Code of Federal Regulations Title-21, and the Harmonized Tripartite Guidelines for Good Clinical Practice (1996). Before screening, all the subjects involved in the SAVE study reviewed and signed informed consent. The SAVE study is registered at ClinicalTrials.gov (Identifier: NCT00908466).
Study Design
The study design, including eligibility criteria and procedures for intraocular and subconjunctival injections, optical coherence tomography (OCT), and data collection and management, have been described previously.12 Briefly, 30 patients with noninfectious intermediate, posterior, and panuveitis were stratified at baseline according to the disease activity and the use of prednisone and/or other immunomodulatory therapeutic (IMT) agents into three categories. Patients in category-1 had active disease at baseline and were receiving no treatment; patients in category 2 had active disease and were receiving prednisone greater than or equal to 10 mg/day (or equivalent dose of another corticosteroid [CS]) and/or at least one other systemic immunosuppressant; and patients in category 3 had inactive disease and were receiving prednisone less than 10 mg/day (or equivalent dose of another CS) and/or at least one other systemic immunosuppressant. Disease activity was defined as having at least 1+ of VH in the study eye at baseline. Patients in each category were randomized in 1:1 ratio to receive sirolimus either intravitreally in a dose of 352 μg or subconjunctivally in a dose of 1320 μg. Each study eye received mandatory treatments at baseline, months 2, and 4. During the follow-up period, months 6 to 12, the study eyes were eligible to receive sirolimus in the same dose and route on an as needed basis and in intervals no less than 2 months between injections.
In patients with bilateral uveitis, the eye with more advanced disease was chosen as the study eye. If both eyes were equally affected, the study eye was chosen at the investigator's discretion prior to randomization. If the standard-of-care local therapies to the fellow eye were contraindicated, proved ineffective, or refused by the patient, then sirolimus injections were administered to the fellow eye at the investigator's discretion and at the same dose and route of administration of the study eye, but at least 14 days apart from the study eye injection.
Prior to the first administration of the study drug at Day 0, all IMT agents were discontinued for at least 30 days. Tapering of corticosteroids started at Day 0 for patients in categories 2 and 3. For patients in category 2, the aim was to reduce the dose of CS to less than 10 mg/day. For patients in category 3, the aim was to discontinue CS or to reduce the dose to levels less than 5 mg/day. Rescue therapy was allowed for all participants at any time when one or more of predefined rescue criteria was met.12
The results at the primary endpoint were previously reported. In this manuscript, we are reporting the 1-year outcomes of the study eyes and fellow eyes treated with sirolimus.
Statistical Analyses
The main outcomes were the bioactivity and ocular tolerability of intravitreal and subconjunctival injections of sirolimus in the treatment of noninfectious uveitis. In patients with active disease at baseline, response to treatment was defined as reduction of VH, as measured using the SUN working group criteria,13,14 at months 6 and 12 by at least two steps when compared with baseline or reduction of a single step to no haze. In patients with inactive disease at baseline (category 3), success (or efficacy) of treatment was assessed by the proportion of patients who maintained quiescent uveitis throughout the 6-month period of the study while tapering or discontinuing their previous CS therapy. In addition, the activity of disease in two patients with punctate inner choroidopathy (PIC), both enrolled in category 3, were also monitored by fluorescein angiography and high-resolution spectral-domain (SD) OCT. Other secondary parameters included change from baseline in best-corrected visual acuity as measured by Early Treatment of Diabetic Retinopathy Study (ETDRS) charts and in macular thickness as measured by SD-OCT.
The safety and tolerability of subconjunctival and intravitreal injection of sirolimus in patients with intermediate, posterior, and panuveitis were evaluated by assessing the frequency and severity of systemic and ocular adverse events as well as their relationship to the study drug. Baseline demographics and disease characteristics were summarized (number and percentage for categorical measures and number, mean, SD, and median for continuous measures) by treatment group and by disease category within treatment group. Nonparametric statistical tests (e.g., Wilcoxon Signed Ranks test and Mann-Whitney U test) were employed to assess the significance of changes from baseline in VH among the different categories of study groups at months 6 and 12. The statistical analysis was run using IBM SPSS Statistical package v. 19 (IBM Corporation, Armonk, NY).
Results
Patients Disposition
The baseline characteristics were demonstrated in our report of primary month 6 results.12 Six patients left the study before the month 12 endpoint. Two of six patients have exited the study before the primary endpoint at month 6 and the reasons of their exit was mentioned in details in our previous report.12 Neither of the two patients has completed the three mandatory injections and hence, bioactivity data from both patients were not included in the analysis of either month 6 or 12 endpoints. The demographic characteristics of the study sample and the discontinuation log are shown in Tables 1 and 2. Twenty-four patients have finished study activities at the secondary endpoint at month 12 with a completion percentage of 80%. Any patient who did not have a month 12 visit, but had a study visit at month 6 and beyond, had their last observations carried forward and included in the month 12 analyses. Subjects who left the study prior to month 6 did not have their bioactivity data carried forward to month 6 or 12 and the baseline data from these subjects were removed when comparing the outcome at month 12 with baseline. Adverse events from such subjects, however, were included in the analysis of safety outcome. The final analysis of bioactivity data included 28 patients; 14 in each treatment arm. Seven patients (three in group 1, IVT; and four in group 2, SCJ) had active uveitis at baseline and were receiving no CS therapy (category 1); 13 patients (eight in group 1 and five in group 2) had active uveitis at baseline and were receiving greater than or equal to 10 mg/day of prednisone (category 2); and eight patients (three in group 1 and five in group 2) had inactive disease at baseline and were receiving prednisone less than 10 mg/day and/or at least one other immunosuppressant (category 3).
Table 1. .
Demographics and Baseline Characteristics of Study Participants
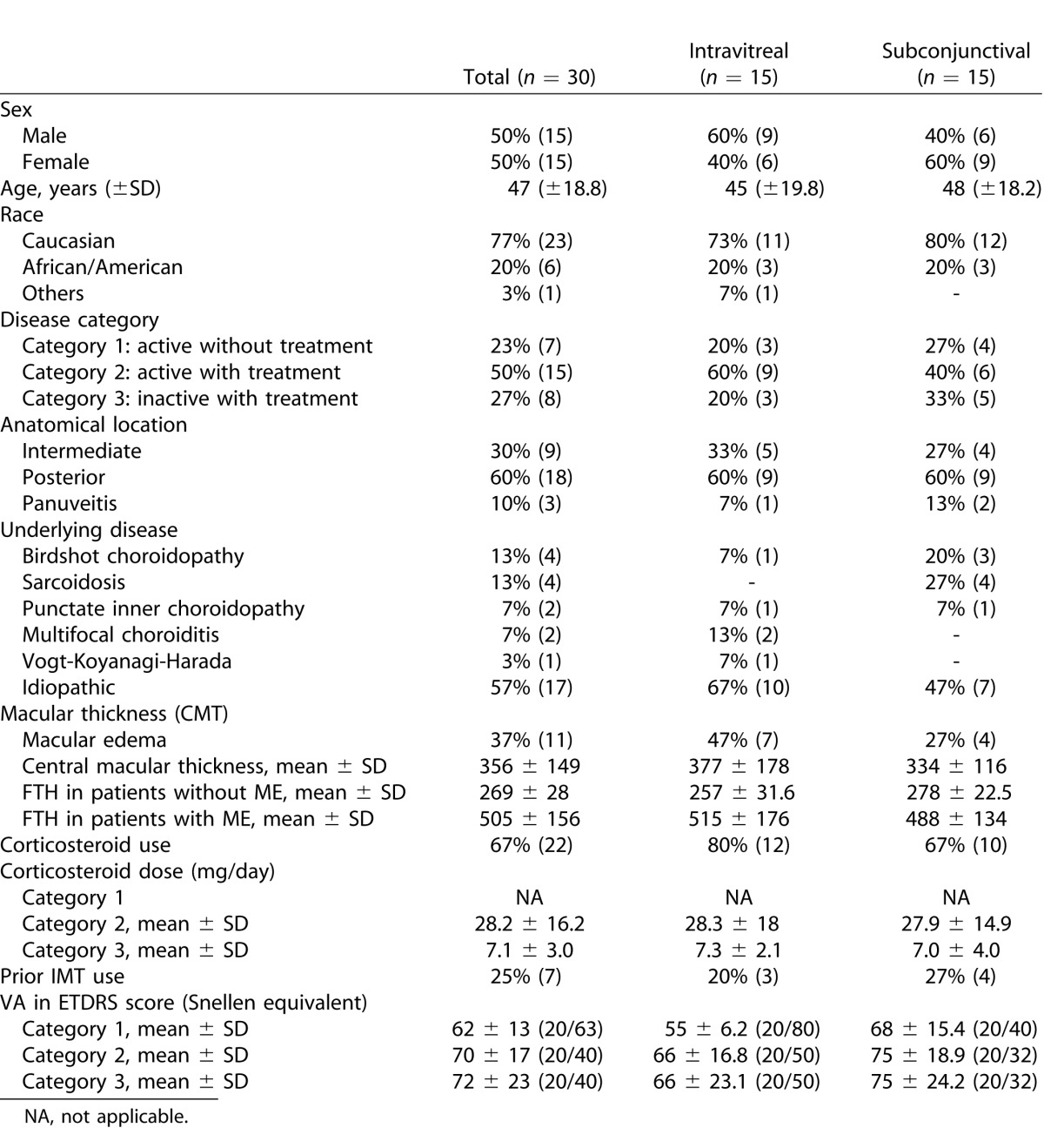
Table 2. .
Log of Subjects Who Have Discontinued From the Study Prior to Month 12

Outcomes at Month 12
Safety Outcome
Intravitreal Injections
Prior to month 6, the study eyes of Group 1, IVT, received 42 injections of sirolimus and the fellow eyes (from nine patients) received 20 injections raising the number of injections in this group to a total of 62 injections. After month 6 and prior to the month 12 endpoint, the study eyes of Group 1 received 28 intravitreal injections of sirolimus and the fellow eyes (from 10 patients) received 15 intravitreal injections with a total number of 43 injections. Similar to the safety outcome in the first 6 months of the study, the adverse events encountered with intravitreal injections of sirolimus were rare with vitreous floaters being the most commonly reported adverse events (four patients/eyes). The other ocular adverse events included single occurrences of pars plana vitrectomy to a study eye to achieve rapid removal of large amount of chronic, vitreous debris, and revision of Ahmed's valve implant in a fellow eye. Systemic adverse events are reported in Table 3. No ocular or systemic adverse event was considered related either to the study drug or to the injection procedure.
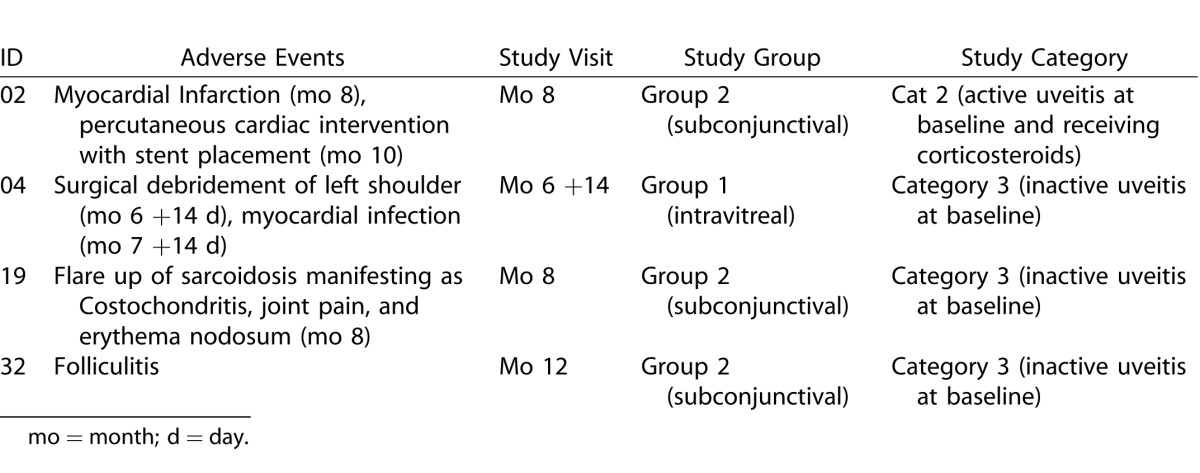
Table 3. .
Systemic Adverse Events Encountered During the Follow-up Period From Months 6 to 12
The average intraocular pressure (IOP) in the study eyes was 15.5 mm Hg (±3.3) at baseline, 13.1 (±2.9) at month 6, and 14 (±3.1) at month 12. Similarly, the average IOP in the fellow eye was 17.3 mm Hg (±5.6), 15.8 (±5.4), and 15.5 (±4.4) at baseline, months 6, and 12, respectively. With the exception of one patient, all other study subjects had IOP less than 25 mm Hg throughout the study.
At month 6, one patient in this group met one of the rescue criteria by losing greater than or equal to 15 ETDRS letters of visual acuity (VA). The patient was rescued and exited from the study. No data other than safety data was recorded beyond month 6 for this patient. Another patient in this group was exited from the study due to the persistence of severe macular edema despite improvement in VH and stabilization of vision, and thus not meeting the rescue criteria; the exit was requested by the patient and the decision was made at the discretion of the principal investigator.
Subconjunctival Injections
Prior to month 6, subjects enrolled in Group 2, SCJ, received a total number of 66 injections of sirolimus; 44 to the study eyes and 22 to the fellow eyes (from 10 patients). After month 6 and up to month 12, subjects enrolled in this group received additional 50 subconjunctival injections of sirolimus; 33 to the study eyes and 17 to the fellow eyes (from 12 patients).
Similar to the first 6 months of the study, the most commonly encountered adverse events were inflammation at injection site. The inflammation manifested as ocular pain and localized tenderness and hyperemia overlying the subconjunctival aggregate of sirolimus. The inflammations were mild to moderate, peaked at approximately 2 weeks after injection, resolved spontaneously without sequelae within additional 2 weeks, and were considered likely related to the study drug. Serious systemic adverse events included an instance of death secondary to complications of hernia surgery and an instance of myocardial infarction that required percutaneous intervention and stent placement.
All systemic adverse events (Table 3) were mild to moderate in severity and none was considered related to the study drug.
Average IOP in the study eyes was 13.4 mm Hg (±3.0) at baseline, 14.4 (±3.3) at month 6, and 14.4 (±4.0) at month 12. Similarly, the average IOP in the fellow eye was 17.3 mm Hg (±5.6), 15.8 (±5.4), and 15.5 (±4.4) at baseline, months 6, and 12, respectively. None of the study participants in this group had an IOP greater than 25 mm Hg throughout the study.
Bioactivity Outcome
Treatment Frequency
During the first 6 months of the study, every study eye received three mandatory injections of sirolimus. During the follow-up period, 25 study eyes received at least one additional injection with a total of 61 additional injections. At month 6, 39% (24/61) of the additional injections were administered, 30% (18/61) at month 8, and 31% (19/61) at month 10. The calculated frequency of retreatments in the IVT group was 0.86 injections/eye at month 6 and 0.57 at both month 8 and 10, with an overall retreatment frequency of two injections/eye over the 6 month follow-up period. Similarly, the calculated frequency of retreatment in study eyes that received sirolimus subconjunctivally was 0.86 injections/eye at month 6, 0.71 at month 8, and 0.79 at month 10; with an overall retreatment frequency of 2.36 injections/eye over the 6 months follow-up period.
The study eyes in category 1 received 13 injections (1.86 injections/eye), eyes in category 2 received 28 injections (2.15 injections/eye), and a total of 20 injections were administered in category 3 (eight eyes), yielding a mean of 2.5 injections/eye, during the follow-up period from month 6 to 12.
Changes in the Inflammatory Indices
Study Categories 1 and 2 (Active Uveitis at Baseline, N=20)
At month 6, eight subjects (40%) showed a reduction of two steps or more of VH (four in each group) compared with baseline and 12 subjects (60%) showed either no change or a reduction of one-step VH (seven in group 1, IVT, and five in group 2, SCJ). At month 12, 70% (n = 14) of subjects showed a reduction of two steps or more of VH (eight in group 1 and six in group 2). Similar to the results at month 6, no patient in either category showed increase of VH of one or more steps at month 12. Figures 1 and 2 demonstrate the change from baseline in VH among the study groups and in patients with active uveitis (category 1 and 2) at month 6 and month 12.
Figure 1. .

Changes from baseline in vitreous haze among study subjects in intravitreal (IVT) and subconjunctival (SCJ) groups.
Figure 2. .

Changes in vitreous haze from baseline (BL) at months 3, 6, and 12 among patients who had active uveitis at BL.
The reduction in VH was statistically significant at month 12, when assessed using Wilcoxon rank sum test with P less than 0.05, in both treatment groups (P = 0.004 and 0.007 for the IVT and SCJ groups, respectively). The difference in the VH outcome between treatment groups (intravitreal versus subconjunctival) was not statistically significant (P = 0.352), when assessed by Mann-Whitney U test.
Comparing both categories at month 12, 86% of subjects in category 1 (6/7; three in each group) showed a reduction of greater than or equal to 2 steps of VH compared with 62% in category 2 (8/13; five in group 1 and three in group 2). Meanwhile, 14% of subjects in category 1 (1/7; in group 2) showed either no change or a reduction less than two steps compared with 38% in category 2 (5/13; three in group 1 and two in group 2).
Study Category 3 (Inactive Uveitis at Baseline; N = 8)
At month 6, 88% (n = 7/8) of patient in this category showed either no change in VH or improvement of one step. The results at month 12 were similar with 88% showing either no change or a reduction of one step of VH. One patient, however, showed worsening of VH from no haze to 1+. The changes in VH at either month 6 or 12 were not statistically significant in any of the study groups (P value = 0.317 and 0.206 Wilcoxon rank sum test, at months 6 and 12, respectively).
Corticosteroids-Sparing Effect
Twenty subjects were receiving systemic CS at baseline, 13 in category 2 (prednisone ≥ 10 mg/day) and 7 in category 3 (prednisone < 10 mg/day and/or IMT). Prior to screening, seven subjects were receiving immunosuppressants other than CS (IMT); two subjects were receiving IMT in conjunction with a prednisone dose less than 10 mg/day, four subjects were receiving IMT in conjunction with prednisone greater than or equal to 10 mg/day, and one subject was receiving IMT as a maintenance monotherapy.
Figure 3 demonstrate the changes in the median dose of CS among the study group and categories. In category 2, the median dose was reduced from 20 mg/day at baseline to 8 mg/day at month 6 and to 2 mg/day at month 12. The dose was reduced to less than 10 mg/day in 11 subjects (six from group 1, IVT; and five from group 2, SCJ) by months 6 and 12, the CS dose was successfully reduced to less than 10 mg/day in one additional patient (group 2). It was not possible to reduce the CS dose to less than 10 mg/day in one patient (group 1), who ended the study receiving 20 mg/day. It was possible to stop the CS completely in five patients (38%).
Figure 3. .

Corticosteroid-sparing beneficial effects of sirolimus. The lines represent the changes in the mean dose of corticosteroids among patients in category 2 (gray line) and category 3 (blue line). The columns represent the change in the proportions of patients receiving 10 mg/day of prednisone or more.
In Category 3, the median dose was reduced from 9 mg/day at baseline to 3 and 2 mg/day at months 6 and 12, respectively. It was not possible to stop CS completely in any patient.
Changes in Visual Acuity
The average VA at baseline was 69 (±17.7) letters (equivalent to 20/40; Table 1). The changes in the mean VA from baseline were not statistically significant at month 6 or 12 (P = 0.48 and 0.39, respectively; Wilcoxon rank sum test), with average total gain of 0.71 (±7.9) letters at month 6 and 0.96 (±6.9) 12. Subgroup and subcategorical analysis did not show any statistically significant differences at either month 6 or 12 as well.
At month 12, 10 subjects (36%) gained one or more lines of VA (3 in group 1 and 7 in group 2). Of the 10 subjects, four gained two or more lines (14%) with no one gaining three lines or more. Eight subjects (29%) lost one or more lines at month 12. Of the eight subjects, two lost two or more lines with one losing three lines. Twelve subjects (43%) showed no change in VA at month 12. A depiction of change in VA in ETDRS lines at months 6 and 12 is shown in Figure 4.
Figure 4. .

Changes in visual acuity in ETDRS scores among the study subjects at months 3, 6, and 12.
Central Macular Thickness (CMT)
At baseline, 37% of subjects had macular edema (n = 11; seven in group 1, IVT, and four in group 2, SCJ), defined as having an average CMT of greater than 315 um,15 using SD-OCT (Spectralis HR+OCT; Heidelberg Engineering, Visa, CA) or viewing of intraretinal fluid on OCT scan passing through the 1-mm circle centered on the fovea. The average CMT of patients with macular edema at baseline was 505 μm (±156) on SD-OCT. CMT in patients without macular edema (n = 17) did not show changes from baseline in any patient, either at month 6 or 12, with an average thickness of 272 μm (±27), 265 μm (±29 μm), and 269 μm (±35 μm) at baseline, months 6, and 12, respectively.
In patients who had ME at baseline, CMT increased in group 1 from an average of 510 μm (±194 μm) at baseline, to 615 (±168 μm) at month 6, and 616 (±165 μm) at month 12 (Fig. 5); a mean change of 105 and 10 6μm, at months 6 and 12, respectively. Group 2 showed reduction of CMT from 481 μm (±131 μm) at baseline to 451 μm (±114 μm) at month 6 and to 434 μm (±122 μm) at month 12; a mean change of −30 and −47 μm, at months 6 and 12, respectively. The changes of CMT from baseline were not statistically significant at either month 6 or 12 (P = 0.169 and 0.182 at month 6 and 12, respectively).
Figure 5. .

Changes in the CMT from BL at months 3, 6, and 12 in study subjects who had macular edema at BL.
Fellow Eyes
Treatment Frequency
At baseline, 12/28 (43%) fellow eyes had active uveitis (four in category 1 and eight in category 2). By month 12, an additional 10 fellow eyes (36%) had uveitic activity that warranted treatment with a mean interval to first treatment of 77 days (±88). At least one injection of sirolimus was administered to 21 of the fellow eyes that had uveitic activity. One fellow eye with active uveitis at baseline did not receive any sirolimus injections because of a history of ocular herpes in that eye. Six fellow eyes (21%) did not have active uveitis at baseline and remained quiescent throughout the study. The fellow eyes received a total of 78 injections. Fifty percent of the injections were delivered intravitreally to 10 fellow eyes (3.9 injection/eye) and the other 50% were delivered subconjunctivally to 11 fellow eyes (3.5 injections/eye), with an overall treatment frequency of 3.7/eye over the 1 year of study duration. Four fellow eyes in category 1 received 17 injections (4.3 injection/eye), 11 eyes in category 2 received 38 injections (3.5/eye), and six eyes in category 3 received 23 injections (3.8/eye).
Changes in the Inflammatory Indices
One month following the first sirolimus injection, two eyes (10%) showed two steps or more reduction in VH, four eyes (20%) showed a reduction of one step to no VH, and the remainder eyes (70%) showed either reduction of one step or no change. The outcome was nearly the same at 2 months following first sirolimus injection; with three eyes showing two steps or more reduction of VH, three eyes showing reduction of one step to no VH, and the remainder eyes showing either reduction of one step or no change. By the end of the study, seven eyes (33%) showed reduction of two steps in VH, six eyes (29%) showed reduction of one step to no VH, seven eyes showed either no change in VH or reduction of one step (33%), or one eye showed worsening of VH of one step. The changes in VH were statistically significant at 1 and 2 months following first administration of sirolimus and at the end of the study (P < 0.05; Wilcoxon rank sum test).
Changes in Visual Acuity
There was an average gain in VA of one letter 1 month after injection (SD = 7.5), three letters (SD = 6.9) 2 months after first injection, and seven letters (SD = 13) by the end of the study at month 12 (Fig. 6).
Figure 6. .

Changes in VA in the fellow eyes that received sirolimus at 1 and 2 months following the first injection of sirolimus and at the end of the follow-up period (month 12). The line represents the changes in the mean VA as measured by the ETDRS letters and the columns represent the proportions of patients who gained one or more lines of VA compared with those who lost one or more lines.
One month following first injection of sirolimus, 33% of fellow eyes gained one or more lines of VA, 10% gained two or more lines, 24% lost one line or more, 10% lost two lines, and 43% showed no change (Fig. 6). Two months following first injection, 43% showed no change, 43% gained one or more lines, 19% gained two or more lines, 14% lost one line, and none showed a loss of two lines or more. By the end of the study, 52% gained one or more lines of VA, 33% gained two or more lines, 24% lost one line or more, 5% lost two line, and 24% showed no change in VA. The change in VA was not statistically significant 1 or 2 months following first sirolimus injection with P equals 0.643 and 0.085, respectively. The improvement in VA was statistically significant at month 12 with P equals 0.046 (Wilcoxon rank sum test).
Discussion
Although the class of mammalian target of mTOR inhibitors (mTOR-Is) seems to be relatively well tolerated and offers exciting new therapeutic opportunities in different disorders, systemic administration of mTOR-Is is associated with a multitude of side effects and adverse events.3–10 Sirolimus inhibits the production of many proinflammatory cytokines such interlukin-8, endothelial-monocyte activating polypeptide II, granulocyte chemotactic protein 2, cyclooxygenase 1 and 2, and inducible nitric oxide synthase through either inhibition of gene expression or interference with intracellular signals.16–19
At the 6-month primary endpoints of the SAVE Study, IVT, or SCJ sirolimus has demonstrated efficacy in reducing VH and need for systemic CS in patients with uveitis. During the follow-up period of the SAVE Study, both repeated intravitreal and subconjunctival injections of sirolimus continued to be well tolerated with similar adverse events to those reported at the primary endpoint. The most encountered adverse event was inflammation at the injection site manifesting as conjunctival hyperemia and chemosis in patients who received sirolimus subconjunctivally, which is consistent with previous studies that investigated the subconjunctival injection of sirolimus,11,20,21 and vitreous floaters in patients who received sirolimus intravitreally. The occurrence of such adverse events during the follow-up period was less frequent compared with the active phase of the study. It is possible that study subjects may be more accustomed and did not report as often (i.e. floaters, during the latter period). The encountered serious adverse events were deemed not related to the study drug. The overall safety and tolerability outcome of the SAVE study is consistent with previous studies of intravitreal11 and subconjunctival11,21 sirolimus.
The SAVE Study is the first study to assess the bioactivity of locally administered sirolimus in patients with noninfectious intermediate, posterior, or panuveitis. The reduction in VH at both the primary endpoint and at the end of the study was statistically significant in both study groups (P < 0.05). Similar to the previously reported results at month 6, no significant differences in response profile were detected based on route of delivery at month 12: both study groups were equally responsive to treatment. Sirolimus administered as an IVT or SCJ injection appeared to have similar ability in reducing VH and systemic dosage of prednisone to control the inflammation in eyes with noninfectious uveitis.
The improvement in the inflammatory indices in category 2 was associated with reduction of the adjunct corticosteroid dose in all patients (n = 13) with the majority (85%) of patients successfully tapered to less than 10 mg/day of CS by month 6 and 92% at month 12. The improvement in the inflammatory indices of category 1 was achieved without the use of CS at any time point during the study. Overall, 88% of the patients with inactive uveitis at baseline (category 3) maintained the quiescence of uveitis at month 6 while the corticosteroid dose was successfully tapered in all patients with an average reduction of 3.9 mg/day at month 6 and 4 mg/day at month 12. As the inclusion criteria for categories 1 and 2 did not require enrolled patients to have 2+ or more VH, not all enrolled subjects had the potential to improve two or more steps. As an exploratory study, SAVE was to evaluate any potential efficacy of sirolimus in uveitis, and thus allowed entry of greater than or equal to 1+ VH. Nevertheless, 40% of subjects showed reduction of two or more steps at month 6, and by month 12 the number has risen to 70%.
Patients with PIC in general do not have VH, and our two patients also did not have haze. These patients were enrolled in category 3 (haze of 0.5+ or less). The activity of disease in these two patients was monitored by fluorescein angiography and high-resolution SD-OCT for the changes in the lesions. Tapering of IMT and/or corticosteroid was another parameter. When we analyzed our data, we analyzed the disease activities in category 3 (inactive uveitis at baseline), which has these two study subjects separately.
In our study, approximately one-third of participants showed improvement of VA at month 6 with one-half of the study participants showing visual stability and 20% losing one or more lines of VA. The numbers were almost identical at month 12, indicating stability of VA over the follow-up period.
The many fellow eyes, which had active diseases requiring treatments confirm the finding that uveitis is often a bilateral process. Despite being treated on as needed basis, the fellow eyes demonstrated statistically significant improvement in VH 1 and 2 months after the first injection of sirolimus and by the end of the study at month 12. The percentages of patients who improved two or more steps of VH, however, were smaller when compared with the study eyes, which can be explained in part by the relative shorter duration of the follow-up following first injection and in part by lower potentials for the fellow eyes to achieve such improvements. Inherent in the study design, eyes with more severe disease were selected as study eyes and, hence had higher potential of improvement when compared with the fellow eyes.
The fellow eyes, however, have demonstrated gain in VA of an average of seven letters by the end of the study, which can be explained perhaps by a different clinical course, nature, and severity of the disease in the fellow eyes. Moreover, the administration of sirolimus in the fellow eyes illustrate that bilateral IVT or SCJ injections of sirolimus are possible and tolerated in patients with intermediate, posterior, and panuveitis.
There are risks associated with local therapy (i.e., intravitreal) such as endophthalmitis. Given that the incidence of endophthalmitis is quite low among the many studies with IVT delivery of pharmacologic agents, fortunately, we did not experience any endophthalmitis among the study subjects. The SAVE Study has illustrated several important points. Local administration of IMT (SCJ or IVT, although IVT appeared to be better tolerated) is possible to control different types of uveitis, without subjecting the patients to adverse events associated with systemic therapy. Patients with uveitis seem to tolerate bilateral IVT injections of sirolimus very well, which certainly help to increase the convenience of IVT delivery. On the other hand, even though local therapy is generally preferred, the frequent clinic visits to receive treatment, the necessity to treat both eyes separately in cases of bilateral uveitis, and the absence of systemic benefits in patients with extraocular manifestations of autoimmune disease are all drawbacks of local therapy that should not be overlooked while assessing the risks/benefits ratio of locally delivered drugs when it is being considered for the management of noninfection uveitis.
There are several limitations with the SAVE Study, including small study sample, relatively short follow-up period, underrepresentation of certain populations, and lack of extensive laboratory and systemic assessments. The relatively high drop-out rate (20% of study sample) after month 6 could be another limitation of the study that might have resulted in potential under- or overestimation of the efficacy of the study drug.
In conclusion, intravitreal and subconjunctival injections of sirolimus appear to provide long-term benefit in patients with noninfectious uveitis up to 1 year. Additional studies with intravitreal injections of sirolimus in the SAVE-2 and other phase 3 trials are needed to assess long-term benefit. mTOR pathway blockade has potential to play an important role in the management of noninfectious uveitis to achieve control of diseases, obtain maximum preservation of vision, and necessitate the least frequent intraocular injection schedule. However, the exact regimen will need to be tailored to individual patients because of the wide range of severity seen in patients with noninfectious uveitis. Although further research and clinical trials are required to establish the appropriate regimens for the different subcategories and underlying pathologies in patients with uveitis, the SAVE Study has provided informative and valuable insights toward the goal of identifying effective local therapy for uveitis and ocular inflammatory diseases.
Acknowledgments
The authors thank Santen, Inc. for providing the study drug.
Supported by grants from Santen, Inc. and the Research to Prevent Blindness to The Johns Hopkins University and the University of Nebraska Medical Center.
Footnotes
Disclosure: M.A. Ibrahim, None; Y.J. Sepah, None; A. Watters, None; M. Bittencourt, None; E.M. Vigil, None; D.V. Do, None; Q.D. Nguyen, Scientific Advisory Board for Santen, Inc., XOMA, Inc., and AbbVie, Inc.; chairs the Study Steering Committees SAKURA, EyeGuard, and VISUAL studies
References
- 1.Napoli KL, Taylor PJ. From beach to bedside: history of the development of sirolimus. Ther Drug Monit. 2001;23:559–586. doi: 10.1097/00007691-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem. 1998;31:335–340. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- 3.Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N. Managing stomatitis in patients treated with Mammalian target of rapamycin inhibitors. Clin J Oncol Nurs. 2011;15:E83–E89. doi: 10.1188/11.CJON.E83-E89. [DOI] [PubMed] [Google Scholar]
- 4.Ravaud A. Treatment-associated adverse event management in the advanced renal cell carcinoma patient treated with targeted therapies. Oncologist. 2011;16((Suppl 2)):32–44. doi: 10.1634/theoncologist.2011-S2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soefje SA, Karnad A, Brenner AJ. Common toxicities of mammalian target of rapamycin inhibitors. Targeted Oncol. 2011;6:125–129. doi: 10.1007/s11523-011-0174-9. [DOI] [PubMed] [Google Scholar]
- 6.Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34:97–115. doi: 10.2165/11585040-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Balagula Y, Rosen A, Tan BH, et al. Clinical and histopathologic characteristics of rash in cancer patients treated with mammalian target of rapamycin inhibitors. Cancer. 2012;118:5078–5083. doi: 10.1002/cncr.27505. [DOI] [PubMed] [Google Scholar]
- 8.Product Information: RAPAMUNE(R) oral solution, oral tablets, sirolimus oral solution, oral tablets. Philadelphia, PA: Wyeth Pharmaceuticals Inc; 2008. [Google Scholar]
- 9.Product Information: RAPAMUNE(R) oral solution, tablets, sirolimus oral solution, tablets. Philadelphia, PA: Wyeth Pharmaceuticals Inc; 2009. [Google Scholar]
- 10.Roberts RJ, Wells AC, Unitt E, et al. Sirolimus-induced pneumonitis following liver transplantation. Liver Transpl. 2007;13:853–856. doi: 10.1002/lt.21141. [DOI] [PubMed] [Google Scholar]
- 11.Dugel PU, Blumenkranz MS, Haller JA, et al. A randomized, dose-escalation study of subconjunctival and intravitreal injections of sirolimus in patients with diabetic macular edema. Ophthalmology. 2012;119:124–131. doi: 10.1016/j.ophtha.2011.07.034. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen Q, Ibrahim M, Watters A, et al. Ocular tolerability and efficacy of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis: primary 6-month results of the SAVE Study. J Ophthalmic Inflamm Infect. 2013;3:32. doi: 10.1186/1869-5760-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature (SUN) Working Group Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140:509–516. doi: 10.1016/j.ajo.2005.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nussenblatt RB, Palestine AG, Chan CC, Roberge F. Standardization of vitreal inflammatory activity in intermediate and posterior uveitis. Ophthalmology. 1985;92:467–471. doi: 10.1016/s0161-6420(85)34001-0. [DOI] [PubMed] [Google Scholar]
- 15.Ibrahim MA, Sepah YJ, Symons RC, et al. Spectral- and time-domain optical coherence tomography measurements of macular thickness in normal eyes and in eyes with diabetic macular edema. Eye (Lond) 2012;26:454–462. doi: 10.1038/eye.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nuhrenberg TG, Voisard R, Fahlisch F, et al. Rapamycin attenuates vascular wall inflammation and progenitor cell promoters after angioplasty. FASEB J. 2005;19:246–248. doi: 10.1096/fj.04-2431fje. [DOI] [PubMed] [Google Scholar]
- 17.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 19.Attur MG, Patel R, Thakker G, et al. Differential anti-inflammatory effects of immunosuppressive drugs: cyclosporin, rapamycin and FK-506 on inducible nitric oxide synthase, nitric oxide, cyclooxygenase-2 and PGE(2) production. Inflamm Res. 2000;49:20–26. doi: 10.1007/PL00000199. [DOI] [PubMed] [Google Scholar]
- 20.Sen HN, Larson TA, Meleth AD, Smith WM, Nussenblatt RB. Subconjunctival sirolimus for the treatment of chronic active anterior uveitis: results of a pilot trial. Am J Ophthalmol. 2012;153:1038–1042. doi: 10.1016/j.ajo.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnadev N, Forooghian F, Cukras C, et al. Subconjunctival sirolimus in the treatment of diabetic macular edema. Graefes Arch Clin Exp Ophthalmol. 2011;249:1627–1633. doi: 10.1007/s00417-011-1694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]