

Abstract
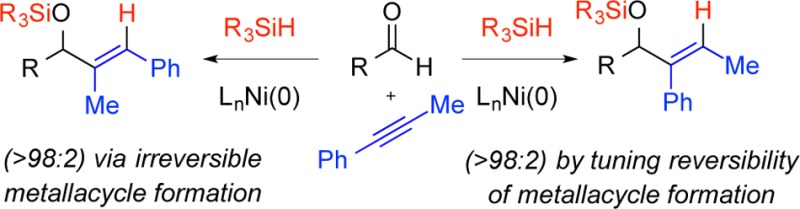
Combinations of ligand, reducing agent, and reaction conditions have been identified that allow alteration in the rate- and regioselectivity-determining step of nickel-catalyzed aldehyde–alkyne reductive couplings. Whereas previously developed protocols involve metallacycle-forming oxidative cyclization as the rate-determining step, this study illustrates that the combination of large ligands, large silanes, and elevated reaction temperature alters the rate- and regiochemistry-determining step for one of the two possible product regioisomers. These modifications render metallacycle formation reversible for the minor isomer pathway, and σ-bond metathesis of the metallacycle Ni–O bond with the silane reductant becomes rate limiting. The ability to tune regiocontrol via this alteration in reversibility of a key step allows highly regioselective outcomes that were not possible using previously developed methods.
Introduction
Reductive couplings of two π-components have been widely developed for numerous substrate combinations utilizing many transition metals across the periodic table.1,2 Numerous highly effective processes have been described that provide control of both regiochemistry, stereoselectivity, and enantioselectivity across a broad range of substrates, but the ability to reverse the regiochemical biases inherent to a particular substrate class is still quite challenging and rare.3 As a result, most regioselective reductive coupling processes employ electronically or sterically biased π-components that naturally favor a single regioisomer. In these cases, access to the opposite regioisomer without modification of the substrate typically cannot be achieved. In the rare cases where regiochemical reversal may be achieved, the most commonly employed strategies involve (a) special alkyne structures that allow directed additions, where the directing effect may be turned on and off by ligands employed, or (b) ligand structures that allow access to either product regioisomer through the development of steric interactions in the oxidative cyclization step.
The nickel-catalyzed reductive coupling of aldehydes and alkynes is a representative reaction class where regiochemical reversals have been demonstrated by these two strategies.4 In alkene-directed reactions reported by Jamison, the ligand environment on nickel may be tuned to allow the directing group either to complex the catalyst or be effectively displaced by altering the ligand structure and concentration. By this strategy, the regiochemical outcome may be reversed.5 In work from our laboratory, rate-determining formation of an oxametallacyclic intermediate is highly sensitive to ligand sterics, and regiochemistry reversals may be directly observed across a range of substrates simply by ligand alteration.6 Computational studies from Houk have been conducted on both of these nickel-catalyzed methods, and significant insight into the factors that govern regioselectivity have been provided.7
Irrespective of strategy employed, the mechanism in all of the prior work on nickel-catalyzed aldehyde–alkyne reductive couplings appears to involve oxidative cyclization of a Ni(0)–aldehyde–alkyne complex 1 to form a five-membered metallacycle 2 as the rate- and regiochemistry-determining step (Scheme 1). σ-Bond metathesis of the silane with the Ni–O bond of 2 then affords intermediate 3 in a fast reaction that follows rate-determining metallacycle formation, and reductive elimination of 3 then provides the observed silyl-protected allylic alcohol product. Both detailed kinetics studies and computational studies have provided evidence in support of this generalization.7,8
Scheme 1. Mechanism of Prior Protocols.

As would be expected for a sequence involving rate- and regiochemistry-determining oxidative cyclization, silane structure and concentration generally has no effect on the regioselectivity outcome. Whereas ligand control or directing group effects influence the regioselectivity outcome, the involvement of silane structural influences on the σ-bond metathesis step has not been developed as a regiocontrol strategy. Herein, we disclose that an interplay of silane structure, ligand structure, and temperature leads to alteration of the kinetic behavior of this system, and the insights provide a new strategy for regiochemistry reversals in this class of reactions.
Results and Discussion
Experimental Factors That Influence Regioselectivity
The nickel-catalyzed coupling of benzaldehyde with phenyl propyne has been reported with a broad range of protocols. Using the most widely used phosphines (i.e., PCy3) or N-heterocyclic carbene ligands (i.e., IMes), near-perfect regioselectivities for structure 6 are typically observed irrespective of reducing agent employed, including a broad range of silanes or Et3B (which provides the free hydroxyl of 6). The outcomes of couplings using IMes or SIMes as ligand with Et3SiH or (i-Pr)3SiH as the reductant are provided as representative examples (Table 1, entries 1–4). In these cases, highly regioselective production of 6 was observed with no influence of silane structure on regioselectivity. Couplings using the bulkier ligand IPr provided nearly equivalent quantities of regioisomers 5 and 6, and a small influence of the silane structure was noted (Table 1, entries 5 and 6). However, couplings using SIPr as ligand surprisingly displayed a much more pronounced influence on silane structure (Table 1, entries 7–11). With SIPr as ligand, couplings proceeded in 58:42 regioselectivity with Et3SiH, whereas increasing the silane size resulted in an increase in regioselectivity up to >98:2 selectivity favoring regioisomer 5 with t-Bu2MeSiH as reductant. This outcome provides a highly selective procedure that completely reverses the outcome seen with more typical protocols, which strongly prefer the production of regioisomer 6.
Table 1. Ligand and Silane Structural Effectsa.

| entry | ligand | R3SiH | 5:6 (% yield) |
|---|---|---|---|
| 1 | IMes | Et3SiH | <2:98 (84) |
| 2 | IMes | (i-Pr)3SiH | <2:98 (83) |
| 3 | SIMes | Et3SiH | 4:96b |
| 4 | SIMes | (i-Pr)3SiH | 4:96b |
| 5 | IPr | Et3SiH | 44:56b |
| 6 | IPr | (i-Pr)3SiH | 56:44b |
| 7 | SIPr | Et3SiH | 58:42 (65) |
| 8 | SIPr | t-BuMe2SiH | 60:40 (83) |
| 9 | SIPr | t-BuPh2SiH | 77:23 (86) |
| 10 | SIPr | (i-Pr)3SiH | 83:17 (89) |
| 11 | SIPr | (t-Bu)2MeSiH | >98:2 (61) |
Ni(COD)2 (0.06 mmol), ligand·HCl (0.05 mmol), and t-BuOK (0.05 mmol) were stirred with 2 mL of THF. Benzaldehyde (0.5 mmol), phenylpropyne (0.5 mmol), and silane (1.0 mmol) were combined, diluted to a total volume of 2 mL, and added to the reaction mixture via syringe drive over 1 h at rt. IMes·Cl = 1,3-bis(mesityl)imidazolium chloride; SIMes·HCl = 1,3-bis(mesityl)-4,5-dihydroimidazolium chloride; IPr·HCl = 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride; SIPr·HCl = 1,3-bis(2,6-diisopropyl-phenyl)-4,5-dihydroimidazolium chloride.
Isolated yield not determined.
We initially rationalized this unexpected finding as potentially being derived from a change in rate-determining step, where metallacycle formation becomes reversible (i.e., the 1 to 2 conversion, Scheme 1),9 and participation of the silane becomes rate-determining (i.e., the 2 to 3 conversion, Scheme 1) as the silane size increases. This hypothesis further suggested additional opportunities for altering the relative rates of metallacycle formation and σ-bond metathesis, thus providing additional experimental handles for adjusting regiocontrol. For example, lowering the concentration of silane should slow the rate of the silane σ-bond metathesis while not affecting the rate of metallacycle formation, therefore favoring the pathway involving reversible metallacycle formation. Additionally, the previous computational studies illustrated a significant entropic penalty associated with the σ-bond metathesis step.7c,7d Such a penalty would be maximized at high temperature, suggesting that increasing temperature would provide another handle for favoring the reversible metallacycle formation pathway. Using the benchmark example of the benzaldehyde–phenylpropyne coupling process, we conducted a series of experiments to evaluate these hypotheses.
As expected for a protocol where metallacycle formation is rate-determining, the silane concentration should not affect the regiochemical outcome. Using the standard protocol with SIPr as the ligand and Et3SiH as the reductant, this outcome was documented, where a 5-fold increase in silane concentration afforded identical outcomes in regioselectivity (Table 2, entries 1 and 2). However, a marked contrast was observed in reactions involving SIPr as ligand but utilizing a bulkier silane (i-Pr)3SiH as reductant (Table 2, entries 3–6). Characteristic improvements in regioselectivity compared with small ligand–small silane protocols were seen as silane concentration was lowered (Table 2, entries 3–5). When 10 equiv of (i-Pr)3SiH was employed, regioselectivities began to approach the lower selectivities observed with Et3SiH (Table 2, entry 5). Even without altering silane stoichiometry, simply diluting the reaction mixture led to significant improvements in regioselectivity in reactions of (i-Pr)3SiH with SIPr as ligand (Table 2, entry 6). It should be noted that yields of protected allylic alcohol products were higher when the aldehyde, alkyne, and silane were all added by syringe drive to the catalyst mixture. Therefore, concentrations vary over the course of the reaction, but the impacts of stoichiometry and concentration were nonetheless essential variables as Table 2 illustrates.
Table 2. Concentration Effectsa.

| entry | silane (equiv) | concb (M) | 5:6 (% yield) |
|---|---|---|---|
| 1 | Et3SiH (2.0) | 0.125 | 58:42 (65) |
| 2 | Et3SiH (10.0) | 0.125 | 58:42 (32) |
| 3 | (i-Pr)3SiH (1.1) | 0.125 | 95:5 (57) |
| 4 | (i-Pr)3SiH (2.0) | 0.125 | 83:17 (89) |
| 5 | (i-Pr)3SiH (10.0) | 0.125 | 68:32 (93) |
| 6 | (i-Pr)3SiH (2.0) | 0.0125c | >98:2 (82) |
Ni(COD)2 (0.06 mmol), SIPr·HCl (0.05 mmol), and t-BuOK (0.05 mmol) were stirred with 2 mL of THF. Benzaldehyde (0.5 mmol), phenylpropyne (0.5 mmol), and silane were combined, diluted to a total volume of 2 mL, and added to the reaction mixture via syringe drive over 1 h at rt.
Concentration refers to the final molarity of the aldehyde and alkyne starting components.
The catalyst was prepared in 38 mL of THF.
Next, to explore the impact of changing temperature, modified outcomes can be judged against the observation that reactions using IMes with (i-Pr)3SiH illustrated no impact of temperature on regioselectivity (Table 3, entries 1 and 2). However, by using the combination of (i-Pr)3SiH with SIPr, a significant temperature effect was seen, with regioselectivities jumping from 68:32 at 0 °C up to 98:2 at 95 °C (Table 3, entries 3–6). Whereas high temperatures provided the best regioselectivities, this comes at the expense of chemical yield, and more modest temperature increases in combination with the concentration effects noted above provide the best strategy for optimizing both yield and regioselectivity.
Table 3. Temperature Effectsa.

| entry | NHC ligand | temp (°C) | 5:6 (% yield) |
|---|---|---|---|
| 1 | IMes | rt | <2:98 (84) |
| 2 | IMes | 50 | <2:98 (77) |
| 3 | SIPr | 0 | 68:32 (81) |
| 4 | SIPr | rt | 83:17 (89) |
| 5 | SIPr | 50 | 94:6 (73) |
| 6 | SIPr | 95 | 98:2 (57)b |
Ni(COD)2 (0.06 mmol), NHC•HCl (0.05 mmol), and t-BuOK (0.05 mmol), were stirred with 2 mL of THF. Benzaldehyde (0.5 mmol), phenylpropyne (0.5 mmol), and silane (1.0 mmol) were combined, diluted to a total volume of 2 mL, and added to the reaction mixture via syringe drive over 1 h.
Toluene was used as the reaction solvent.
While previous efforts had shown some successes in regioselectivity reversals of aldehyde–alkyne reductive couplings, we sought to evaluate the above findings against a broader range of substrate combinations that led to modest regioselectivities or required noncommercial ligands in prior studies. To address this goal of a more general and convenient regioselective process, two optimized protocols were developed that could be applied to a wide range of alkynes. As shown in Tables 1–3, several different conditions could be used to access high regioselectivity with internal alkynes; however, heating the reaction to 50 °C and using (i-Pr)3SiH (method A, Table 4) proved most efficient and versatile. With terminal alkynes, excellent regioselectivities upon heating came at the expense of chemical yield. Therefore, a second general procedure (method B, Table 4) was developed for terminal alkynes using commercially available (t-Bu)2MeSiH and diluted reaction conditions while maintaining a room temperature protocol.
Table 4. Reaction Scope with Optimized Protocol.

| entry | R1 | R2 | R3 | methoda | 5:6 (% yield) |
|---|---|---|---|---|---|
| 1 | Ph | Ph | Me | Ab | >98:2 (82) |
| 2 | 4-FC6H4 | Ph | Me | A | 93:7 (85) |
| 3 | n-Hept | Ph | Me | A | >98:2 (77) |
| 4 | c-Hex | Ph | Me | A | >98:2 (90) |
| 5 | Ph | i-Bu | Et | A | 94:6 (86) |
| 6 | n-Hept | i-Bu | Et | A | 93:7 (66) |
| 7 | Ph | n-Pr | Et | A | 68:32 (56) |
| 8 | 2-furyl | n-Pr | Me | A | 93:7 (76) |
| 9 | Ph | i-Pr | Me | A | >98:2 (78) |
| 10 | c-Hex | i-Pr | Me | A | >98:2 (75) |
| 11 | Ph | i-Pr | H | B | >98:2 (61) |
| 12 | Ph | n-Hex | H | B | 95:5 (69) |
Method A: i-Pr3SiH as reductant, reaction conducted at 0.125 M in the aldehyde and alkyne at 50 °C. Method B: t-Bu2MeSiH as reductant, reaction conducted at 0.0125 M in the aldehyde and alkyne with 22 mol % Ni(COD)2 at rt.
Conducted at 0.0125 M in the aldehyde and alkyne.
Using optimized method A, phenyl propyne was coupled with a variety of aromatic and aliphatic aldehydes to produce regioisomer 5 with high selectivity in all instances (Table 4, entries 1–4). Steric influences in the homopropargylic position were sufficient to allow for excellent regiocontrol (Table 4, entries 5 and 6). However, when steric branching was decreased in the homopropargylic position (i.e., comparing ethyl to n-propyl), a large erosion in regioselectivity was observed (Table 4, entry 7). This very challenging case defines the limits of the current strategy where very modest biases in alkyne substitution are present.
Not surprisingly, when the steric differences were increased closer to the alkyne, excellent selectivity was maintained. For example, internal alkynes bearing a methyl substituent were well controlled as the aldehyde and large alkyne substituent were varied (Table 4, entries 8–10). Terminal alkynes, which previously required the use of a noncommercial ligand to obtain regioisomer 5 in high selectivity, could be coupled efficiently using commercially available SIPr (Table 4, entries 11 and 12), although catalyst loading needed to be increased to achieve good chemical yields. It should be noted that for all of the illustrations in Table 4 regioisomer 6 would be expected using standard protocols, as has been previously reported for a number of the cases described.
Origin of Regioselectivity Reversals
As documented above, experimental conditions were identified that provide a new handle for regioselectivity reversals across a broad range of substrates. The unique capabilities of this regioselectivity reversal strategy are well illustrated in couplings of aromatic alkynes. Phenyl propyne, for example, can now be converted to either regioisomer with very high selectivity as the above optimization studies illustrate (Tables 1–3). The simplest explanation for the effects is that the rate-determining step for the standard protocol involves metallacycle formation (1 to 2, Scheme 2a) leading to the preferred formation of 6, whereas the rate-determining step for the new protocols using a large ligand, large silane, high temperature, and low concentration involves silane σ-bond metathesis (2 to 3, Scheme 2b) leading to the preferred formation of 5. In related processes, the reversibility of metallacycle formation has been demonstrated, which would be required for the σ-bond metathesis step to be rate-determining.9 This hypothesis of changing rate-determining step between the different experimental protocols suggested the evaluation of the experimental variations described above (Tables 1–3).
Scheme 2. Initial Mechanistic Hypothesis.

Given that silane σ-bond metathesis had not been previously documented to be rate-determining in any of the prior experimental or computational studies of this reaction type, we sought to gain direct evidence for this mechanistic feature. Kinetic isotope effects using (i-Pr)3SiH and (i-Pr)3SiD in the above protocols were small and were little changed between protocols.10 However, initial rates experiments involving variations of silane concentration provide much more useful information in this context.11
The dependence of initial rates on silane concentration was thus conducted with the experimental protocol described in this work following method A (Table 4), except at an overall concentration of 0.0125 M and without slow addition of any reagents. To our surprise, initial rates for the generation of products illustrated only a small rate dependence on the silane concentration. However, the origin of this effect became clear by analyzing the silane dependence in the generation of regioisomers 5 and 6 separately. As the plots of the rates of formation of the major product 5 (Figure 1a) and minor product 6 (Figure 1b) depict, the rate dependence varied sharply between the two product regioisomers. Upon varying silane concentration from 2.0 to 6.0 equiv at constant volume, the increase in rate of formation of the major regioisomer 5 was very small, with a near-zero rate dependence. Alternatively, upon tracking the initial rates of the formation of the minor isomer 6, a significant rate dependence was noted with a near-first-order rate dependence. Plotting the ratios of initial rates of the product formation (initial rate of 6/initial rate of 5) across the range of silane concentrations provides a clear demonstration of the changes in rate dependence for each regioisomer (Figure 1c). Furthermore, the trend visible in this latter graph shows that regioselectivity should increase as silane concentration decreases, which explains the observation that syringe drive addition protocols and experiments at high dilution lead to exceptionally high regiocontrol.
Figure 1.

(a) Initial rates for formation of 5. (b) Initial rates for formation of 6. (c) Ratio (6/5) of initial rates.
The above initial rate data suggests, at least for the fast addition protocols, that the origin of the effects described above (Tables 1–3) is that the rate-determining step is different for the two regioisomeric pathways (Scheme 3). The formation of major regioisomer 5 follows the previously observed mechanistic feature of rate-determining metallacycle formation (1a to 2a), while the minor regioisomer 6 follows a mechanism that involves rate-determining σ-bond metathesis (2b to 3b). This difference in behavior of 2a and 2b can be explained by the more crowded nickel center of 2b, due to the proximal position of the bulkier alkyne substituent (Ph in this case). The concentration effects described above (Table 2) can be rationalized by a mechanism involving different rate-determining steps for the major and minor isomers (Scheme 3), since only formation of product 6 strongly depends on silane concentration. Similarly, the temperature effects (Table 3) can be rationalized, since the rate-determining step for the production of 5 (i.e., 1a to 2a) is a unimolecular rearrangement, whereas the rate-determining step for the production of 6 (i.e., 2b to 3b) is a bimolecular process involving two bulky components (metallacycle 2b and (i-Pr)3SiH), which thus proceeds with a large entropic penalty. The modification of reaction kinetics for only one of two regioisomeric products thus provides an unusual but effective handle for rational reversal of regioselectivity in a catalytic process.
Scheme 3. Mechanism Invoking Different Rate-Determining Steps for Production of 5 and 6.

While we are unaware of direct precedent for this regiocontrol strategy in reductive couplings, Ohmura and Suginome demonstrated a strategy with reversal of regioselectivity in alkyne silaborations by switching between reversible and irreversible alkyne insertion pathways using ligand control.3a In other conceptually related advances, Waymouth demonstrated control in the reversibility of metallacycle formation as a strategy for controlling diastereoselectivity of zirconocene-catalyzed diene cyclomagnesiations.12 However, the simultaneous operation of differing kinetic descriptions for two regioisomeric pathways in a single reaction has not previously been described in reactions of this type.
While it is conceivable that a completely different mechanism involving direct oxidative additive of silane to Ni(0) could explain the silane rate dependence, several pieces of evidence argue against this. First, the increasing silane bulk required to introduce the silane rate dependence would disfavor silane oxidative addition on steric grounds.13 Second, the differing kinetic descriptions for formation of the two regioisomers, as documented in Figure 1, would not be expected by mechanisms involving silane oxidative addition to nickel. Third, silane oxidative addition pathways typically afford the products of aldehyde hydrosilylation8,14a or alkyne hydrosilylation,14b but not three-component coupling of the aldehyde, alkyne, and silane. The hydrosilylation of neither the aldehyde nor the alkyne proceeds efficiently under the conditions (Table 4, method A) where the three-component coupling efficiently occurs. Finally, the aldehyde hydrosilylation processes proceed with a significant inverse kinetic isotope effect,8 which is not seen under the conditions developed in this study. For these reasons, the data reported herein are best interpreted as described as following the mechanism outlined in Scheme 3.
Conclusions
In summary, this study illustrates that a rational change in the regioselectivity- and rate-determining step of aldehyde–alkyne reductive couplings for one of the two possible regioisomers leads to a significantly improved regiocontrol strategy using commercially available ligands and silanes. The improvement in selectivity arises from a change in mechanism such that the silane participates in the rate- and regiochemistry-determining step of the reaction for minor regioisomer production. This methodology possesses broad scope and improves the regioselectivity outcome for numerous substrate combinations by selecting for addition to the more hindered alkyne terminus.
Acknowledgments
We thank Dr. Hasnain Malik, Dr. Grant Sormunen, and Prof. Ryan Baxter for helpful suggestions and discussions. We are grateful to the National Institutes of Health (GM57014) for financial support.
Supporting Information Available
Experimental details, kinetics analysis, and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For processes involving nickel catalysis:; a Montgomery J. Organonickel Chemistry. In Organometallics in Synthesis; Lipshutz B. H., Ed.; John Wiley & Sons, Inc.: Hoboken, 2013; pp 319–428. [Google Scholar]; b Montgomery J. Angew. Chem., Int. Ed. 2004, 43, 3890. [DOI] [PubMed] [Google Scholar]; c Moslin R. M.; Miller-Moslin K.; Jamison T. F. Chem. Commun. 2007, 4441. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tanaka K.; Tajima Y. Eur. J. Org. Chem. 2012, 3715. [Google Scholar]
- For processes involving other metals, see:; a Ketcham J. M.; Shin I.; Montgomery T. P.; Krische M. J. Angew. Chem., Int. Ed. 2014, 53, 9142. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reichard H. A.; McLaughlin M.; Chen M. Z.; Micalizio G. C. Eur. J. Org. Chem. 2010, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bower J. F.; Kim I. S.; Patman R. L.; Krische M. J. Angew. Chem., Int. Ed. 2009, 48, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Huddleston R. R.; Jang H.-Y.; Krische M. J. J. Am. Chem. Soc. 2003, 125, 11488. [DOI] [PubMed] [Google Scholar]; e Jang H.-Y.; Huddleston R. R.; Krische M. J. J. Am. Chem. Soc. 2004, 126, 4664. [DOI] [PubMed] [Google Scholar]; f Barchuk A.; Ngai M.-Y.; Krische M. J. J. Am. Chem. Soc. 2007, 129, 8432. [DOI] [PubMed] [Google Scholar]; g Liu P.; Krische M. J.; Houk K. N. Chem.—Eur. J. 2011, 17, 4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For notable catalytic processes that demonstrate regiochemistry reversal:; a Ohmura T.; Oshima K.; Taniguchi H.; Suginome M. J. Am. Chem. Soc. 2010, 132, 12194. [DOI] [PubMed] [Google Scholar]; b Gao F.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10961. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jang H.; Zhugralin A. R.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 7859. [DOI] [PubMed] [Google Scholar]; d Park J. K.; Ondrusek B. A.; McQuade D. T. Org. Lett. 2012, 14, 4790. [DOI] [PubMed] [Google Scholar]; e Han L.-B.; Zhang C.; Yazawa H.; Shimada S. J. Am. Chem. Soc. 2004, 126, 5080. [DOI] [PubMed] [Google Scholar]; f Wu J. Y.; Moreau B.; Ritter T. J. Am. Chem. Soc. 2009, 131, 12915. [DOI] [PubMed] [Google Scholar]; g Tekavec T. N.; Arif A. M.; Louie J. Tetrahedron 2004, 60, 7431. [Google Scholar]; h Miller Z. D.; Li W.; Belderrain T. R.; Montgomery J. J. Am. Chem. Soc. 2013, 135, 15282. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Xu K.; Thieme N.; Breit B. Angew. Chem., Int. Ed. 2014, 53, 2162. [DOI] [PubMed] [Google Scholar]; j Miller Z. D.; Montgomery J. Org, Lett. 2014, 16, 5486. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Ding S. T.; Song L. J.; Chung L. W.; Zhang X. H.; Sun J. W.; Wu Y. D. J. Am. Chem. Soc. 2013, 135, 13835. [DOI] [PubMed] [Google Scholar]
- For early illustrations including those that involve regiocontrol through substrate bias, see:; a Montgomery J.; Oblinger E.; Savchenko A. V. J. Am. Chem. Soc. 1997, 119, 4911. [Google Scholar]; b Tang X.-Q.; Montgomery J. J. Am. Chem. Soc. 1999, 121, 6098. [Google Scholar]; c Huang W.-S.; Chan J.; Jamison T. F. Org. Lett. 2000, 2, 4221. [DOI] [PubMed] [Google Scholar]; d Mahandru G. M.; Liu G.; Montgomery J. J. Am. Chem. Soc. 2004, 126, 3698. [DOI] [PubMed] [Google Scholar]; e Malik H. A.; Chaulagain M. R.; Montgomery J. Org. Lett. 2009, 11, 5734. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Saito N.; Katayama T.; Sato Y. Org. Lett. 2008, 10, 3829. [DOI] [PubMed] [Google Scholar]
- a Miller K. M.; Jamison T. F. J. Am. Chem. Soc. 2004, 126, 15342. [DOI] [PubMed] [Google Scholar]; b Miller K. M.; Luanphaisarnnont T.; Molinaro C.; Jamison T. F. J. Am. Chem. Soc. 2004, 126, 4130. [DOI] [PubMed] [Google Scholar]; See also:; c Bahadoor A. B.; Flyer A.; Micalizio G. C. J. Am. Chem. Soc. 2005, 127, 3694. [DOI] [PubMed] [Google Scholar]; d Bahadoor A. B.; Micalizio G. C. Org. Lett. 2006, 8, 1181. [DOI] [PubMed] [Google Scholar]
- a Malik H. A.; Sormunen G. J.; Montgomery J. J. Am. Chem. Soc. 2010, 132, 6304. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knapp-Reed B.; Mahandru G. M.; Montgomery J. J. Am. Chem. Soc. 2005, 127, 13156. [DOI] [PubMed] [Google Scholar]
- a Liu P.; McCarren P.; Cheong P. H.-Y.; Jamison T. F.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 2050. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McCarren P. R.; Liu P.; Cheong P. H.-Y.; Jamison T. F.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 6654. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu P.; Montgomery J.; Houk K. N. J. Am. Chem. Soc. 2011, 133, 6956. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Haynes M. T.; Liu P.; Baxter R. D.; Nett A. J.; Houk K. N.; Montgomery J. J. Am. Chem. Soc. 2014, 136, 17495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter R. D.; Montgomery J. J. Am. Chem. Soc. 2011, 133, 5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ogoshi S.; Tonomori K.; Oka M.; Kurosawa H. J. Am. Chem. Soc. 2006, 128, 7077. [DOI] [PubMed] [Google Scholar]; b Park B. Y.; Montgomery T. P.; Garza V. J.; Krische M. J. J. Am. Chem. Soc. 2013, 135, 16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Primary kinetic isotope effects of silane σ-bond metathesis can be very small, for example, with the kH/kD as small as 1.15 in some well-defined σ-bond metathesis processes. See:Sadow A. D.; Tilley T. D. J. Am. Chem. Soc. 2005, 127, 643. [DOI] [PubMed] [Google Scholar]; b For an outstanding review on the interpretation of kinetic isotope effects, see:Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. [DOI] [PubMed] [Google Scholar]; For a comprehensive overview of KIE effects including those involving silanes, see:; c Gómez-Gallego M.; Sierra M. A. Chem. Rev. 2011, 111, 4857. [DOI] [PubMed] [Google Scholar]
- Previous computational work7c illustrated that the σ-bond metathesis step (silane addition to the metallacycle) proceeds by coordination of silane to nickel, followed by the development of an interaction between silicon and oxygen during the transition state. During these processes, the Si–H bond length is only very slightly elongated, and cleavage of the Si–H bond occurs after the transition-state energy maximum. For this reason, even if the silane is involved in the rate-determining step, a primary kinetic isotope effects would be expected to be small. If the silane is involved in the rate-determining step, the concentration of the silane will influence the initial rates irrespective of the extent and timing of Si–H cleavage during the σ-bond metathesis. For this reason, evaluating initial rates avoids the limitations of KIE experiments.
- Knight K. S.; Wang D.; Waymouth R. M.; Ziller J. J. Am. Chem. Soc. 1994, 116, 1845. [Google Scholar]
- a Corey J. Y.; Braddock-Wilking J. Chem. Rev. 1999, 99, 175. [DOI] [PubMed] [Google Scholar]; b Ampt K. A. M.; Duckett S. B.; Perutz R. N. Dalton Trans. 2007, 2993. [DOI] [PubMed] [Google Scholar]; c Hester D. M.; Sun J.; Harper A. W.; Yang G. K. J. Am. Chem. Soc. 1992, 114, 5234. [Google Scholar]; d Hill R. H.; Wrighton M. S. Organometallics 1987, 6, 632. [Google Scholar]
- a Lage M. L.; Bader S. J.; Sa-ei K.; Montgomery J. Tetrahedron 2013, 69, 5609. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chaulagain M. R.; Mahandru G. M.; Montgomery J. Tetrahedron 2006, 62, 7560. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.