

Abstract
The glial excitatory amino acid transporter 2 (EAAT2) mediates a majority of glutamate re-uptake in human CNS and, consequently, is associated with a variety of signaling and pathological processes. While our understanding of the function, mechanism and structure of this integral membrane protein is increasing, little if any mass spectrometric (MS) data is available for any of the EAATs specifically, and for only a few mammalian plasma membrane transporters in general. A protocol to express and purify functional EAAT2 in sufficient quantities to carry out MS-based peptide mapping as needed to study ligand–transporter interactions is described. A 6×HIS epitope was incorporated into the N-terminus of human EAAT2. The recombinant protein was expressed in high levels in mammalian HEK 293T cells, where it exhibited the pharmacological properties of the native transporter. EAAT2 was purified from isolated cell membranes in a single step using nickel affinity chromatography. In-gel and in-solution trypsin digestions were conducted on the isolated protein and then analyzed by MALDI-TOF and LC-MS/MS mass spectrometry. Overall, 89% sequence coverage of the protein was achieved with these methods. In particular, an 88 amino acid tryptic peptide covering the presumed substrate binding domains HP1, TMD7, HP2, and TMD8 domains of EAAT2 was also identified after N-deglycosylation. Beyond the specific applicability to EAAT2, this study provides an efficient, simple and scalable approach to express, purify, digest and characterize integral membrane transporter proteins by mass spectrometry.
Keywords: Glutamate, Membrane transporter, Excitatory amino acid transporter, Integral membrane protein, Protein digestion, Mass spectrometry, Peptide mapping, MALDI-TOF
Introduction
Excitatory amino acid transporters (EAATs)2 are members of the SLC1 solute carrier family of proteins that also include the sodium-dependent neutral amino acid transporters ASCT1 and ASCT2 [1]. In the CNS, the EAATs serve to regulate extracellular concentrations of l-glutamate. As the primary excitatory neurotransmitter in the CNS, l-glutamate-mediated signaling contributes to a wide range of processes, including: fast synaptic transmission, synaptic plasticity, development, learning and memory, and even neuropathology [2]. Indeed, l-glutamate-mediated neuronal injury (i.e., excitotoxicity) is now well-recognized for contributing to the primary or secondary mechanisms underlying the pathology observed in growing number of acute insults to the CNS (e.g., ischemia, hypoglycemia, spinal cord injury, traumatic brain injury), as well as chronic neurological and neuropsychological disorders (e.g., amyotrophic lateral sclerosis, epilepsy, schizophrenia, Huntington’s disease, Alzheimer’s disease) [3–9]. The rapid and efficient clearance of extracellular l-glutamate into neurons and glia by the EAATs is believed to contribute to the termination of its excitatory signal, the recycling of the neurotransmitter, and the prevention of excitotoxicity [10]. Five EAAT subtypes (EAAT1–5) have been identified and each has been shown to exhibit a characteristic anatomical and cellular distribution, as well as pharmacology [10,11]. Among the subtypes, the glial transporter EAAT2, is accepted as being responsible for more than 90% of total glutamate uptake in the brain [12]. Alterations in the expression and function of EAAT2 have also been reported to be associated with neurodegenerative diseases such as Amyotrophic lateral sclerosis (ALS) [13] and Alzheimer’s [14].
Given the central role that the EAATs play in excitatory transmission and excitotoxicity, considerable effort has been directed toward a better understanding of transporter structure and function. The EAATs utilize the electrochemical gradients across the cell membrane to drive the uptake of l-glutamate against a significant concentration gradient. Stoichiometrically, the uptake of one molecule of l-glutamate is coupled with the inward movement of three Na+ ions and a H+, while one K+ is transported out of the cell to reorient the substrate binding domains and complete the cycle [15]. Based upon a number of approaches, including surface cysteine scanning [16–18], a transporter protein model emerged that consisted of eight prominent transmembrane domains in combination with two less well-defined reentrant loops near the C-terminal region [19,20]. Site-directed mutagenesis studies also demonstrated that several residues around these reentrant loops are critical to transport activity [18–20]. During this same time period, considerable progress was also made in the development of EAAT inhibitors and the generation of SAR-based pharmacophore models [11]. These two fields essentially merged with crystallization of a homologous glutamate/aspartate transporter GltPh from the archaeal thermophile Pyrococcus horikoshii. Sharing 37% sequence homology with human (h)EAAT2, the GltPh structure has eight transmembrane regions (TMD1–TMD8) and two hairpin loops (HP1 and HP2) (See Fig. 1). Solved structures have been proposed for an outward facing substrate-bound (i.e., aspartate) “closed” state, a non-substrate inhibitor-bound (i.e., threo-β-benzyloxyaspartate, TBOA) “occluded” state and an inward facing state which capture the differential positioning of the molecular “gates” responsible for the alternating access mechanism of the transporter [21–23]. Based on these structures, potential permeation pathways for aspartate through GltPh have been proposed by molecular dynamics simulations [24]. With both a growing library of potent ligands and an EAAT homology model in hand, the stage is set to begin exploiting ligand-based structural approaches (e.g., R-group modification, photoaffinity labeling, etc.) to examine substrate binding and translocation by the EAATs in greater detail. Such studies are often dependent upon the development of mass spectrometric (MS), expression and purification protocols specifically relevant to the proteins in question. To our knowledge, little or no MS data is available for any of the EAATs specifically, and for only a few mammalian plasma membrane transporters. The lack of such data is not surprising considering the inherent problems associated with the MS-based characterization of integral membrane proteins, including: (i) low abundance, (ii) poor solubility in aqueous buffers, (iii) poor stability in-solution, and (iv) complex post-translational modifications. Despite these difficulties, improved strategies for membrane solubilization, purification and a growing analytical database, have led to successful MS studies on an increasing number of integral membrane proteins [25–28]. In the present study we add to this effort with the isolation and MS characterization of hEAAT2. Using a mammalian HEK293-based cellular expression system to maximize consistency in terms of protein processing, activity, and pharmacological specificity, hEAAT was isolated as hexahistidine-tagged construct (6×HIS hEAAT2). A combination of in-gel and in-solution methods followed by MS analysis yielded an overall sequence coverage of the protein of 89%, including two glycosylation sites that appear to influence the access of trypsin to presumed active site regions. Importantly, the reported protocols yielded about 98% coverage of the 155 amino acid region of hEAAT2 that includes HP1, TMD7, HP2, and TMD8 domains that are particularly relevant to the proposed substrate binding site on the transporter.
Fig. 1.

Upper image: Amino acid sequence of the N-terminal 6×HIS-tagged human EAAT2. The transmembrane domains are marked in bold and the hairpin loops are marked in italic. Solid lines represent regions identified by in-gel digestions, dotted lines represent regions identified by in-solution digestions (corresponding to Table 1). Lower image: Sequence coverage of hEAAT2 by MALDI-TOF MS analysis aligned to secondary structure predictions derived from sequence alignment of hEAAT2 against crystal structure of GltPh [23] (PDB entry: 2NWW). Amino acids identified in in-gel and in-solution analyses are labeled in blue and green, respectively. Peptide fragments identified by both methods are labeled in red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Materials and methods
General chemicals and buffers were obtained from Sigma Chemical Co. (St. Louis, MO) and Fisher Chemical (Pittsburgh, PA). Protease inhibitor cocktail was obtained from Roche Applied Science (Indianapolis, IN). HisTrap chelating columns were obtained from GE Healthcare (Piscataway, NJ). Sequencing-grade modified trypsin was obtained from Promega (Madison, WI). Peptide calibration standards were obtained from Bruker Instruments (Billerica, MA) and Invitrogen (Carlsbad, CA).
Construction of hexahistidine-tagged hEAAT2
hEAAT2 cDNA was PCR amplified from pBlueScripthEAAT2 (kindly provided by M. Kavanaugh, University of Montana) using primer pairs (forward; 5′-ATTAGGATCCATGGCATCTACGGAAGG TG-3′ and reverse 5′-TATTGATATCTTATTTCTCACGTTTCCA-3′). The primer pair introduced BamHI sites at the 5′ end and EcoRV sites at the 3′ end of the amplified fragment. The PCR fragment was then subcloned into the BamHI and EcoRV sites within the polylinker of the AAV vector pAM-CAG-WPRE (kindly provided by M. During, the Ohio State University) to create pAM-CAG-EAAT2-WPRE. Final clones were confirmed by double stranded sequencing. A 110 b.p. HIS-Xpress insert was PCR amplified from pBAD/HIS B (Invitrogen) using primer pairs 5′-TTT GGA TCC ACC ATG GGG GGT TCT CAT-3′ and 5′-ATT GGA TCC CTT ATC GTC ATC GTC GTA-3′ introducing BamHI sites into the 5′ and 3′ ends of the fragment. The PCR fragment was subcloned into pCR-BLUNT-TOPO (Invitrogen) according to the manufacturer’s protocol. Plasmid DNA was isolated from positive clones and the reading frame and sequence were confirmed by double strand sequencing analysis (Murdock Lab sequencing facility, University of Montana). The fragment was isolated by BamHI digestion, purified and subcloned into pAM/CAG-EAAT2 plasmid at the 5′-end of the gene, generating an N-terminal fusion protein. The correct orientation was confirmed by restriction analysis and PCR amplification.
Cell culture and transfection
HEK 293T cells (ATCC, Manassas, VA) between passages 10 and 20 were cultured at 37 °C in a humidified atmosphere containing 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids solution, and 0.05% penicillin–streptomycin (5000 U/ml) and gentamicin sulfate (0.05 mg/ml). Cells were seeded at 1 × 105 cells/well in 12-well plates for transporter activity experiments and 1.5 × 106 cells/dish in 100 mm2 dishes for membrane preparations. At 48 h after plating, cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or FuGENE 6 Transfection Reagent (Roche, Indianapolis, IN) at a ratio of 2 μl of to 2 μg and 4 μl of to 3 μg of purified plasmid DNA, respectively, in accordance with the manufacturer’s instructions.
Transporter activity
Approximately 24 h after transfection, near-confluent HEK 293T cells were rinsed with a physiological buffer (138 mM NaCl, 11 mM d-glucose, 5.3 mM KCl, 0.4 mM KH2PO4, 0.3 mM Na2HPO4, 1.1 mM CaCl2, 0.7 mM MgSO4, 10 mM HEPES, pH 7.4) and allowed to preincubate at 37 °C for 5 min. Uptake was initiated by replacing the pre-incubation buffer with buffer containing the indicated amount of d-[3H]-aspartate essentially as described [34]. Following a 5 min incubation, the media was removed by rapid suction and the cells rinsed three times with ice-cold buffer. The cells were dissolved in 0.4 N NaOH for 24 h and analyzed for radioactivity by liquid scintillation counting. Protein concentrations were determined by the BCA (Pierce) method. Transport rates were corrected for background, i.e., radiolabel accumulation at 4 °C. Initial studies confirmed that uptake quantified in this manner was linear with time and protein levels and that uptake in untransfected HEK 293T cells was indistinguishable from background. In those experiments examining the concentration dependence of d-[3H]-aspartate uptake, kinetic constants were determined by non-linear curve fitting (KaleidaGraph 3.6, Synergy Software).
Membrane preparation and immobilized metal affinity chromatography (IMAC)
Approximately 24 h after transfection, near-confluent HEK 293T cells were rinsed with DPBS (MediaTech, Manassas, VA) after the culture medium was aspirated. Cells were harvested in DPBS with 5 mM dithiothreitol (DTT) and EDTA-free complete protease inhibitor (Roche, Indianapolis, IN). The cell suspension was homogenized in 0.2 M sucrose with 5 mM DTT and then centrifuged at 500g for 5 min (low speed spin). The pellet was discarded and the membrane fraction was centrifuged at 13,000g for 30 min (high speed spin). The membrane pellet was resuspended in 1 ml IMAC suspension buffer containing 0.5 M NaCl, 0.02 M sodium phosphate, 1% n-dodecyl-β-d-maltoside (DDM), 30% glycerol, 5 mM DTT and 2 mg/ml asolectin. The membrane suspension was incubated at 4 °C for 8 h, then centrifuged at 14,000g for 3 min and the supernatant applied to a 1 ml HisTrap High Performance nickel-Sepharose column (GE Healthcare, Piscataway, NJ). The flow through fractions were collected and the column was washed with 15 ml of washing buffer containing 0.5 M NaCl, 0.02 M sodium phosphate, 20 mM imidazole, 0.1% DDM, 30% glycerol and 2 mg/ml asolectin. The 6×HIS hEAAT2 was eluted with the same wash buffer containing an increasing gradient of imidazole (50–300 mM final concentration), or a single-step elution of 300 mM imidazole in the wash buffer.
Western blotting
For detection of the hEAAT2 by Western immunoblotting, samples were mixed with 2× Laemmli buffer and resolved by SDS–PAGE using 4–15% gradient gels, then transferred to PVDF membranes (Bio-Rad, Hercules, CA). Membranes were blocked for 1 h in 0.1% dried milk, 20 mM Tris base, 130 mM NaCl, 0.1% Tween 20, pH 7.6, followed by overnight incubation at 4 °C with rabbit anti-GLT-1 polyclonal antibody (Affinity Bioreagents, Golden, CO). Membranes were then washed and incubated with anti-rabbit IgG Horseradish Peroxidase (HRP)-linked antibody (Cell Signaling, Danvers, MA) for 1 h at room temperature and developed using ECL Western blot chemiluminescence reagent (Cell Signaling, Danvers, MA). Fujifilm LAS 3000 imaging system (Bio-Rad, Hercules, CA) was used to capture chemiluminescence.
SDS–PAGE for in-gel digestion
Fractions containing the purified hEAAT2 from the HisTrap column were concentrated from 500 to 20 μl using Microcon YM-50 filters (Millipore, Billerica, MA). The concentrated sample was mixed with 2× Laemmli buffer and loaded onto a 4–15% SDS–PAGE gel, then stained with Biosafe Coomassie (Bio-Rad). Gel images was taken using a Bio-Rad Molecular Imager ChemiDoc XRS system.
Intact protein MALDI-TOF analysis
Intact purified hEAAT2 protein was not reduced or alkylated before being precipitated from IMAC fractions by adding 10% trichloroacetic acid (TCA) and kept at 4 °C for 30 min. The protein pellet was collected by centrifugation at 15,000g for 5 min. The protein pellet was then washed three times with 1 ml of cold 1:1 ethanol:ether (v:v) solution. The washed pellet was briefly air-dried and resuspended in 50% acetonitrile and 0.1% TFA. An aliquot of the solution was mixed with an equal volume of sinapic acid matrix solution in 50% acetonitrile and 0.3% TFA before being spotted onto a MALDI plate. A bovine serum albumin MALDI-MS standard (Sigma–Aldrich, St. Louis, MO) was used for calibration.
Protein deglycosylation
Purified hEAAT2 protein from HisTrap column was concentrated by Millipore Microcon YM-50 filter to a concentration of 1.1 mg/ml. A denaturant solution containing 2% octyl-β-d-glucopyranoside and 100 mM 2-mercaptoethanol was added, followed by the addition of 500 U/ml PNGase F (from Elizabethkingia meningoseptica, Sigma–Aldrich, St. Louis, MO) at a ratio of 50 U/mg of protein. The solution was mixed and incubated at 37 °C for 1 h. This enzyme recognizes the N-glycosylation sites of glycoproteins and cleaves the linkage between Asn and the carbohydrate chain [29]. The reaction was stopped by adding appropriate volume of 2× Laemmli sample buffer and reaction products were analyzed on SDS/PAGE or Western immunoblotting.
In-solution digestion
Aliquots of purified hEAAT2 from HisTrap column were precipitated in 10% TCA at 4 °C for 30 min. The protein pellet was collected by centrifugation at 15,000g for 5 min and then washed three times with 1 ml of cold 1:1 ethanol:ether (v:v) solution. The washed pellet was briefly air-dried and resuspended in 50 mM ammonium bicarbonate buffer, pH 8.0 (AMBIC). Sequence grade trypsin (Promega, Madison, WI) was added at a 1:20 ratio, and digestion was allowed to proceed at 37 °C for 20 h. Tryptic peptides were analyzed from the digest solution.
In-gel digestion
The SDS–PAGE gel fragments were diced into 1 mm3 pieces and destained twice with 200 μl 50% acetonitrile/25 mM AMBIC, for 1 h to remove the Coomassie blue dye. Gel pieces were then dried in a SpeedVac (Savant) at room temperature. Prior to digestion, gel pieces were reduced with 10 mM DTT in 100 mM AMBIC at 56 °C for 1 h, then alkylated with 55 mM iodoacetamide in ammonium bicarbonate for 1 h at room temperature in the dark. Gel pieces were washed with 50% acetonitrile/25 mM ammonium bicarbonate and dehydrated in SpeedVac. Tryptic digestion was started with the addition of 50 μg reconstituted trypsin per mg of EAAT2 protein in 25 mM NH4HCO3 at pH 8.0. The concentration of the trypsin solution was 12.5 ng/μl. After reswelling, excess trypsin solution was removed and the gel pieces were covered with an overlay of ~20 μl of 25 mM ammonium bicarbonate. The protein was digested for 20 h at 37 °C.
MALDI-TOF MS analysis
MALDI-TOF analysis was performed on a Voyager DE PRO (Applied Biosystems, Foster City, CA). For whole protein analysis, 0.5 μl of sample was spotted onto the target plate followed by spotting an equal volume of sinapic acid matrix solution in 50% acetonitrile and 0.3% TFA. A bovine serum albumin MALDI-MS standard (Sigma–Aldrich, St. Louis, MO) was used for calibration. For peptide analysis, an aliquot of sample was mixed with an equal volume of CHCA matrix solution in 50% acetonitrile and 0.1% TFA, and 1 μl was spotted onto the target plate. A Bruker peptide standard was used for calibration. For average masses obtained in linear mode, mass accuracy was set at 200 ppm; for monoisotopic masses obtained in reflectron mode, mass accuracy was set at 50 ppm. Masses were searched against the NCBI protein database or a hEAAT database using Mascot (www.matrixscience.com). Modifications for carbamidomethylated cysteines and partial methionine oxidation were included.
Nano-LC-Q-Tof MS analysis
The trypsin-digested samples were vacuum-dried and resuspended in 10 μl of 5% acetonitrile /0.1% formic acid solution. An aliquot of the sample (2 μl) was injected into a nanoLC system (Waters, Milford, MA). The sample was separated on a C18 BEH column (100 μm i.d., 10 cm long; Waters, Milford, MA) coupled with an electrospray ionization (ESI)-Q-Tof mass spectrometer (Waters, Milford, MA). The gradient profile started with 95% solvent A (0.1% FA) for 10 min. This was followed by a 20 min linear gradient from 5% solvent B (0.1% FA in ACN) up to 60% solvent B. The system remained at 60% solvent B for 10 min. The flow rate was maintained at 800 nl/min during the entire run. Peptides were detected in the positive ion mode. Upon detection of target m/z values, MS/MS analysis was performed.
Results
Expression and transporter activity
Our long-term goal is to carry out structural studies on human EAAT2 (hEAAT2) that provide greater mechanistic insight into the how substrates and inhibitors interact with this transporter during the binding and translocation processes. Our intent in the present study was to express and purify hEAAT2 in a manner that would facilitate a thorough MS-based characterization. This transporter belongs to the solute carrier family 1 (SLC1) proteins that also include EAATs 1, 3–5, ASCT1 and ASCT2 [1]. Owing to the inherent problems associated with the isolation and structural characterization of integral membrane proteins, it is not surprising that few, if any, MS studies have been carried out on members of this family. The challenges are readily exemplified by reports that hEAAT2 is a protein without a defined molecular weight [30], is extremely unstable in-solution and easily forms SDS-resistant oligomers [31]. Previously, efforts have been made to purify the rat homologue of hEAAT2, GLT-1. Danbolt et al. fractionated solubilized brain membranes with lectin affinity chromatography, followed by chromatography on hydroxyapatite and DEAE-cellulose [32]. More recently, Semliki Forest virus particles carrying a HIS-tagged GLT-1 gene were used to infect BHK-21 cells from which the transporter protein was subsequently purified through IMAC [33]. In the present work we chose to employ a routinely used mammalian cell line (HEK 293T) to express the recombinant protein, as it not only allowed for more direct comparisons of SAR data, but also simplified the protein isolation to a one-step process for fast MS characterization.
Based upon numerous biochemical characterizations, and ultimately confirmed in the GltPh crystal structures, both termini of the hEAAT2 are localized to the cytoplasmic side of the plasma membrane. These, as well as site-directed mutagenesis studies, also led to the conclusion that that the substrate and ion binding domains are located near the C-terminus of hEAAT2 [18–20]. To minimize any potential effect on ligand binding, a 6×HIS hEAAT2 vector was constructed with the epitiope located on the N-terminus of hEAAT2. HEK 293T cells transiently transfected with 6×HIS hEAAT2 cDNA were assayed for transporter activity by quantifying the uptake of d-[3H]-aspartate 24 h after transfection. d-aspartate is commonly used as a substrate in these assays, as it reduces the complications associated with radiolabel metabolism. We found that 6×HIS hEAAT2 expressed in HEK 293T cells is fully functional and closely mimics the activity of native hEAAT2 (Fig. 2). The Vmax and Km for d-Aspartate uptake were calculated to be 4318 ± 734 pmol/min per mg protein and 140 ± 49 μM, respectively, for the 6×HIS hEAAT2 and 3594 ± 267 pmol/min per mg protein and 106 ± 18 μM, respectively, for the native transporter. It is also worth noting that expression of hEAAT2 in HEK 293T cells yielded a much higher level of active protein than other established mammalian cell expression systems. For example, hEAAT2 expressed in neural progenitor cell line C17.2 exhibited a Vmax of 501 ± 72 pmol/min per mg protein under the same assay conditions (data not shown) [34]. The pharmacological specificity of the 6×HIS-tagged transporter was also confirmed by characterizing its sensitivity to well-known blockers. Dihydrokainate (DHK) and l-glutamate reduced the levels of hEAAT2-mediated uptake of d-[3H]-aspartate (25 μM) to 55% and 45% of control, respectively, when included in the assays at 100 μM (data not shown).
Fig. 2.
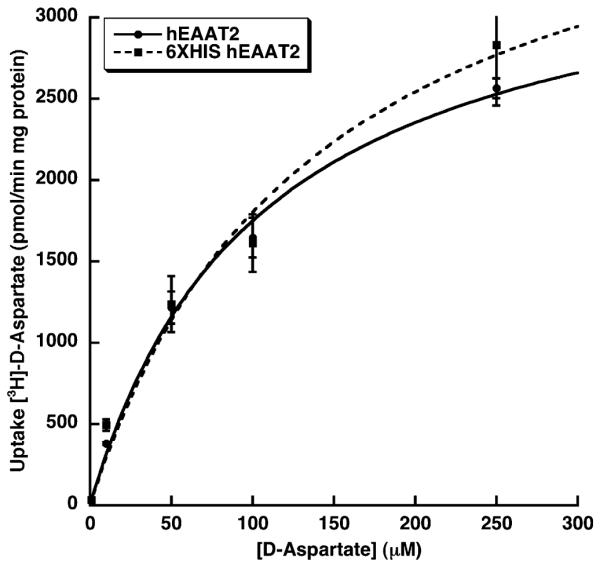
Concentration dependence of the uptake of [3H]-d-aspartate into HEK 293T cells expressing either 6×HIS hEAAT2 or native hEAAT2. The transport rates were determined as described in Materials and methods and have been corrected for non-specific uptake and leakage (4 °C). Data are reported as means ± SEM from a minimum of 4 determinations. Kinetic constants (reported in the text) were determined by non-linear curve fitting (KaleidaGraph 3.6).
Membrane solubilization and purification
Solubilization of membrane protein pellets often requires high salt concentrations, strong detergents, organic solvents [35] and/or organic acids [36]. However, these methods are often not compatible with downstream IMAC purification and MS analysis. In light of this, we developed a membrane isolation method that effectively separated and enriched the 6×HIS hEAAT2 proteins as indicated by hEAAT2 immunoreactivity (Fig. 3, lanes 2–5). To obtain sufficient amounts of membrane protein for the purification of 6×HIS hEAAT2, transfected HEK 293T cells were plated in 100 mm2 dishes at a density of 2 × 106 cells/dish and allowed to reach confluency. Two dishes were used for each membrane preparation. Dithiothreitol (DTT) was included in the lysis and suspension buffers at a concentration of 5 mM to prevent cysteine crosslinking. We attempted to solubilize the membrane pellets with several solvent systems and detergents. It was found that at least 500 mM NaCl was required to effectively solubilize the protein. Among the detergent solutions tested (1% SDS, 2% CHAPS, 2% octyl-β-d-glucoside and 1% DDM), only 1% DDM effectively solubilized the 6×HIS hEAAT2 membrane pellet and was able to prevent the non-specific binding of other cellular proteins to the IMAC column. DDM is a gentle non-ionic detergent that has been shown to enhance protein delipidation and protein mobility in the solubilization and purification of several membrane proteins including glycerol-3-phosphate transporter GlpT [37] and the human glucose transporter, Glut1 [38]. The improved mobility of the transporter was readily observed in our column purification experiments, as the membrane pellets solubilized with the other detergents had trouble eluting through the columns. Consistent with previous observations of the instability of hEAAT2 in-solution and its tendency to form aggregates [31], a significant amount of oligomeric protein was observed in our initial membrane preparations (see Western blot in Fig. 3, lanes 2 and 3). The addition of asolectin (2 mg/ml) [33] to the suspension buffer greatly increased the monomer/oligomer ratio as illustrated by Western blot analysis (Fig. 3, lane 6).
Fig. 3.
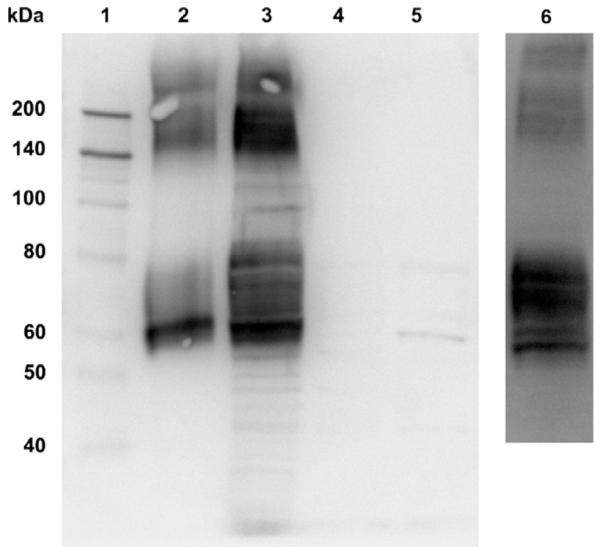
Western blot analysis of hEAAT2 immunoreactivity in HEK 293T cell membrane preparations. Samples collected and analyzed from the membrane preparations include: resuspended pellet (1× Laemmli buffer) from first low speed spin, lane 5; aliquot of supernatant from first low speed spin, lane 2; resuspend pellet (10 μg) from second high speed spin, lane 3; aliquot of supernatant from second high speed spin, lane 4. Molecular weight standards were included in lane 1. The addition of asolectin in the suspension buffer (lane 6) significantly improved monomer/oligomer ratio under the same conditions. Approximate molecular weight range of monomeric 6×HIS hEAAT2 is from 60 to 80 kDa. The molecular weight of oligomeric 6×HIS hEAAT2 ranges from 150 to higher than 200 kDa.
The solubilized membrane fraction containing the 6×HIS hEAAT2 was applied to a HisTrap Ni2+ column. Following a 15 ml wash, the column was eluted with sequential applications of 4 ml volumes of elution buffer containing imidazole with an increasing concentration gradient (50–300 mM). The 6×HIS hEAAT2 eluted in the fractions containing 150 mM and 200 mM imidazole (Fig. 4A, lanes 5 and 6). Isolated protein was quantified by Bradford assay and BCA assay (Pierce). In average, the isolated 6×HIS hEAAT2 protein from 2 confluent 100 mm2 dishes of transfected cells was about 300 μg. As has been previously reported for native glutamate transporters [30], Western blot analysis of the 6×HIS hEAAT2 yielded a wide, fuzzy band that lacked defined edges and had an apparent mass of 70–80 kDa. Given that the calculated molecular weight of 6×HIS hEAAT2 is 66 kDa and that hEAAT2 is known to be a glycoprotein, we treated an aliquot of the suspended protein with peptide-N-glycosidase (PNGase) F. This enzyme cleaves the linkages between Asn and carbohydrate chain at N-glycosylation sites. PNGase F treatment resulted in a shift of the monomer 6×HIS hEAAT2 band to around 65 kDa (Fig. 4B). The observed ~10 kDa change in size is in agreement with the masses of the N-linked glycans on both fully-glycosylated GLAST [39] and GLT-1 [40]. This suggested the majority of 6×HIS hEAAT2 expressed in HEK 293T cells were fully-glycosylated, “mature” transporters. Isolated fractions containing 6×HIS hEAAT2 were concentrated by Microcon filters and resolved on SDS–PAGE gels. When gels were stained with Coomassie, a predominant band around 75 kDa was also observed (Fig. 4C, lane 1), consistent with Western blot analysis, deglycosylation with PNGase F produced a band around 65 kDa (Fig. 4C, lane 2). Although the elution buffer contains DTT and asolectin, the isolated 6×HIS hEAAT2 remained relatively unstable in-solution by either precipitation or aggregate formation on gels. We also observed that freezing precipitated the protein out of solution such that it could not be re-dissolved. Similarly, storage of the protein solution for more than a week at 4 °C resulted in the extensive SDS-resistant aggregation of 6×HIS hEAAT2.
Fig. 4.

IMAC purification of 6×HIS hEAAT2. (A) Western blot to detect recombinant 6×HIS hEAAT2. Lane 1: resuspend pellet from second high speed spin prior to application to IMAC column. Lane 2: 1:200 dilution of 1st column eluent (50 mM imidazole) from the Ni2+-chelating column. Lane 3: 1:200 dilution of 2nd column eluent (100 mM imidazole) from the Ni2+-chelating column. Lane 4: biotin ladder standard. Lane 5: 1:200 dilution of 3rd column eluent (150 mM imidazole) from the Ni2+-chelating column. Lane 6: 1:200 dilution of 4th column eluent (200 mM imidazole) from the Ni2+-chelating column. (B) Western blot probed with anti-GLT-1 antibody to show the glycosylation of 6×HIS hEAAT2. Lane 1: 1:200 dilution of 200 mM imidazole fraction eluted from the Ni2+-chelating column. Lane 2: deglycosylated 6×HIS hEAAT2 treated by PNGase F. (C) Coomassie blue stained SDS–PAGE gel (20 μl of concentrated samples from 200 μl 150 mM imidazole fraction eluted from the Ni2+-chelating column). Lane 1: control. Lane 2: PNGase F deglycosylation.
Intact 6×HIS hEAAT2 protein analysis by MALDI-TOF
In order to determine an accurate mass for the isolated protein, MALDI-TOF analysis was performed on the intact protein following its elution from IMAC. Protein was precipitated with 10% TCA (final concentration) to desalt and remove detergents and washed with ethanol:ether 1:1 before being dissolved into 50% acetonitrile and 0.1% TFA. An aliquot of the solution was then mixed with equal volume of sinapinic acid matrix and spotted on the target plate. Sonication was used when proteins could not be completely dissolved. The MALDI spectrum of the intact protein displayed a singly charged monomer peak with a molecular mass of 76,451 ± 20 Da (Fig. 5A). No dimer or multimer peaks were observed. The experimental mass determined by MALDI-TOF matched the apparent mass observed on the Western blot and Coomassie-stained SDS–PAGE. The single glycoprotein peak also suggested that the 6×HIS hEAAT2 expressed in HEK 293T cells is fully glycosylated at both sites. In contrast, only partial glycosylation of the transporter was observed when GLT-1 was expressed in BHK-21 cells [33].
Fig. 5.

Representative MALDI-TOF mass analyses of native (A) and in-gel trypsin-digested (B–D) 6×HIS hEAAT2. (B) Mass spectrum obtained in reflectron mode. The monoisotopic masses are shown. (C) Mass spectrum obtained in linear mode. The average masses are shown.
In-gel trypsin digestion and MALDI-MS
An in-gel approach was first used to characterize 6×HIS hEAAT2 protein isolated from IMAC. Briefly, gel pieces were destained in 25 ammonium bicarbonate (AMBIC) solution in 50% acetonitrile, reduced in 10 mM DTT in 100 mM AMBIC and alkylated in 55 mM iodoacetamide in 100 mM AMBIC. Diluted trypsin solution (final concentration 12.5 ng/μl, in 25 mM AMBIC) was added to Speed-Vac-dried pieces of the cut gel band representing 6×HIS hEAAT2 and allowed to react for 20 h in 37 °C. Tryptic peptides were extracted by 100 μl of 0.1% TFA in 60% acetonitrile. Peptide solutions were vacuum-dried in a SpeedVac and resuspended in 0.1% TFA in 50% acetonitrile solution for MALDI-TOF. Both linear mode and reflectron mode of the MALDI-TOF produced similar spectra, with more peptides >2000 Da observed in linear mode (Fig. 5B and C). Experimental masses were searched against the human database of the protein sequences in NCBInr using MASCOT. Peptides containing cysteine residues were identified as the carbamidomethylated species, consistent with a complete reduction and alkylation. A few methionine oxidations were also observed, together with a proline to pyro-glutamate modification in the peptide SADCSVEEEPWKR (1592.70 Da, 1606.70 Da in modified peptide). The source of the identified peptides from the in-gel digestions was evenly distributed between transmembrane domains and intracellular/extracellular regions. Tryptic fragments NDEVSSLDAFLDLIR (1706.9 Da, GRAVY = 0.073) and TSVNVVGDSFGAGIVYHLSK (2050.1 Da, GRAVY = 0.500) generated the strongest ion signals.
Predictions based on both hEAAT2 sequence and the glycosylation states of GLT-1 suggested that there are two N-glycosylation sites on hEAAT2, both of which were found on the same tryptic peptide: VLVAPPPDEEANATSAVVSLLNETVTEVPEETK (3448.8 Da; GRAVY = −0.073) [18]. This peptide was not observed following the in-gel digestion of the glycosylated protein, but was readily observed in the spectra of deglycosylated 6×HIS hEAAT2. Overall, the in-gel trypsin digestion of 6×HIS hEAAT2 yielded about 40% sequence coverage of the 66 kDa protein (Table 1). Considering the hydrophobic nature of this integral membrane protein (GRAVY = 0.332), the resulting sequence coverage is much higher than that of most in-gel digestions reported. For example, 18% sequence coverage was achieved following the in-gel digestion of VGLUT1 [25].
Table 1.
Tryptic peptides of 6 × HIS hEAAT2 observed by MALDI-TOF MS.
| Peptide position | m/z observed (Da) | In-gel | In-solution | Missed cleavages | Modification |
|---|---|---|---|---|---|
| 001–025 | 2610.48 mi | X | |||
| 001–052 | 5562.39 avg | X | 3 | ||
| 026–047 | 2405.68 mi | X | 1 | ||
| 053–064 | 1366.42 mi | X | |||
| 053–071 | 2238.16 avg | 2 | |||
| 076–122 | 4931.46 avg | 2 | |||
| 101–122 | 2438.49 avg | X | 2Met-ox | ||
| 101–125 | 2811.03 avg | X | 1 | ||
| 101–150 | 5317.59 avg | X | 3 | 1Met-ox | |
| 123–146 | 2558.32 avg | X | 1 | ||
| 155–183 | 3056.42 avg | X | |||
| 184–192 | 969.71 avg | X | 2 | ||
| 194–208 | 1706.88 mi | X | X | ||
| 209–228 | 2378.10 mi | X | 1CAM | ||
| 230–262 | 3449.10 avg | X | |||
| 263–272 | 1210.68 avg | X | 2 | ||
| 315–334 | 2372.41 avg | X | 2CAM, 2Met-ox | ||
| 315–376 | 6940.20 avg | 3 | 2Met-ox | ||
| 347–408 | 6765.80 avg | 2 | |||
| 409–419 | 1346.85 avg | X | 1CAM | ||
| 423–511 | 9013.93 avg | X | |||
| 423–513 | 9300.75 avg | 1 | |||
| 514–533 | 2050.03 mi | X | X | ||
| 534–544 | 1300.60 mi | X | |||
| 534–553 | 2400.63 avg | X | X | 1 | 1Met-ox |
| 554–562 | 1100.18 mi | X | |||
| 563–606 | 4835.07 avg | X | 3 | ||
| 566–586 | 2424.22 mi | X | 2CAM | ||
| 595–607 | 1592.73 mi | X | 1 | 1CAM |
mi = observed as a monoisotopic mass; avg = observed as an average mass; CAM = carbamidomethyl cysteine.
While the extent of overall sequence coverage was higher than expected, this protocol fell somewhat short with respect to our goal of visualizing the peptides from the proposed substrate binding regions. An alignment of the hEAAT2 sequence with the crystal structure of the homologous bacterial glutamate/aspartate transporter GltPh suggests that substrates and inhibitors bind to the region traversing HP1, TMD7, HP2 and TMD8 of hEAAT2 (Fig. 1). However, our in-gel trypsin digestion protocol yielded only 20% coverage of this 153 amino acid sequence (Fig. 1). A computationally-based mock digestion (ProteinProspector, UCSF) of hEAAT2 predicted an 88 amino acid fragment covering TMD7, HP2 and part of TMD8. The length and hydrophobic nature of this peptide may have precluded its extraction from the gel matrix, even if it had been liberated during the digestion.
In-solution trypsin digestion and MS analysis
As suggested above, peptide recovery from in-gel digestions can be low because of the difficulties in extracting large, hydrophobic peptides from gel matrices. For this reason, in-solution methods have become a preferred option for membrane protein MS characterizations as exemplified by studies on the human cannabinoid CB2 receptor [28], bacteriorhodopsin [35], and vesicular glutamate transporter VGLUT1 [25]. Further, the in-solution digestion performed on lens aquaporin 0 (AQP0) suggested that the addition of organic solvents, such as acetonitrile, could improve the digestion efficiency for integral membrane proteins [41]. The in-solution trypsin digestion of 6×HIS hEAAT2 was carried out following direct precipitation with 10% TCA from the appropriate IMAC eluant fraction. The resulting pellet was washed with ethanol:ether to remove detergents and residual TCA. The protein pellet was collected by centrifugation before being dissolved into one of the three digestion buffers: 50 mM AMBIC, 50 mM AMBIC with 10% acetonitrile or 50 mM AMBIC in 60% methanol. Trypsin was added at an enzyme to protein ratio of 1:20. The reaction was allowed to proceed at 37 °C for 20 h, after which the digestion solution was vacuum-dried in a SpeedVac. The peptides were resuspended in 0.1 TFA, 50% acetonitrile. Trypsin digestion in 50 mM AMBIC with 10% acetonitrile yielded a spectrum with slightly more peptides than 50 mM AMBIC alone. In contrast, digestion in 60% MeOH produced very few identifiable peptides. This may be attributable to the differential solubility of TCA-precipitated protein pellets in each of the solvent systems. Alternatively, purified proteins were reduced in 5 mM TCEP and alkylated in 100 mM iodoacetamide prior to TCA precipitation. The resulting peptides did not differ in sequence from the peptides identified from native conditions. This suggested reduction and alkylation did not improve the sequence coverage of in-solution digestion method in our study.
In contrast to peptides identified from in-gel digestion, the in-solution digestion generated many more long, hydrophobic peptides. Searching the human database of the protein sequences in NCBInr using MASCOT, we were able to identify multiple 6×HIS hEAAT2 tryptic fragments from MALDI-TOF m/z scans ranging from 500 to 3000 Da, 2000 to 6000 Da and 4000 to 10,000 Da (Fig. 6). The identified peptides covered 7 out of 8 transmembrane domains with the exception of TMD4. In the region of the presumed binding pocket, we were able to identify HP1 as a single peptide KNPFSFFAGIFQAWITALGTASSAGTLPVTFR (3405.9 Da, GRAVY = 0.559). Moreover, the 88 amino acid fragment predicted from the in silico tryptic digestion was identifiable only following in-solution digestion of the 6×HIS hEAAT2. The possible origin of this peptide was examined in greater detail using PNGase F. Comparisons between digestions in the presence and absence of PNGase F confirmed with high confidence that the 9013 Da peptide was indeed this tryptic fragment in 6×HIS hEAAT2. These findings indicate that the spatial locations of postulated glycans on the two Asn residues (Asn241 and Asn251) in extracellular loop 2 of 6×HIS hEAAT2 may have limited the entry of trypsin to a specific region of the protein containing TMD7, HP2 and TMD8.
Fig. 6.

Representative MALDI-TOF mass analyses of in-solution trypsin digestion of purified 6×HIS hEAAT2. (A) Mass spectrum of small, hydrophilic peptides in 50% acetonitrile, 0.1% TFA. (B) Mass spectrum of large, hydrophobic peptides identified after N-deglycosylation by PNGase F. All m/z values shown are average masses.
In-solution digests were vacuum-dried in a SpeedVac and resuspended in several other solvents including: 5% formic acid in 50% acetonitrile, 50% formic acid and FAPH (50% formic acid, 25% acetonitrile, 15% isopropanol, and 10% water) buffer followed by MALDI-MS. No additional peptides were identified using these solutions. Indeed, none of the tested solutions produced more peptides than 0.1% TFA in 50% acetonitrile. The sequence coverage of the 6×HIS hEAAT2 by in-solution digestion after deglycosylation was 77% (Table 1, Fig. 1). It is noteworthy that the coverage of the hypothesized hEAAT2 substrate binding domain markedly increased from 20% via in-gel digestion to 91% via in-solution method. Overall, combining in-gel and in-solution methods produce a 98% coverage of the 155 amino acid region of the hEAAT2 that is particularly relevant to substrate binding (Fig. 1).
In addition to MALDI-TOF characterization, preliminary MS/MS analysis was performed on selected peptides from in-solution digests. The peptides in digestion solutions were vacuum-dried and resuspended in 5% ACN/0.1% FA solution. The sample was then applied to a nanoLC-coupled ESI-Q-Tof and peptides were eluted from the LC column by a gradient concentration of ACN/H2O/FA solution described in Materials and methods. Two representative peptides with monoisotopic masses of 1299.47 and 1705.66 were eluted at minute 10 and 33, respectively. The ions were subjected to MS/MS analysis and yielded spectra shown in Fig. 7.
Fig. 7.

NanoHPLC-ESI-Q-TOF MS analysis of tryptic peptides from in-solution digestion of the 6×HIS hEAAT2. Shown are MS/MS spectra of m/z 1299.47 (A) and at m/z 1705.66 (B). Fragment ion notations: b or y, unmodified b or y ions, with single charge; ++, doubly charged ion; *, lost ammonia; 0, lost water.
Discussion
Accumulating evidence indicates that the EAATs play a critical role in excitatory transmission and excitotoxic pathology by regulating the access of the l-glutamate to synaptic and extrasynaptic EAA receptors [10,11]. In order to advance our understandings of the structure of hEAAT2 and lay a foundation for future ligand-based probes, functional hEAAT2 was expressed in HEK 293T cells as a His-tagged construct and purified in a single step using IMAC in quantities sufficient for detailed MS characterization. Kinetic assays indicate the 6×HIS epitope engineered into the N-terminus of the transporter did not affect its ability to bind and transport substrates. Similarly its specificity appeared unaffected, as tested with two well-characterized inhibitors (e.g., DHK and l-glutamate) [11]. Recombinant 6×HIS hEAAT2 expressed in much higher levels in HEK 293T cells than other tested mammalian cell lines, and thereby provided sufficient amounts of membrane material for IMAC purification and MS characterization. Using this protocol we were able to isolate about 300 μg purified 6×HIS hEAAT2 protein in a single preparation from two 100 mm2 culture dishes of HEK 293T cells (≈4 × 106 cells). Using an estimated molecular weight of 76 kDa, this would equate to approximately 6 × 108 transporters/cell, which is markedly greater than that observed when Glt-1 from Rattus norvegicus was expressed and purified from BHK cells using a Semlike Forest virus expressing system [33]. The high levels of expression may be attributable to the vector employed, which contained the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) [42,43], the use of the HEK293 cells or a combination of both. The present protocol is also readily scalable for larger preparations.
Isolated 6×HIS hEAAT2 was digested with trypsin by both in-gel and in-solution methods. In-gel digestion resulted in a 40% of sequence coverage of the whole protein, but lacked coverage of the hypothesized substrate and inhibitor binding domains that would be of interest in mechanistic studies (e.g., HP1, TMD7, HP2 and TMD8, [21–23]). In-solution digestion with the addition of organic solvent, however, greatly increased the number of hydrophobic peptides identified from MALDI-TOF spectra. A 77% sequence coverage was achieved and 91% of the amino acids in the binding domains were identified through this in-solution method (see Fig. 1). We also hypothesize that glycosylation of the protein limited the access of trypsin and consequently prevented the identification of a large peptide from the substrate binding domains. This would suggest that the glycans localized to the external loop between TMD3 and TMD4 [18] are very close to the binding pocket in 3-D space. It has also been reported that glycosylation of GLT-1 does not appear to directly affect its activity, but could possibly stabilize transporters in-solution and the plasma membrane [33]. The spatial proximity of these glycans to the binding pocket raise interesting questions as to the possible functional influences that glycosylation may have on the more subtle aspects of substrate/inhibitor interactions with the transporters.
Overall, combining the in-gel and in-solution methods, 89% of the sequence coverage of the protein was obtained with only TMD4 remaining unidentified in our study. The successful identification of the peptides from the putative substrate binding domains provide a foundation for future studies of ligand–protein complexes and covalent modifications of this binding pocket. In this respect, it is also significant that the 6×HIS hEAAT2 expressed in HEK 293T cells is fully functional and pharmacologically consistent with the WT transporter. Lastly, the expression system, protein solubilization, purification and MS methods we developed in this work should be adaptable to the structural analysis of other mammalian plasma membrane transporters that exhibit similar topologies.
Acknowledgments
This work has been supported by grants from the National Institute of Health: NINDS NS30570 and NCRR COBRE P20 RR15583. We also thank Dr. Chih-Kai Chao and Beverley Parker for technical assistance, as well as Dr. S.A. Patel for his valuable discussions during the preparation of this manuscript.
Footnotes
Abbreviations used: ACN, acetonitrile; AMBIC, ammonium bicarbonate buffer; CHCA, α-cyano-4-hydroxycinnamic acid; DDM, n-dodecyl-β-d-maltoside; DHK, dihydrokainate; FA, formic acid; GLT-1, glutamate transporter subtype 1 (mus musculus); GRAVY, grand average hydropathy; hEAAT2, human excitatory amino acid transporter 2; HEK 293T, human embryonic kidney 293T; IMAC, immobilized metal affinity chromatography; NBI-59159, β-2-fluorene aspartylamide; SDS, sodium dodecyl sulfate; TBOA, threo-β-benzyloxyaspartic acid; TCA, trichloroacetic acid; TCEP, tris(2-carboxyethyl)phosphine; TFA, trifluoroacetic acid, WT; wild type.
References
- [1].Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- [2].Balazs R. Trophic effect of glutamate. Curr. Top. Med. Chem. 2006;6:961–968. doi: 10.2174/156802606777323700. [DOI] [PubMed] [Google Scholar]
- [3].Natale NR, Magnusson KR, Nelson JK. Can selective ligands for glutamate binding proteins be rationally designed? Curr Top. Med. Chem. 2006;6:823–847. doi: 10.2174/156802606777057535. [DOI] [PubMed] [Google Scholar]
- [4].Foster AC, Kemp JA. Glutamate- and GABA-based CNS therapeutics. Curr. Opin. Pharmacol. 2006;6:7–17. doi: 10.1016/j.coph.2005.11.005. [DOI] [PubMed] [Google Scholar]
- [5].Waxman SG, Dib-Hajj S. Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol. Med. 2005;11:555–562. doi: 10.1016/j.molmed.2005.10.004. [DOI] [PubMed] [Google Scholar]
- [6].Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- [7].Olney JW. Excitotoxicity, apoptosis and neuropsychiatric disorders. Curr. Opin. Pharmacol. 2003;3:101–109. [PubMed] [Google Scholar]
- [8].Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Coyle JT. The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochem Pharmacol. 2004;68:1507–1514. doi: 10.1016/j.bcp.2004.07.034. [DOI] [PubMed] [Google Scholar]
- [10].Danbolt NC. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- [11].Bridges RJ, Esslinger CS. The excitatory amino acid transporters: pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacol. Ther. 2005;107:271–285. doi: 10.1016/j.pharmthera.2005.01.002. [DOI] [PubMed] [Google Scholar]
- [12].Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- [13].Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- [14].Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- [15].Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- [16].Slotboom DJ, Konings WN, Lolkema JS. Glutamate transporters combine transporter- and channel-like features. Trends Biochem. Sci. 2001;26:534–539. doi: 10.1016/s0968-0004(01)01925-9. [DOI] [PubMed] [Google Scholar]
- [17].Seal RP, Amara SG. A reentrant loop domain in the glutamate carrier EAAT1 participates in substrate binding and translocation. Neuron. 1998;21:1487–1498. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- [18].Grunewald M, Bendahan A, Kanner BI. Biotinylation of single cysteine mutants of the glutamate transporter GLT-1 from rat brain reveals its unusual topology. Neuron. 1998;21:623–632. doi: 10.1016/s0896-6273(00)80572-3. [DOI] [PubMed] [Google Scholar]
- [19].Grunewald M, Menaker D, Kanner BI. Cysteine-scanning mutagenesis reveals a conformationally sensitive reentrant pore-loop in the glutamate transporter GLT-1. J. Biol. Chem. 2002;277:26074–26080. doi: 10.1074/jbc.M202248200. [DOI] [PubMed] [Google Scholar]
- [20].Seal RP, Leighton BH, Amara SG. A model for the topology of excitatory amino acid transporters determined by the extracellular accessibility of substituted cysteines. Neuron. 2000;25:695–706. doi: 10.1016/s0896-6273(00)81071-5. [DOI] [PubMed] [Google Scholar]
- [21].Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- [23].Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- [24].Gu Y, Shrivastava IH, Amara SG, Bahar I. Molecular simulations elucidate the substrate translocation pathway in a glutamate transporter. Proc. Natl. Acad. Sci. USA. 2009;106:2589–2594. doi: 10.1073/pnas.0812299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cox HD, Chao CK, Patel SA, Thompson CM. Efficient digestion and mass spectral analysis of vesicular glutamate transporter 1: a recombinant membrane protein expressed in yeast. J. Proteome Res. 2008;7:570–578. doi: 10.1021/pr070452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee BK, Jung KS, Son C, Kim H, VerBerkmoes NC, Arshava B, Naider F, Becker JM. Affinity purification and characterization of a G-protein coupled receptor, Saccharomyces cerevisiae Ste2p. Protein Expr. Purif. 2007;56:62–71. doi: 10.1016/j.pep.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takayama H, Chelikani P, Reeves PJ, Zhang S, Khorana HG. High-level expression, single-step immunoaffinity purification and characterization of human tetraspanin membrane protein CD81. PLoS ONE. 2008;3:e2314. doi: 10.1371/journal.pone.0002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zvonok N, Yaddanapudi S, Williams J, Dai S, Dong K, Rejtar T, Karger BL, Makriyannis A. Comprehensive proteomic mass spectrometric characterization of human cannabinoid CB2 receptor. J. Proteome Res. 2007;6:2068–2079. doi: 10.1021/pr060671h. [DOI] [PubMed] [Google Scholar]
- [29].Tarentino AL, Plummer TH., Jr. Enzymatic deglycosylation of asparagine-linked glycans: purification, properties, and specificity of oligosaccharide-cleaving enzymes from Flavobacterium meningosepticum. Methods Enzymol. 1994;230:44–57. doi: 10.1016/0076-6879(94)30006-2. [DOI] [PubMed] [Google Scholar]
- [30].Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J. Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- [32].Danbolt NC, Pines G, Kanner BI. Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry. 1990;29:6734–6740. doi: 10.1021/bi00480a025. [DOI] [PubMed] [Google Scholar]
- [33].Raunser S, Haase W, Bostina M, Parcej DN, Kuhlbrandt W. High-yield expression, reconstitution and structure of the recombinant, fully functional glutamate transporter GLT-1 from Rattus norvegicus. J. Mol. Biol. 2005;351:598–613. doi: 10.1016/j.jmb.2005.06.036. [DOI] [PubMed] [Google Scholar]
- [34].Esslinger CS, Agarwal S, Gerdes J, Wilson PA, Davis ES, Awes AN, O’Brien E, Mavencamp T, Koch HP, Poulsen DJ, Rhoderick JF, Chamberlin AR, Kavanaugh MP, Bridges RJ. The substituted aspartate analogue l-beta-threobenzyl-aspartate preferentially inhibits the neuronal excitatory amino acid transporter EAAT3. Neuropharmacology. 2005;49:850–861. doi: 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- [35].Blonder J, Goshe MB, Moore RJ, Pasa-Tolic L, Masselon CD, Lipton MS, Smith RD. Enrichment of integral membrane proteins for proteomic analysis using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2002;1:351–360. doi: 10.1021/pr0255248. [DOI] [PubMed] [Google Scholar]
- [36].Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- [37].Auer M, Kim MJ, Lemieux MJ, Villa A, Song J, Li XD, Wang DN. High-yield expression and functional analysis of Escherichia coli glycerol-3-phosphate transporter. Biochemistry. 2001;40:6628–6635. doi: 10.1021/bi010138+. [DOI] [PubMed] [Google Scholar]
- [38].Boulter JM, Wang DN. Purification and characterization of human erythrocyte glucose transporter in decylmaltoside detergent solution. Protein Expr. Purif. 2001;22:337–348. doi: 10.1006/prep.2001.1440. [DOI] [PubMed] [Google Scholar]
- [39].Schulte S, Stoffel W. UDP galactose:ceramide galactosyltransferase and glutamate/aspartate transporter, copurification, separation and characterization of the two glycoproteins. Eur. J. Biochem. 1995;233:947–953. doi: 10.1111/j.1432-1033.1995.947_3.x. [DOI] [PubMed] [Google Scholar]
- [40].Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled l-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- [41].Han J, Schey KL. Proteolysis and mass spectrometric analysis of an integral membrane: aquaporin 0. J. Proteome Res. 2004;3:807–812. doi: 10.1021/pr049945w. [DOI] [PubMed] [Google Scholar]
- [42].Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory elements: implications for gene therapy. Hum. Gene Ther. 1999;10:2295–2305. doi: 10.1089/10430349950016942. [DOI] [PubMed] [Google Scholar]
- [43].Paterna JC, Moccettii T, Mura A, Feldon J, Bueler H. Influence of promoter and WHV post-transcriptional regulatory element on AAV-mediated transgene expression in rat brain. Gene Ther. 2000;15:1304–1311. doi: 10.1038/sj.gt.3301221. [DOI] [PubMed] [Google Scholar]