

Abstract
Despite decades of basic and clinical research, treatments to improve outcomes after traumatic brain injury (TBI) are limited. However, based on the recent recognition of the prevalence of mild TBI, and its potential link to neurodegenerative disease, many new and exciting secondary injury mechanisms have been identified and several new therapies are being evaluated targeting both classic and novel paradigms. This includes a robust increase in both preclinical and clinical investigations. Using a mechanism-based approach the authors define the targets and emerging therapies for TBI. They address putative new therapies for TBI across both the spectrum of injury severity and the continuum of care, from the field to rehabilitation. They discuss TBI therapy using 11 categories, namely, (1) excitotoxicity and neuronal death, (2) brain edema, (3) mitochondria and oxidative stress, (4) axonal injury, (5) inflammation, (6) ischemia and cerebral blood flow dysregulation, (7) cognitive enhancement, (8) augmentation of endogenous neuroprotection, (9) cellular therapies, (10) combination therapy, and (11) TBI resuscitation. The current golden age of TBI research represents a special opportunity for the development of breakthroughs in the field.
Keywords: head injury, concussion, neurocritical care, treatment, drugs, excitotoxicity, inflammation, oxidative stress, regeneration, rehabilitation, secondary injury
Despite decades or basic and dinical research, treatments to improve outcomes after traumatic brain injury (TBI) are limited. In severe TBI, with few exceptions, supportive measures such as controlling brain swelling using osmolar therapy or barbiturates, and/or surgical interventions remain the standard of care. In severe TBI, development or novel therapies targeting secondary injury has been the topic or considerable research, but failures in translation continue to mount with the recently published clinical trial on erythropoietin1 and recent halting of the PRO-TECT trial of progesterone therapy (personal communication, David Wright, MD). Only treatment with amantadine in the subacute phase has translated-from the controlled cortical impact (CCI) model of TBI in rats to moderate-severe TBI in humans.2 In mild TBI (mTBI), empiric therapies targeting sequelae such as cognitive impairment and posttraumatic stress disorder are often used. However, there has been little research on therapies in mTBI, secondary injury pathways, or the link between mTBI and neurodegenerative disease. Given the increased recognition of the scope of the problem, the growth in funding for TBI research, and the expanding discussion of therapies,3 an acceleration of research into the treatment of TBI across the injury spectrum is emerging and there is cause for optimism. Using a mechanism-based approach we will define the targets and emerging therapies for TBI. We will address emerging therapies for TBI across the spectrum of severity and the continuum of care, from the field to rehabilitation. We will discuss TBI therapy using 11 categories, namely, (1) excitotoxicity and neuronal death, (2) brain edema, (3) mitochondria and oxidative stress, (4) axonal injury, (5) inflammation, (6) ischemia and cerebral blood flow (CBF) dysregulation, (7) cognitive enhancement, (8) augmentation of endogenous neuroprotection, (9) cellular therapies, (10) combination therapy, and (11) TBI resuscitation.
Therapies Targeting Excitotoxicity and Neuronal Death
Excitotoxicity and its link to neuronal death pathways in TBI has been richly explored, and yet after 45 years of research most antiexcitotoxic therapies have failed to translate.4,5 New understanding of glutamatergic neurotransmission inspires optimism that future drugs will be more effective.
Excitotoxicity after Traumatic Brain Injury
Presynaptic glutamate release depolarizes post-synaptic neurons by opening ion channels such as N-methyl-o-aspartate receptors (NMDARs) that permit Na+ and Ca2+ to surge into cells. Excitotoxic events occur early after TBI and trigger apoptosis, necrosis, necroptosis, autophagy, or pyroptosis (none of which are mutually exclusive). Ca2+ overload is a key feature that is upstream to cell-death signaling.6 Na+ overload is less significant, but may contribute to neuronal swelling.7 After TBI, there is a surge in extracellular glutamate followed by persistent elevations.8 Extracellular glutamate increases intracellular Ca2+ (iCA2+) by activating neuronal glutamate receptors (GluRs) like NMDARs and a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs). Release of Ca2+ from intracellular stores in organelles also contributes to excitotoxicity.9
Necrosis and Apoptosis
Nuclear Ca2+ promotes neuronal survival by inducing protective genes like brain-derived neurotrophic factor (BDNF). In contrast, cytoplasmic Ca2+ is toxic. Necrotic death is triggered in part by calpains Ca2+ activated cysteine proteases that destroy survival substrates.10 Traumatic brain injury induces rapid and sustained iCa2+ elevations and calpain activation.10,11 Blocking calpain activity in TBI models reduces tissue loss.12 Increased iCa2+ also overwhelms mitochondria resulting in oxidative stress, mitochondrial permeability transition pore (MPTP) opening and cytochrome c release.13 This activates the intrinsic pathway (capase-9 dependent).14 Apoptotic cell death has been characterized after TBI.14, 15 Therapies that block executioner caspases reduce tissue loss and improve outcome.16, 17
Additional Excitotoxic Death Pathways
Necroptosis (programmed necrosis) is regulated by receptor-interacting serine/threonine-protein kinase (RIPKl/3) and mixed lineage kinase domain-like.18 Necroptosis is induced by co-incubation of tumor necrosis factor a (TNFa) with Z-VAD FMK (a pan caspase inhibitor). Alone, TNFa stimulates the extrinsic cell death receptor-mediated apoptotic pathway (caspase-8 dependent). Caspase-8 cleaves and destroys RIPK preventing its activation.19 The addition of Z VAD during TNFa induced apoptosis in neurons stabilizes RIPKl/3 activating necroptosis.20 The necroptotic inhibitor necrostatin-1 inhibits neuronal death after TBI.21 However, excitotoxicity induces necroptosis in cultured neurons, but accounts for a fraction of the cell death.22 Autophagy is a homeostatic mechanism that regulatescatabolism of damaged organelles(macroautophagy). Loss of autophagy may contribute to neurodegenerative disease,23 whereas overactivation may promote cell death. There is marked upregulation of autophagy after TBI.24 Mice treated with the autophagy inhibitor 3-methyladenine show reduced neuronal death after TBI.25 However, autophagy may also be beneficial late after TBI by helping to "clean-up" injured brain.26 Microglia can also release glutamate, enhance excitoxicity,27 and promote inflammasome-mediated cell death (pyroptosis) linked to caspase-1. All of these cell death pathways are targets for TBI therapy.
Antiexcitotoxic Therapy
Excitotoxicity plays a key role in tissue damage after TBI. Vespa et al28 reported early posttraumatic subclinical seizure activity and its deleterious effects after severe TBI. Most strategies to reduce excitotoxicity include averting Ca2+ accumulation, or inhibiting downstream death signaling (caspases/apoptosis or calpains/necrosis). Historically NMDARs have been key targets. Nonselective NMDAR antagonists like MK801 are neuroprotective in TBI models. However, they failed in clinical TBI due to psychosomatic side effects, inadvertent death of select brain regions (cingulate and retrosplenial cortex), and limited therapeutic window.5 Progress in NMDAR-mediated neurotransmission has revealed greater complexity then previously appreciated-and may better inform therapy. N-Methyl-o-aspartate receptors consist of heterodimeric glutamate receptor (GluR) subunits including NR1, NR2A, and NR2B. NR2A containing NMDARs are enriched in the synapse (synaptic NMDARs).29 NR2B containing NMDARs are enriched at extrasynaptic sites (extrasynaptic NMDARs).29 Spatial distribution of NMDARs greatly affects excitotoxic signaling. Activation of synaptic NMDARs is neuroprotective. They increase nuclear Ca2+,activate CREB, BDNF, protein kinase B (AKT), phosphorylated-JACOB (pJACOB), and upregulate antioxidants.30-33 In con trast, activation of extrasynaptic NMDARs by glutamate spillover after TBI has the opposite effect. Extrasynaptic NMDARs increase cytoplasmic ca2+, inhibit CREB, AKT, p-JACOB, BDNF, active calpain, stimulate death-associated protein kinase (DAPK), and activate autophagy.30,32-35 Extrasynaptic NMDARs play a role in cell death in TBI. The selective NR2B antagonist Ro 25 6981 inhibits induction of autophagy after TBI.34 Stretch inj ury increases NR2B/NMDAR currents, which open AMPARs. That deleterious cascade is prevented by the NR2B antagonists Ro 25 6981 and memantine (a lipophilic form of amantadine).36 Memantine (Namenda) was tested in rodent TBI many years ago.37 Recently it was discovered to block extrasynaptic NMDARs while sparing synaptic NMDAR function.38 It is approved by the U.S. Food and Drug Administration (FDA) to treat dementia and has proved a more tolerable NMDAR antagonist than MK801.39 Memantine and other next-generation NR2B-selective antagonists deserve additional study.40
Considerations for Chronic Recovery after Traumatic Brain Injury
Long-term blockade of NMDARs may limit recovery of brain function. Even mTBI disturbs synaptic processes resulting in impaired network connectivity.41,42 Abnormal synaptic connectivity may reflect aberrant decreases in glutamatergic neurotransmission or overactivation of GABAergic inhibitory input.43,44 Levetiracetam (Keppra; UCB, Brussels, Belgium) is often used to control seizures in severe TBI patients.45 Although the mechanisms of action are not fully understood they involve GABAergic activation and inhibition of presynaptic glutamate release.46,47 Cognition remains largely intact in patients on levetiracetam.48 It is curious to speculate if levetiracetam may help balance long-term excitatory/inhibitory disturbances in neurotransmission after TBI. Zou et al49 recently reported benefit from chronic treatment with levetiracetam after CCI in rats. Thus, one approach might be to use a potent NMDAR antagonist like memantine early after TBI, to block excitotoxicity and then transition to therapies that fine-tune glutamatergic activity during recovery such as levetiracetam...Fig. 1 provides an overview of excitotoxicity and its link to the neuronal death pathways along with emerging therapies.
Therapies Targeting Brain Edema
Brain edema has been a TBI target for decades. It is identified and continuously monitored in patients with severe TBI by imaging, clinical examination, and intracranial pressure (ICP) monitoring. Cerebral edema is caused by two main mechanisms-cellular (traditionally called cytotoxic) and vasogenic-resulting from a disturbance in the blood-brain barrier (BBB). There are no therapies in clinical practice designed to prevent edema, rather than simply to treat it once it has occurred. Treatments for brain edema are limited to the use of osmolar agents (mannitol, hypertonic saline), which aid in water removal, sedatives like barbiturates, which can lower the cerebral metabolic rate and reduce brain swelling (via a coupled reduction in cerebral blood volume), cerebrospinal fluid (CSF) drainage via a ventriculostomy, and craniectomy. These guidelines-based therapies are routinely used in severe TBI.50,51 However, they have toxicities. For example, increasing serum sodium to > 170 mEq/L to manage refractory brain swelling after severe TBI caused an increased rate of acute renal failure, thrombocytopenia. and acute respiratory distress syndrome.52 Also, the decompressive craniectomy (DECRA) trial failed to show improved outcome in adults with severe TBI (although it decreased ICP).53 Some have thus even begun to question the use of ICP monitoring in patients with severe TBI; this remains controversial.54 A recent randomized controlled trial (RCT) by Chesnut et al55 showed that outcomes after severe TBI did not differ between patients managed with ICP monitoring versus clinical exam/imaging-although the use of therapies for brain swelling was similar or greater in the patients treated based on the clinical exam/imaging. In contrast, other recent clinical studies have shown that even short periods of increased ICP unfavorably affect outcome,56 and new preclinical work suggests that very modest levels of raised ICP (< 20 mm Hg) may be deleterious.57 Brain edema might even contribute to secondary damage in mTBI if astrocyte swelling at the cellular level compromises astrocyte function.
Novel Pathways of Edema Formation
New understandings of the molecular underpinnings of brain edema are revealing new targets and therapies. Laird et al58 outlined molecular events that could contribute to the development of brain edema after TBI (fig. 2). After TBI, neuronal necrosis can induce release of the danger signal high-mobility box protein 1 (HMGB-1). HMGB-1 binds to toll-like receptor-4 (TLR-4) on microglia and triggers interleukin-6 (IL-6) mediated aquaporin-4 (AQP4) channel upregulation in astrocytes, mediating edema formation. AQP4 is a membrane channel that regulates water transport.59 Consistent with this hypothesis, IL-6 and HMGB-1 are increased in human CSF after severe TBI.60,61 HMGB-1 is also linked to brain edema in TBI models.58 In addition to binding to TLR4, HMGB-1 interacts with the receptor for advanced glycation end product (RAGE); the RAGE pathway may play a role in vasogenic edema/breakdown of BBB,62 while TLR4 may mediate cellular edema.58 Cerebrospinal fluid levels of HMGB-1 correlate with unfavorable outcome after TBI.61 More discussion of this pathway is provided later in this review. Finally, Simard et al63 reported that sulfonylurea receptor 1 (SURl) contributes to the development of brain edema. SURl channels conduct monovalent cations, are upregulated after TBI, and function independent of Na+-K+ ATPase activity.63
Fig. 2.
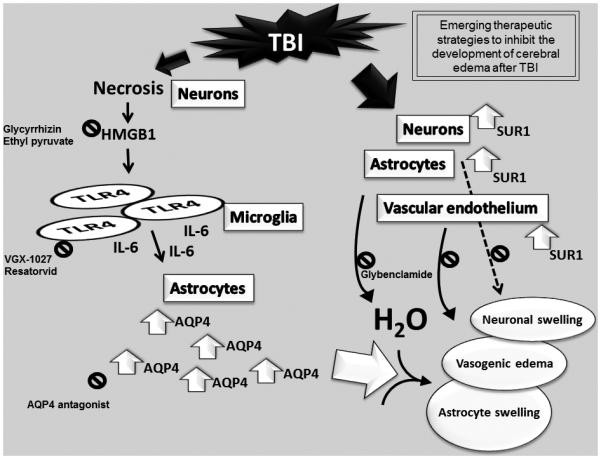
Emerging therapies to inhibit the development of brain edema after traumatic brain injury (TBI). On the left, the novel molecular pathway of brain edema formation proposed by Laird et al,58 which hypothesizes that in areas of necrosis, HMGBl release from neurons binds to TLR4 receptors on microglia and leads to elaboration of IL 6. IL 6 triggers upregulation of AQP4 water channels in astrocytes with water uptake across the blood brain barrier and astrocyte swelling. On the right, a second pathway involves upregulation of the SUR1 channel on neurons, astrocytes, and cerebrovascular endothelium. This monovalent cation channel is normally absent but is upregulated after TBI. Emerging therapies targeting both pathways (identified by 0). For the HMGB1 TLR4 cascade (left), potential therapies include (1) the HMGBl binding agent glycyrrhizin or release inhibitor ethyl pyruvate, (2) inhibitors of TLR4 signaling (VGX 1027 or resatorvid), or (3) AQP4 antagonists. The prototype therapy targeting the SURl channel pathway (right) is glibenclamide. HMGB-1, high mobility box protein 1; TLR4, toll like receptor 4; IL 6, interleukin 6; AQP4, aquaporin 4; SURl, sulfonylurea receptor 1.
Emerging Therapies to Prevent Brain Edema
Effort has been directed at developing new therapies for brain edema in TBI. The anti-inflammatory drug glycyrrhizin inhibits HMGB-1 from binding to RAGE and prevents BBB breakdown and vasogenic edema.58,62 Similarly, Okuma et al64 reported a reduction of edema in a rodent model using an anli-HMGBt monoclonal antibody. Edema could also be reduced by inhibiting TLR4 with VGX, l027. which is in clinical t1ials tor inflammatory diseases.58 Other therapies targeting HMGB-1 /TLR4 are discussed later. AQP4 is anothe1 edema target in TBI. Injection of small-interfe1ing RNA target ing AQP4 1educed brain edema in rats65 and selective AQP4 antagonists are in development (personal communication. Marc Pelletie1). The SURl 1eceptor blocke1 glibenclamide (Glyburide) is another a promising therapy that 1educes edema in TBI models.66 It is in a phase II clinical trial (NCT-01132703) in TBI. These new agents give hope for improved management of b1ain edema. Studies in preclinical models are also needed to determine if reducing edema blunts secondary injury independent of ICP or reduced CBF, including studies in mTBI.
Mitochondrial Targeting Therapies and Oxidative Stress
Mitochondria have impmtant functions ranging from generation of ATP to production of reactive oxygen species (ROS). which are important in the 1egulation of life and death decisions in cells.67 Dysfunctional mitochondria can also gene1ate inf ammatory and vasoactive mediators.58 Timely elimination of dysfunctional mitochondria via macroautophagy is essential particularly in postmitotic cells such as neurons.69 Mitochond1ial dysfonction has been reported in experimental models and humans after TBl,70,73 and is a robust target because alterations in mitochondrial function persist days after injury.74
Role of Oxidative Stress in Traumatic Brain Injury
Oxidative stress plays a key role after TBJ.75 It is classically described as a misbalance between the generation of free radicals and the body's ability to detoxify them.76 However, this definition fails to describe the essential roles free radicals play in normal neuronal function, such as long-term potentiation.77 A more contemporary definition, "disruption of redox signaling and control," recognizes the compartmentalized nature of these events.78 For a thorough evaluation of oxidative stress, a battery of studies, including (1) assessment of generation of free radicals; (2) quantification of oxidation products of lipids, proteins, and DNA; and (3) evaluation of radical scavenging capacity, should be performed. All of these components of oxidative stress occur after experimental and clinical TBI.79-83 Several sacrificial antioxidants and free radical scavengers have been explored in TBI, such as PEG-SOD, tirilazad, and edaravone.84-86 Although they have shown efficacy in TBI models, clinical trials failed to confirm efficacy.87,88 The limitations of general antioxidant strategies include narrow therapeutic window, inability to cross the BBB, and lack of targeting of the specific causes of oxidative stress. Without a targeted approach, the use of sacrificial antioxidants and free radical scavengers will fail.
Mitochondrial Failure after Traumatic Brain Injury
Mitochondria are a major intracellular source of ROS with production of superoxide and its dismutation product hydrogen peroxide.89 Other sources of oxidative stress after TBI include NADPH oxidase, nitric oxide (NO) synthases, xanthine oxidase, and transition metals, which can be released by hemorrhage.90,91 Underlying mechanisms of increased production of ROS by mitochondria include dysfunction of electron transport92 and impairment in ca2+ buffering.93 Mitochondria are also targets of ROS, which can promote MPTP opening, leading to apoptosis.94 Mitochondrial DNA, which encodes elements for electron transport, is also a target for free radical damage.95 Thus, oxidative stress can impair mitochondrial function, which in turn generates more oxidative stress in a vicious cycle.
Targeting Mitochondrial after Traumatic Brain Injury
Several strategies have been designed to combat mitochondrial dysfunction, including alternative fuels and MPTP inhibitors.96,97 Preliminary studies in adults describe the safety of cyclosporine, a MPTP inhibitor, when given after severe TBI.98 These strategies target different components of mitochondrial dysfunction, but fail to localize into mitochondria. Recently small molecules have been discovered99 that can selectively accumulate in mitochondria and bind targets in the organelle to exert their effects. Several strategies have been used including (1) conjugation to lipophilic cations such as triphenylphosphonium that ta ke advantage of negative membrane potential of mitochondria.99 and (2) binding to a specific mitochondrial target such as cardiolipin (CL).a phospholipid exclusively found in the inner mitochondrial membrane.67 Both strategies are effective at delivering thera pies into mitochondria. Compounds in the first category have not been tested inTBI. One of them.Mito-Q (a ubiquinone moiety linked to triphenyl phosphonium) was used in recent human trials in Parkinson disease and hepatitis c.100 Compounds in the second category include Szeto-Schiller (SS) peptides.101 Their uptake into mitochondria is thought to be independent of membrane potential. with a high affinity binding to inner membrane. 101 They contain four alternating aromatic amino acids and some have antioxidant activity. One of these peptides, SS-31. protects mitochondria. accelerates ATP recovery,and reduces infarct size in the hearr 101 Another class of compounds in the second category promising forTBI are thehemigramicidin nitroxides (GS-nitroxide), inspired by the shared ancestry between mitochondria and bacteria, taking advantage of chemical moieties used in antibacterial agents (the antibiotic gramicidin S) with high affinity for the inner membrane. 102 One GS-nitroxide, XJB-5–131, was shown co partition almost exclusively into neuronal mitochondria in vitro. penetrate the BBB. prevent TBl-induced Cl oxidation and caspase activation and improve lesion volume and neurocognitive outcome after TBI.70 This is an exciting targeted strategy for TBI.
In summary, there is promising experimental success in application of mitochondria targeted redox regulators in treatment of TBI. and these approaches deserve significant future efforts.
Therapies Targeting Neuroinflammation
Evidence suggests the inflammatory response. including cytokines. chemokines. microglial activation, and recruitment of circulating leukocytes. mediates secondary injury and /or repair after TBI. Traumatic brain injury causes the release of endogenous danger signals (i.e.. extracellular ATP and HMGB 1),58,61 which bind to pattern recognition receptors such asTLR4 on neurons and glia to activate the immune response. Activated microglia undergo a phenotypic shift from an anti inflamma tory (M2) state to a proinflammatory and procytotoxic (M 1) state. M 1 microglia proliferate and migrate to the injury site and (1) form a barrier between damaged and healthy tissue.103 (2) increase expression of proinflamma torycytokines such asTNFa104 and IL-β,105 and (3) release ROS and reactive nitrogen species. Chronic microglial activation develops and may mediate chronic traumatic encephalopathy (CTE) and neurodegenerative diseases.106
Dual Role of Inflammation InTBI
Studies of the role ofTNFa after TBI reveal the dual effects cf inflammation in secondary injury and repair. Scherbel et at 107 using a TNF KO mouse reported evidence of early neuroprotection in the KO at 48 hour ; however. lNF KO mice had persistent motor deficits and more tissue loss at 4 weeks compared with wi Id type.Balancing neurotoxicitywith repair must be considered for therapies that modulate inflammation. Preclinical work with thalidomide ana logs (TNFa synthesis inhibitor) and etanercept (fusion protein that binds to and inhibits TNFα) have shown benefit early after TBI.108,109
HMGB-1/TLR4 Pathway Inhibition
HMGB1/TLR4 pathway inhibitors were previously discussed. They also block secondary injury due to immune activation. Ethyl pyruvate.an inhibitor of HMGBl secretion.and resatorvid a small molecule inhibitor of TIR4, improved outcome and reduced levels of TNFa and IL-1p in rodent TBI models. 110-111 These drugs have not yet been evaluated in dinicaltrials for TBI.
Other Anti-lnftammatory Agents
Minocycline, a lipophilic tetracycline antibiotic with several proposed mechanisms of action.including inhibition ofmicroglial activation reduces IL-1β production, lesion volume, and functional deficits in TBI models.112 A phase 1 clinical trial of minocycline in TBI is recruiting patients. IL-1β antagonism via intraventricular injection of anti-IL-113 antibody113 and transgenic overexpression of IL 1 receptor antagonist (IL-1ra)114 reduced lesion volume and improved outcomes in TBI models. A recent phase 2 trial in adults with severe TBl115 randomized 20 patients to receive 100 mg recombinant human IL-1ra (Anakinra) for 5 days, an FDA-approved dose for rheumatoid arthritis. Adverse eventsdid not differ in treatment and control groups. Cerebral microdialysis showed increased levels of IL lra with treatment and a shift in the cytokine/chemokine profile. HMG-CoA reductase inhibitors (statins) have several proposed mechanisms of action after TBI (antiapoptotic, anti-oxidant,increase CBF.and neurogenesis): however.the primary mechanism islikely anti-inflammatory. Two clinical studies of statins have been conducted-a RCr of rosuvastatin in 20 adults with TBI that showed improved memory with treatment,115 and a retrospective study showing a 76% relative risk reduction for mortality in patients treated with statins before injury.117 urger RCTs are needed.
Promoting lnftammatlon-Medlated Regeneration
Another approach is to promote shifting microglia from the Ml to the M2 phenotype. Currently under investigation for treating multiple sderosis,118 therapies such as glatiramer acetate, interferon-p, or dimethyl fumarate may promote the beneficial aspects of neuroinflammation-neurogenesis and repair-while reducing cytotoxic mediators. A similar ap proach is being adapted from spinal cord injury research with the use of "pro-inflammatory" therapy :G-CSF alone 119 or in combination with mesenchymal stem cells.120 These therapies may also be useful latel to mitigate CTE.121
Therapies Targeting Traumatic Axonal Injury
Therapies targeting traumatic axonal injury (TAI) is a prominent feature of l:BI and represents a vital target across the spectrum of injury severity. In classic studies, Povlishock122,123 showed that TAI is a fundamental component of secondary injury after TBI and thus a key therapeutic target. Progression of TAI involves TBI-induced dysregulation of Na+ channels, in turn causing increased Ca2+ influx into axons, calpain activation with loss of microtubules, neurofilament impaction with impaired axoplasmic transport,124 and mitochondrial failure with permeability transition pore opening and oxidative stress.125 The aforementioned novel strategies targeting mitochondrial failure may be particularly efficacious in TAI. Smith et al124 recently identified categories of therapies for TAI based on work in TBI models: (1) cytoskeleton stabilization, (2) ion homeostasis, (3) protease inhibition, (4) mitochondrial protection, (5) mild hypothermia, and (6) other therapies. We will review and update those categories (Fig. 3).
Cytoskeleton Stabilization
Loss of microtubule function devastates axonal transport. Unfortunately, data are limited on approaches to block this mechanism. The chemotherapeutic drug Taxol can inhibit chemical depolymerization of microtubules during stretch.126 Taxol has not been tested in TBI in vivo, but recently produced axonal preservation after spinal cord injury in rats.127
Ion Homeostasis
There has also been limited work targeting Ca2+ accumulation linked to TAI. Rather, focus has been on downstream signaling cascades such as calpain activation. Therapies targeting major breaches in membrane disruption such as Kollidon VA64 show promise in TBI models.128
Protease Inhibition
Beneficial effects of calpain inhibition on TAI have been shown in rodent TBI models for over a decade, including use of MDL281 70, AK295, and SJA-601 7.124,129 However, lack of brain bioavailability and target specificity have limited the development of calpain inhibitors. Recent studies have suggested that citicoline suppresses calpain activation after TBI. However, the failed COBRIT clinical trial with citicoline argues against this agent.130 Given the evidence supporting a role for calpain in TAI, it is disappointing that additional calpain inhibitors are not available.
Mitochondrial Protection
Mitochondrial failure may worsen Ca2+ overload and exacerbate TAI. Preclinical work125 has shown benefit from cyclosporine A (CsA) on TAI. Inhibition of MPTP opening is suggested as the mechanism for this effect. However, clinical studies with CsA have been equivocal.3 The aforementioned GS-nitroxides70 or N-acetyl cysteine (NAC) amide131 are logical candidates to study.
Hypothermia
Preclinical work shows that mild hypothermia can attenuate TAI.129 Sadly clinical trials in TBI have failed.132 Surprisingly, hypothermia failed to attenuate the increase in CSF levels of myelin basic protein after severe TBI in children.133 Mild hypothermia was recently shown to markedly attenuate TAI after repetitive mTBI in rats.134 Mild hypothermia deserves exploration in mTBI.
Other
In addition to CsA, the calcineurin inhibitor FK506 has been shown to reduce TAI, particularly in unmyelinated axons.135 Jt is unclear whether this is independent of effects on mitochondria.124 Therapies targeting oxidative damage have also been suggested to reduce TAI134 these were discussed previously.
Therapies Targeting Cerebral Blood Flow Dysregulation and lschemia
After TBI, CBF dysregulation develops and may contribute to secondary damage. In mTBI, vascular dysregulation could mediate vulnerability to a second hit.136 Cerebral blood flow is often reduced early after severe TBI.137 This may result from a coupled reduction in brain activity, but can be pathologic during excitation. Cerebrovascular resistance (CVR) is controlled at the macro- and microvascular level. At the microvascular level, it is coupled to brain metabolic activity. Recently, Hall et al138 showed that pericytes positioned around brain capillaries mediate dilation that produces a much greater change in CBF (19% vs. 3%) than dilation of arteries alone-implicating pericyte-mediated dilation as the major contributor to changes in CBF. Key metabolites identified included the vasodilators nitric oxide (NO) and prostaglandin-E2 (PGE) and the vasoconstrictor 20-hydroxyeicosatertraenoic acid (20-HETE). They implicated the relative production of these metabolites in regulating pericyte dilation and thus CBF. The potent vasoconstrictor endothelin-1 (ET-1) may also contribute to CBF dysregulation after TBI.
Modulating Nitric Oxide
Nitric oxide is a potent vasodilator and may be useful to treat ischemia after TBI. Conversely, NO at high levels can be converted to peroxynitrite, which can worsen damage. This paradox has yielded interventions that augment NO delivery or block NO production after TBI. The primary clinical strategy to increase NO delivery is with inhaled NO. Inhaled NO is FDA-approved for neonatal respiratory failure. Studies suggest that inhaled NO improves collateral circulation and outcomes in TBI models.139 However, in newborn piglets, the NO donor sodium nitroprusside did not prevent impaired autoregulation during hypotension after TBI, implying that NO augmentation may only be beneficial in normotensive states.140 This is important, given that NO augmentation can produce hypotension that may mitigate benefit. Whether inhaled NO is a viable approach remains to be determined. Nitric oxide synthase inhibitors are also being explored in TBI. A phase II placebo controlled trial with the NO synthase inhibitor VAS 203 was just completed in 32 adults with TBI (NOSTRA; NCT 02012582). It suggested improved outcomes, but renal injury was a concern. It is unknown if NO augmentation or inhibition is preferred. A targeted approach to NO delivery to maximize microvascular affects coupled with selective inhibition of inducible NOS (iNOS) to prevent nitrosative stress might be considered. However, sustained inhibition of iNOS may be deleterious given that iNOS KO mice exhibit marked impairments in cognitive outcome.141
Modulating ET-1
In TBI, ET-1 levels in CSF are increased and linked to unfavorable outcomes.142 However, in a phase 2b trial, the ET receptor A antagonist clazosentan failed to improve outcomes after subarachnoid hemorrhage (SAH).143 New ET-1 antagonists remain to be tested in TBI.
Statins
HMG-CoA reductase inhibitors (statins) are used for cholesterol reduction. As discussed, they have anti-inflammatory effects, but also upregulate eNOS and increase NO production, leading to improved capillary patency.144 Clinical trials with statins suggest a potential benefit on outcome in TBI, although the role of effects on CBF is unclear.145 In stroke, an RCT of lovastatin is in phase 2 trials (NeuSTART, NCTOl 976936).146 The optimal choice of statin and dosing for BBB penetration also remain unclear. Results from larger RCTs in TBI are needed to define the utility of these agents in TBI.
Cytochrome P450 Metabolites
Cytochrome P450 produces two classes of arachidonic acid metabolites with opposing microvascular effects. Hydroxylation produces 20-HETE, a potent vasoconstrictor, while epoxidation produces epoxyeicosatrienoic acids (EETs), which are vasodilatory. Both 20-HETE and EETs are autoregulatory mediators. Inhibition of 20-HETE formation by NO is an essential pathway of PGE2-mediated pericyte dilation.138 Inhibition of 20-HETE formation or prevention of EET metabolism can reduce lesion volume in stroke and SAH models.147,148 20-HETE is involved in reduced CBF in cortical spreading depression1 49-a secondary injury mechanism implicated in TBI.150 Thus, cytochrome P450 arachidonic acid metabolites affect CBF after TBI. It remains to be determined whether targeting these pathways can improve outcome after TBI.
Enhancing Oxygen Delivery
Another approach to reduce ischemic damage is to improve oxygen delivery despite CBF reductions-such as with perfluorocarbon-enhanced oxygen delivery. A safety/efficacy RCT is evaluating the pertluorocarbon Oxycyte (STOP-TBI; NCT00908063). Mild hypothermia has also been shown to reduce CBF dysregulation after TBI.151 This includes benefit injury in repetitive mTBI.134
In summary, an emerging area TBI is mitigating microvascular dysregulation. These approaches and others deserve further investigation in TBI, including testing in mTBI.
Cognitive Enhancement
Many rehabilitation strategies have been used to enhance cognitive function after TBI. Some of the most effective preclinical rehabilitative strategies in TBI have been neurostimulant pharmacotherapies, and as discussed below, suc cessful clinical translation of this approach in an RCT was recently acheived.2 Thus, we will focus on these agents as emerging TBI therapies.
Catecholamine Agonists
Catecholamine agonists promote functional recovery after TBI. This was confirmed in weight drop cortical contusion or ablation models in rats and/or cats.151,154 Because norepinephrine antagonists block or reinstate deficits, the noradrenergic system has been implicated. However, clinical155,156 and experimental157,158 research shows that the dopamine (DA) system is also involved in both injury and rehabilitative processes. Methylphenidate, a psychostimulant and DA transporter inhibitor, exhibits pharmacological properties similar to amphetamine, but without undesirable sympathomimetic effects. In a study assessing motor function after sensorimotor cortex injury in rats, a single dose of methylphenidate followed by symptom relevant experience (beam walking) enhanced beam-walk ability.154 This work supports the importance of an interaction between pharmacotherapy and symptom-relevant experience (rehabilitation) in promoting functional recovery after TBI. Moreover, daily treatment with methylphenidate beginning as late as 24 hours after CCI in rats revealed less spatial memory deficits versus controls.159 Wagner et al160 has showed that methylphenidate exhibits some restorative capacity for striatal DA neurotransmission after experimental TBI; their additional work suggests potential sex differences in methylphenidate treatment effects and dosing for male versus female rats.161
Amantadine-A Translational Success
With success in preclinical studies as well as phase 2 and phase 3 clinical trials, amantadine (Symmetrel; Endo Phar maceuticals, Malvern, PA) is a promising drug for TBI rehabilitation.162 Daily treatment with amantadine (for 20 d) after CCI in rats revealed improvements in spatial memory perfor mance deficits versus saline-treated counterparts.163 Higher doses also benefit cognition after fluid percussion.164 Clinically, amantadine accelerates the rate of functional recovery in vegetative or minimally conscious patients during the subacute phase after TBI.2 It exerts its effects by increasing extracellular DA by blocking reuptake and by facilitating synthesis.165,166 In addition to its presynaptic actions, amantadine increases the density of postsynaptic DA receptors 166 or alters their conformation, which may be clinically important.167 Because the mechanism of action of amantadine differs from other DA-releasing drugs,168 it is likely that the dopaminergic effects of amantadine are a combination of presynaptic and postsynaptic effects. It also blunts NMDAR activation.
Bromocriptine
Delayed or chronic treatment with the D2 receptor agonist bromocriptine also improves the acquisition of spatial and working memory in rats after CCI.169 Bromocriptine (5 mg/kg) has been shown to increase extracellular DA levels in rats,170 suggesting that enhanced presynaptic DA neurotransmission may have mediated the benefit at this dose. Bromocriptine also attenuated lipid peroxidation, suggesting antioxidant effects.169 Further support for dopaminergic activity in restoring functional recovery after TBI comes from a preclinical study showing that selegiline (L-deprenyl), which enhances the action of DA by inhibiting its main catabolic enzyme in brain, monoamine oxidase-B, improved cognitive outcome when given daily for 7 days after fluid percussion injury.171 Clinical studies showing benefits of DA augmentation after TBI also exist.172,173
Thus enhancing catecholamine neurotransmission during the chronic postinjury phase may be a useful adjunct in ameliorating the neurobehavioral sequelae of TBI in humans. Additional studies are warranted.
Augmenting Endogenous Neuroprotectants
Evolution has provided several naturally occurring neuroprotective mechanisms. Perhaps the "low-hanging fruit" for increasing therapeutic options for TBI patients resides in augmenting the mechanisms that allowed the brain to evolve in the first place.
Adenosine as an Archetype for Endogenous Neuroprotection
Adenosine is released by tissue injury via several pathways. Breakdown of ATP is one source of adenosine in the injured brain (the traditional route). Recently, an alternative 2',3' cAMP pathway was discovered, and involves production of adenosine from mRNA breakdown via 2',3'cAMP. This latter pathway appears to play a major role after TBI.174 Adenosine acts on cell surface receptors (A1, A2A, A2B, and A3) and engages signal transduction that is neuroprotective in TBI.175 Activation of Al receptors attenuates post-TBI excitotoxicity. Mice null for A1 receptors suffer lethal status epilepticus after TBJ,176 and variants in the Al receptor genes associate with posttraumatic seizures in TBI patients.177 Moreover, A1 receptor KO mice exhibit enhanced microglial proliferation after TBI.178 However, systemic effects of A1 agonists (bradycardia, hypotension) limit their use in TBI. A more effective strategy might be to enhance adenosine levels in the brain or increase A1 receptor numbers and/or signaling. One approach would be to upregulate enzymes that produce adenosine. For example, isoflurane increases activity of the adenosine-form ing enzyme ecto-5'-nucleotidase (CD73) by stimulating release of microparticles,179 which may contribute to its neuroprotection.180 Another approach would be to administer chronically and prophylactically an A1 receptor blocker with a short half-life. This would upregulate A1 receptor numbers and signaling in the brain, yet post-TBI the antagonist would dissipate rapidly, leaving enhanced adenosine signaling at the time of greatest need. Improved outcomes are seen in TBI patients with caffeine (a short half-life adenosine receptor antagonist) in their CSF at the time of injury.181 Finally, adenosine kinase (ADK) is a key enzyme in the breakdown of adenosine. Adenosine kinase increases markedly in the astrocyte scar and limits adenosine availability chronically after TBI.182 Given the anticonvulsant effects of A1 receptors, blocking ADI< or using other strategies to overexpress ad enosine may represent a therapy for posttraumatic seizures. Huber et al183 used grafts of adenosine-releasing fibroblasts to suppress seizures in rats. A1 receptor gene polymorphisms are strongly associated with posttraumatic seizures.177 Thus, it may be possible to use a personalized medicine approach to define patients who might benefit from adenosine augmentation therapy.
Other Endogenous Neuroprotectants
There are many other endogenous neuroprotectants emerging as therapies for TBI. Augmentation of trophic factors such as BDNF, or immediate early gene products such as HSP, could improve outcome after TBJ.1 84 It was recently shown that remote preconditioning using tourniquet inflation/deflation may mediate benefit in ischemia/reperfusion via local elaboration of nitrite-which is converted to NO in regions of tissue hypoxia.185 Systemic nitrite therapy might yield similar effects.186 An endogenous neuroprotectant receiving attention in Parkinson disease is uric acid, which has antioxidant effects.187 Finally, regulators of cold stress such as RNA-binding motif 3 (RBM3) stabilize mRNAs and may underlie benefit in hypothermia.188 New drugs are targeting RBMs.189
Cellular Therapies
Therapies designed to replenish cells Jost after TBI may help improve outcome. Methodologies include supplementation of exogenous stem cells or therapies that enhance endogenous neurogenesis in the adult brain. Both approaches have promise based on studies in TBI models. New approaches are being used to enhance the regenerative capacities of these cellular therapies in hopes of developing interventions that can ultimately promote recovery in patients.
Administration of Exogenous Cells
Traumatic brain injury can be associated with loss of neurons and other cells in multiple brain regions. Administration of exogenous bone marrow stromal cells in rats, via intracranial,190 intra-arterial,191 or intravenous delivery,192 results in a portion of transplanted cells migrating into the brain parenchyma, and is associated with improved motor function after TBI.190,193 Survival of stromal cells is associated with increased production of BDNF and nerve growth factor.192 In other studies, transplantation of neural stem cells194,195 and the coadministration of neural stem cells and olfactory ensheathing cells similarly improve motor performance after TBI.196 However, it is unclear whether the regenerative capacity of transplantation is dependent upon the incorporation of the exogenous cells, the production of soluble growth permissive factors, or their combination. Two studies provide evidence for complex dynamics between the contributions of exogenous cell survival and the release of soluble factors that promote regeneration after TBI. Intracranial administration of human bone marrow stromal cells in a collagen scaffold matrix enhances the survival of the stromal cells in the cortex and improves motor function compared with administration of stromal cells alone.52 Coadministration of the collagen scaffold also enhanced corticospinal tract sprouting in the denervated spinal cord,197 suggesting contributions from both the surviving stromal cells and soluble factors that promote regeneration after TBI. Tajiri et al198 studied soluble factors released from transplanted human adipose-derived stem cells that may play a role in recovery after TBI. Delivery of either the adipose-derived stem cells or conditioned media improved outcome in rats after TBI. However, knockdown of two long noncoding RNAs, important for cellular differentiation, blunted the recovery. These data indicate that regeneration can be enhanced with soluble factors in the absence of transplanted cells. They reveal the potential of transplantation approaches and highlight the complex dynamics between stem cells and growth permissive factors in promoting recovery.
Enhancing the Generation and Survival of Newborn Cells
Therapies that enhance the generation of newborn neurons are also attractive after TBI. Enhancement of neurogenesis after TBI with growth factors, neuroprotective agents, and hypothermia promotes cellular proliferation and increases the generation of newborn neurons in neurogenic regions of the injured brain.199-202 These studies have also shown that an enhancement of posttraumatic neurogenesis in the weeks after TBI is associated with improved neurobehavioral performance. Hippocampal immature neurons are particularly sensitive to brain injury in the days postinjury.202 Promoting survival of neural stem cells and immature neurons is a promising target to improve outcome.203 Blaya et al204 evaluated the efficacy of the neuroprotective agent P730-A20 to promote the survival of immature neurons, as this drug blocks apoptosis in immature neurons. Treatment with P730-A20 improved immature neuron density, increased the number of newly generated neurons, and improved cognitive performance. This highlights the promise of therapies promoting newborn neuron survival and incorporation into the injured brain.
Clinical Trials of Cellular Therapy
There are several clinical trials exploring cellular therapy in TBI. Recently a study addressing "Safety of Autologous Stem Cell Treatment for TBI in Children" (NCT00254722) was completed. The objective of that phase 1 study was to determine if bone marrow precursor cell harvest and autologous transplantation (within 36 h of injury and by intravenous route) is safe in children after TBI. The study was completed; a phase II trial is recruiting (NCT01851083). There is also an open label study of "Autologous Bone Marrow Mononuclear Cells in TBI" (NCT0202810), in which bone marrow-derived mononuclear cells are given intrathecally. That study is also recruiting.
Combination Therapy
Combination therapy is attractive to overcome translational challenges. Incomplete understanding of dose-response relationships and poor central nervous system (CNS) penetration of therapies are factors widely acknowledged to contribute to failed clinical trials.205-207 The heterogeneity of TBI and its complex pathophysiology suggest that it is unlikely that any single agent can address all of the secondary injury mechanisms.205 The success of combination therapies that enhance drug exposure or have complementary mechanisms of action in cancer and human immunodeficiency virus has further increased enthusiasm for this approach in TBI research.
Emerging Combination Therapies
In 2008, the NIH convened a workshop on multidrug combinations for TBI. The recommendation was to combine therapies with complementary targets and effects rather than focus on a single target with multiple therapies.207 Several combination therapies meeting this definition are being investigated. One of the most promising combines the antiinflammatory agent minocycline and the glutathione precursor NAC. Effects of minocycline were discussed previously. N-acetyl cysteine is a precursor for synthesis of the antioxidant glutathione, impacts glutamatergic transmission, and despite poor CNS penetration improved outcomes in some TBI models and in blast-induced mTBI in humans.208-210 When given together, benefits of the combination exceed that of the single agents studied in CCI and mTBI models.211,212 N-acetyl cysteine is used in another combination therapy designed to improve drug exposure. Our group is testing coadministration of the FDA-approved organic acid transporter and multidrug resistance-associated protein inhibitor, probenecid, with NAC in preclinical and phase I pediatric studies (NCT01322009). The aim is to overcome membrane barriers, such as the BBB, to synergistically improve NAC bioavailability and antioxidant reserves after TBI. Preclinical pharmacokinetic (PK) data show that probenecid increases NAC brain penetration in juvenile rats two- to threefold as early as 1 hour after injury.213 Outcome studies are underway. Other combinations such as progesterone plus vitamin D are in early stages of preclinical investigation.214
Addressing Unique Challenges of Combination Therapy
Interactions between therapies may alter PK (dose-concentra tion relationships) or pharmacodynamic (concentration-effect relationships) properties of either therapy. They may be additive. synergistic, or antagonistic. Thus, studies using full-factorial designs at multiple dosing levels are ideal.207 These data are used to identify combinations and sequences of therapies that achieve greater efficacy and lower toxicity than either therapy alone. Specific statistical approaches to identify synergism are advocated.215 Coadministered therapies should also be evaluated for physiochemical incompatibilities to ensure systemic bioavailability. As with single drugs, it is imperative to measure brain concentrations of therapies used in combination to optimize their potential for success.
Therapies Targeting TBI Resuscitation in Polytrauma
Traumatic brain injury is often accompanied by secondary insults (hypotension, hemorrhage, hypoxemia) that worsen outcome.216 However, given their complexity, clinical studies commonly exclude these patients, and few animal models have been developed to investigate therapies. Optimal resuscitation of the TBI patient with polytrauma continues to present unique challenges and remains understudied.
Resuscitation Fluids
The mainstay of resuscitation involves fl uids: crystalloids, colloids, and/or blood products. Crystalloids are the initial therapy; however, large volumes are often needed. which can exace rbate brain edema.217 Colloids, given their improved ability at maintaining intravascular volumes, are attractive; however, in the SAFE study, TBI patients resuscitated with albumin had raised ICPs and greater mortality compared with saline-treated patients.218 Traumatic brain injury-induced BBB permeability may have allowed extravasation of albumin into brain-potentiating rebound brain edema.219 Thus, small molecule colloids may be problematic early after TBI. Blood products are not available for prehospital use.
Emerging Resuscitation Agents
Given the risk of exacerbation of brain edema with current resuscitation fluids, new therapies are being investigated, including novel. ultra-small-volume resuscitation agents. Polynitroxylated pegylated hemoglobin (PNP H) is one agent that may represent an out-of hospital bridge to transfusion. Polynitroxylated pegylated hemoglobin is a bovine-based hemoglobin that, in an effort to eliminate toxicity of cell-free hemoglobin, is covalently bonded with antioxidant nitroxide moieties and polyethylene glycol side chains. Polynitroxylated pegylated hemoglobin is being developed as a small-volume resuscitation solution. In a model of TBI plus hemorrhage, it dramatically reduced resuscitation fl uid requirements compared with crystalloid.220,221 It also reduced ICP, brain edema, and neuronal death.220,221 Unlike conventional free hemoglobins, it has surprising in vitro neuroprotective effects.221 It is in preclinical development. Traditional resuscitation approaches focus on improving tissue perfusion by increasing circulating blood volume. An alternative might entail modification of microcirculatory blood flow. Drag-reducing polymers (DRPs) at nM levels markedly reduce the resistance of microvascular fl ow, improving tissue perfusion.222,223 Drag-reducing polymers, such as long-chain polyethylene glycol (kDa > 106), improve perfusion and reduce mortality in models of hemorrhagic shock.224 Drag-reducing polymers thus could maintain brain perfusion despite using a volume-limited resuscitation.
Conventional Resuscitation Plus Antiedema Therapies
Although volume-limiting resuscitation strategies hold promise in TBI, treatment with the aforementioned novel drugs targeting brain edema (Kollidon VA64, glibenclamide, AQP4 antagonists) could reduce the deleterious effects of resuscitation fluids on edema after TBI. Preclinical studies of these drugs are needed in models of TBI plus hemorrhage/polytrauma.
Other Therapies
Many more emerging therapies are on the horizon. including the use of transcranial low level laser.225 neutraceuticals.226,227 lithium.228 modulating cell cycle229 and targeting microhemorrhage,230 among many others. This brief list illustrates the level of creativity in our dynamic research field.
Conclusion
This review is far from comprehensive. However, given our goal to address mechanism-based emerging therapies across the injury-severity spectrum, and from the field to rehabili tation,we chose to provide a “survey” across key mechanisms. There are many other promising agents worthy of investiga tion. We also believe that the recent surge in interest in mTBI may identify new targets in severe TBI particularly given the fact that in patients with severe TBI. brain regions outside of areas of major disruptions are likely to be plagued by the pathomechanisms seen in mTBI. Thus. new investigations into therapies for mTBI may provide new opportunities for treatment of severe TBI. For example. emerging mechanisms in mTBI. such as disturbances in the balance between excitatory and inhibitory pathways or disturbances in synchronization, 42,231 could be important in severe TBI-layered upon the classical injury paradigms (…Fig. 4). Finally. to complement rehabilitation and cognitive enhancing therapies currently used chronically afterTBI. new approaches to break the link between TBI and chronic neurodegenerative diseases including crE are needed (…Fig. 4). The current golden age of TBI research thus represents a special opportunity for the development of breakthroughs in the field.
Fig. 4.

A contemporary view of therapeutic targets across the spectrum of traumatic brain injury (TBI) from mid to severe includes classical secondary injury pathways along with novel mechanisms such asexcitation inhibition imbalance and functionalasynchronythat have come to the forefront in mild TBI. Therapies to embellish rehabilitation based str.i:egies to breakthe link between TBI and chronic neurodegenerative diseases such as chro11ic traumatic encephalopathy (CTE) are also needed. Ttlerapies are identified by 0. ALS. amyotrol)hic lateral sclerosis.
Fig. 1.

Strategies to modulate traumatic brain injury (T81-) induced excitotoxicity while maintaining beneficial glutamatergic activation. Glutamate is an essential neurotraosmitter to brain function. Normal release of glutamate from presynaptic oeuroos activates synaptic NMDA Rs (NR2A enriched) on postsynaptic neurons. Synaptic NMDAR activation promotes nuclear CA2+ influx. Synaptic to nuclear CA2+ communication transmits prosurvival signals (CaMKIV and CREB). The transcription factor CREB induces neuroprotective BNDf. Also. synapllc NMDAR activity stlmulales expression of other protective AIDs. Traumatic brain injury alters CA2+ biochemistry to favor toxic cylop lasmk signal. Extra cellular glutamate activates distal exttasynapllc NMDARs (NR2B enriched). Extr.asynaptic NMDARs promote cell death (top left). They oppose synaplk NMDAR/CR EB prosurviva Iresponses and actlvale ca Ipaios and DAPK, aod inhibit AKTsurvival signalin. Drugs .approved bythe U.S. food and Drug Administration targeting excitotoxicity include (ideotitied by \l=O/\): (1) Memaotine-blocks extrasyn.apticfllMDAR, (2) celtriaxooeand raloxifene increases expression of 9lulama1e uplake transporters io aslrocytes. aod (3) levetiracetam-may inhibit high presynaptic glutamate release by modulating inhibitoryGABAe<gic input to excitatory neurons. ALS, amyotrophic lateral sclerosis; CBF. cerebral blood flow; CTE. chronic trauma Ile encephalopathy; NMDAR. N-methyl.o.aspamte receptor: Glufll2A/NR2A. glutamate N2A subunit: GluN2B{NR2B. glutamate N2:8 subunil; CREB, cAMP response element binding protein; GLT 1, glial glutamate transporter 1: GLAST. glutamale/aspirate transporter; AKT. protein kinase DAPK. death associated protein kinase: BDNF. brain-derived neurothic factor; AID. activity.regulated inhibitor of death: CaMK. CA2+ calmodulin dependent protein kinase.
Fig. 3.

Emerging therapies to limit traumatic axonal injury (TAI) after traumatic brain injury (TBl).124 Cellular and molecular events in the TAI cascade include Ca2+ accumulation with calpain activation and resultant microtubule proteolysis, MPTP opening, and oxidative stress. Direct membrane poration can also mediate injury. Emerging therapies (identified by 0) include calpain antagonists (MDL28170, AK295, SJA 6017), and taxol, which may prevents microtubule polymerization. Therapies targeting mitochondria include CsA that blocks MPTP, FK506 that may target calcineurin induced translocation of BAD, and XJB 5 131 and NACA that target oxidative stress in mitochondria. Kolloidan VA64 may directly reseal membranes. Mild hypothermia may reduce TAI by multiple mechanisms. CsA, Cyclosporin A; NACA, N acetyl cysteine amide.
Acknowledgments
Supported by U.S. Army grants WS1XWH-10–1-0623 (PMK) and W81XWH-14-2-0018 (PMK). NIH grants NS088145 (TJ). NS087978 (PMK, EKJ), and T32HD040686 (NF, EB, SC); and KL2-TR000146 from the NCATS as part of the Multidisciplinary Clinical Research Scholars Program (PE).
References
- 1.Robertson CS, Hannay HJ, Yamal JM, et al. Epo Severe TBI Trial Investigators. Efrect or erythropoietin and transfusion threshold on neurological recovery aftertraumaticbrain injury: a randomized clinical trial. JAMA. 2014;312(1):36–47. doi: 10.1001/jama.2014.6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giacino JT, Whyte J, Bagiella E, et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med. 2012;366(9):819–826. doi: 10.1056/NEJMoa1102609. [DOI] [PubMed] [Google Scholar]
- 3.Diaz-Arrastia R, Kochanek PM, Bergold P, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report or the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31(2):135–158. doi: 10.1089/neu.2013.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164(3880):719–721. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 5.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1(6):383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 6.Randall RD, Thayer SA. Glutamate induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12(5):1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7(2):369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chamoun R, Suki D, Gopinath SP, Goodman JC, Robertson C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J Neurosurg. 2010;113(3):564–570. doi: 10.3171/2009.12.JNS09689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber JT, Rzigalinski BA, Ellis EF. Traumatic injury of cortical neurons causes changes in in tracellular calcium stores and capacitative calcium influx. J Biol Chem. 2001;276(3):1800–1807. doi: 10.1074/jbc.M009209200. [DOI] [PubMed] [Google Scholar]
- 10.Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, Mcintosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55(7):850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Sun DA, Deshpande LS, Sombati S, et al. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intra-cellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008;27(7):1659–1672. doi: 10.1111/j.1460-9568.2008.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saatman KE, Murai H, Bartus RT, et al. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A. 1996;93(8):3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumgartner HK, Gerasimenko JV, Thorne C, et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284(31):20796–20803. doi: 10.1074/jbc.M109.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoblach SM, Nikolaeva M, Huang X, et al. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19(10):1155–1170. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- 15.Conti AC, Raghupathi R, Trojanowski JQ, Mcintosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post traumatic period. J Neurosci. 1998;18(15):5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark RS, Kochanek PM, Watkins SC, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74(2):740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- 17.Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24(10):1119–1132. doi: 10.1097/01.WCB.0000138664.17682.32. [DOI] [PubMed] [Google Scholar]
- 18.Chen W, Zhou Z, Li L, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) in teraction in necroptotic signaling. J Biol Chem. 2013;288(23):16247–16261. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaiser WJ, Upton JW, Long AB, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471(7338):368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Qiu J, Liang M, et al. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 2014;5:e1084. doi: 10.1038/cddis.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You Z, Savitz SI, Yang J, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28(9):1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Yang X, Ma C, Qiao J, Zhang C. Necroptosis contributes to the NMDA induced excitotoxicity in rat's cultured cortical neurons. Neurosci Lett. 2008;447(23):120–123. doi: 10.1016/j.neulet.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 23.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4(1):88–90. doi: 10.4161/auto.5173. [DOI] [PubMed] [Google Scholar]
- 25.Luo CL, Li BX, Li QQ, et al. Autophagy is involved in traumatic brain injury induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Zhang YB, Li SX, Chen XP, et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull. 2008;24(3):143–149. doi: 10.1007/s12264-008-1108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210(1-2):3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Vespa PM, Miller C, McArthur D, et al. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit Care Med. 2007;35(12):2830–2836. [PMC free article] [PubMed] [Google Scholar]
- 29.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19(10):4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufman AM, Milnerwood AJ, Sepers MD, et al. Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J Neurosci. 2012;32(12):3992–4003. doi: 10.1523/JNEUROSCI.4129-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadia S, Soriano FX, Leveille F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11(4):476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NM DARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5(5):405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 33.Karpova A, Mikhaylova M, Bera S, et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell. 2013;152(5):1119–1133. doi: 10.1016/j.cell.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Bigford GE, Alonso OF, Dietrich D, Keane RW. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma. 2009;26(5):703–720. doi: 10.1089/neu.2008.0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tu W, Xu X, Peng L, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrario CR, Ndukwe BO, Ren J, Satin LS, Goforth PB. Stretch injury selectively enhances extrasynaptic, GluN2B-containing NMDA receptor function in cortical neurons. J Neurophysiol. 2013;110(1):131–140. doi: 10.1152/jn.01011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao VLR, Dogan A, Todd KG, Bowen KK, Dempsey RJ. Neuro-protection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Res. 2001;911:96–100. doi: 10.1016/s0006-8993(01)02617-8. [DOI] [PubMed] [Google Scholar]
- 38.Xia P, Chen HSV, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autopsies. J Neurosci. 2010;30(33):11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rammes G, Danysz W, Parsons CG. Pharmacodynamics of memantine: an update. Curr Neuropharmacol. 2008;6(1):55–78. doi: 10.2174/157015908783769671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies DJ, Crowe M, Lucas N, et al. A novel series of benzimidazole NR2B-selective NMDA receptor antagonists. Bioorg Med Chem Lett. 2012;22(7):2620–2623. doi: 10.1016/j.bmcl.2012.01.108. [DOI] [PubMed] [Google Scholar]
- 41.Niogi SN, Mukherjee P, Ghajar J, et al. Structural dissociation of attentional control and memory in adults with and without mild traumatic brain injury. Brain. 2008;131(Pt 12):3209–3221. doi: 10.1093/brain/awn247. [DOI] [PubMed] [Google Scholar]
- 42.Bashir S, Vernet M, Yoo WK, Mizrahi I, Theoret H, Pascual-Leone A. Changes in cortical plasticity after mild traumatic brain injury. Restor Neurol Neurosci. 2012;30(4):277–282. doi: 10.3233/RNN-2012-110207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schumann J, Alexandrovich GA, Biegon A, Vaka R. Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma. 2008;25(8):945–957. doi: 10.1089/neu.2008.0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A, Zou L, Yuan X, Long Y, Yang K. N-methyl-D-aspartate receptors: transient loss of NR1/NR2A/NR2B subunits after traumatic brain injury in a rodent model. J Neurosci Res. 2002;67(6):781–786. doi: 10.1002/jnr.10181. [DOI] [PubMed] [Google Scholar]
- 45.Szaflarski JP, Meckler JM, Szaflarski M, Shutter LA, Privitera MD, Yates SL. Levetiracetam use in critically ill patients. Neurocrit Care. 2007;7(2):140–147. doi: 10.1007/s12028-007-0042-8. [DOI] [PubMed] [Google Scholar]
- 46.Lee CY, Chen CC, Liou HH. Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus. Br J Pharmacol. 2009;158(7):1753–1762. doi: 10.1111/j.1476-5381.2009.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakita M, Kotani N, Kogure K, Akaike N. Inhibition of excitatory synaptic transmission in hippocampal neurons by levetiracetam involves zn2+-depen denl GABA type A receptor-mediated pre-synaptic modulation. J Pharmacol Exp Ther. 2014;348(2):246–259. doi: 10.1124/jpet.113.208751. [DOI] [PubMed] [Google Scholar]
- 48.Benge JF, Phenis RA, Bernett A, Cruz-Laureano D, Kirmani BF. Neurobehavioral effects of levetiracetam in patients with traumatic brain injury. Front Neurol. 2013;4:195. doi: 10.3389/fneur.2013.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou H, Brayer SW, Hurwitz M, Niyonkuru C, Fowler LE, Wagner AK. Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil Neural Repair. 2013;27(9):878–888. doi: 10.1177/1545968313491007. [DOI] [PubMed] [Google Scholar]
- 50.Brauon SL, Chestnut RM, Ghajar J, et al. Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons; Joint Section on Neurotrauma and Critical Care, AANS/CNS Guidelines for the management of severe traumatic brain injury. II. Hyperosmolar therapy. J Neurotrauma. 2007;24(Suppl 1):S14–S20. doi: 10.1089/neu.2007.9994. [DOI] [PubMed] [Google Scholar]
- 51.Kochanek PM, Carney N, Adelson PD, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents – second edition. Pediatr Crit Care Med. 2012;13(Suppl 1):S1–82. doi: 10.1097/PCC.0b013e31823f435c. [DOI] [PubMed] [Google Scholar]
- 52.Gonda DD, Meltzer HS, Crawford JR, et al. Complications associated with prolonged hypertonic saline therapy in children with elevated intracranial pressure. Pediatr Crit Care Med. 2013;14(6):610–620. doi: 10.1097/PCC.0b013e318291772b. [DOI] [PubMed] [Google Scholar]
- 53.Cooper DJ, Rosenfeld JV, Murray L, et al. DECRA Trial Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364(16):1493–1502. doi: 10.1056/NEJMoa1102077. [DOI] [PubMed] [Google Scholar]
- 54.Shafi S, Diaz-Arrastia R, Madden C, Gentilello L. Intracranial pressure monitoring in brain-injured patients is associated with worsening of survival. J Trauma. 2008;64(2):335–340. doi: 10.1097/TA.0b013e31815dd017. [DOI] [PubMed] [Google Scholar]
- 55.Chesnut RM, Temkin N, Carney N, et al. Global Neurotrauma Research Group A trial of intracranial pressure monitoring in traumatic brain injury. N Engl J Med. 2012;367(26):2471–2481. doi: 10.1056/NEJMoa1207363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein DM, Hu PF, Brenner M, et al. Brief episodes of intracranial hypertension and cerebral hypoperfusion are associated with poor functional outcome after severe traumatic brain injury. J Trauma. 2011;71(2):364–373. doi: 10.1097/TA.0b013e31822820da. [DOI] [PubMed] [Google Scholar]
- 57.Lafrenaye AD, McCinn MJ, Povlishock JT. Increased intracranial pressure after diffuse traumatic brain injury exacerbates neuronal somatic membrane poraiion but not axonal injury: evidence for primary intracranial pressure-induced neuronal perturbation. J Cereb Blood Flow Metab. 2012;32(10):1919–1932. doi: 10.1038/jcbfm.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laird MD, Shields JS, Sukumari-Ramesh S, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62(1):26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22(4):367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Bell MJ, Kochanek PM, Doughty LA, et al. Interleukin-6 and interleukin-10in cerebrospinal fluid after severe traumatic brain injury in children. J Neurotrauma. 1997;14(7):451–457. doi: 10.1089/neu.1997.14.451. [DOI] [PubMed] [Google Scholar]
- 61.Au AK, Aneja RK, Bell MJ, et al. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J Neurotrauma. 2012;29(11):2013–2021. doi: 10.1089/neu.2011.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okuma Y, Liu K, Wake H, et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMG61-RAGE interaction. Neuropharmacology. 2014;85:18–26. doi: 10.1016/j.neuropharm.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 63.Simard JM, Woo SK, Schwartibauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab. 2012;32(9):1699–1717. doi: 10.1038/jcbfm.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okuma Y, Liu K, Wake H, et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol. 2012;72(3):373–384. doi: 10.1002/ana.23602. [DOI] [PubMed] [Google Scholar]
- 65.Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33(10):1621–1632. doi: 10.1038/jcbfm.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zweckberger K, Hackenberg K, Jung CS, et al. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience. 2014;272:199–206. doi: 10.1016/j.neuroscience.2014.04.040. [DOI] [PubMed] [Google Scholar]
- 67.Kagan VE, Wipf P, Stoyanovsky D, et al. Mitochondrial targeting of electron scavenging antioxidants: regulation of selective oxidation vs random chain reactions. Adv Drug Deliv Rev. 2009;61(14):1375–1385. doi: 10.1016/j.addr.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tyurina YY, Poloyac SM, Tyurin VA, et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014;6(6):542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ji J, Kline AE, Amoscato A, et al. Lipidomics identities cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15(10):1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okonkwo DO, Povlishock JT. An intrathecal bolus or cyclosporin A before injury preserves mitochondrial integrity and attenuaies axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19(4):443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 72.Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma. 1997;14(1):23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- 73.Robertson CL. Mitochondrial dysfunction contributes to cell death following traumatic brain injury in adult and immature animals. J Bioenerg Biomembr. 2004;36(4):363–368. doi: 10.1023/B:JOBB.0000041769.06954.e4. [DOI] [PubMed] [Google Scholar]
- 74.Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications fer neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26(11):1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- 75.Abdul-Muneer PM, Schuetz H, Wang F, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumaticbr ain injury induced by primary blast. Free Radie Biol Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sies H. Oxidative Stress. Academic Press; London, England: 1985. Oxidative stress, introductory remarks. [Google Scholar]
- 77.Thiels E, Urban NN, Gonzalez-Burgos GR, et al. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2000;20(20):7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9-10):1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 79.Opii WO, Nukala VN, Sultana R, et al. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J Neurotrauma. 2007;24(5):772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 80.Mendez DR, Cherian L, Moore N, Arora T, Liu PK, Robertson CS. Oxidative DNA lesions in a rodent model of traumatic brain injury. J Trauma. 2004;56(6):1235–1240. doi: 10.1097/01.ta.0000130759.62286.0e. [DOI] [PubMed] [Google Scholar]
- 81.Bayir H, Tyurin VA, Tyurina YY, et al. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62(2):154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 82.Cristofori L, Tavazzi B, Cambin R, et al. Early onset of lipid peroxidation after human traumatic brain injury: a fatal limitation for the free radical scavenger pharmacological therapy? J Investig Med. 2001;49(5):450–458. doi: 10.2310/6650.2001.33790. [DOI] [PubMed] [Google Scholar]
- 83.Bayir H, Kagan VE, Tyurina YY, et al. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51(5):571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 84.Hamm RJ, Temple MD, Pike BR, Ellis EF. The effect of postinjury administration of polyethylene glycol-conjugated superoxide dismutase (pegorgotein, Dismutec) or lidocaine on behavioral function following fluid percussion brain injury in rats. J Neuro trauma. 1996;13(6):325–332. doi: 10.1089/neu.1996.13.325. [DOI] [PubMed] [Google Scholar]
- 85.Hall ED, Andrus PK, Smith SL, et al. Pyrrolopyrimidines: novel brain-penetrating antioxidants with neuroprotective activity in brain injury and ischemia models. J Pharmacol Exp Ther. 1997;281(2):895–904. [PubMed] [Google Scholar]
- 86.Wang GH, Jiang ZL, Li YC, et al. Free-radical scavenger edaravone treatment confers neuroprotection against traumatic brain injury in rats. J Neurotrauma. 2011;28(10):2123–2134. doi: 10.1089/neu.2011.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marshall LF, Maas AI, Marshall SB, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89(4):519–525. doi: 10.3171/jns.1998.89.4.0519. [DOI] [PubMed] [Google Scholar]
- 88.Muizelaar JP, Kupiec JW, Rapp LA. 1995;83(5):942. doi: 10.3171/jns.1995.83.5.0942. [DOI] [PubMed] [Google Scholar]
- 89.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bayir H, Kochanek PM, Kagan VE. 2006;28(4-5):420–431. doi: 10.1159/000094168. Review. [DOI] [PubMed] [Google Scholar]
- 91.Hall ED, Wang JA, Miller DM. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp Neurol. 2012;238(2):176–182. doi: 10.1016/j.expneurol.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang F, Franco R, Skotak M, Hu G, Chandra N. Mechanical stretch exacerbates the cell death in SH-SY5Y cells exposed to paraquat: mitochondrial dysfunction and oxidative stress. Neurotoxicology. 2014;41:54–63. doi: 10.1016/j.neuro.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu A, Ying Z, Gomez-Pinilla F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J Neurotrauma. 2007;24(10):1587–1595. doi: 10.1089/neu.2007.0313. [DOI] [PubMed] [Google Scholar]
- 94.Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mito chondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79(12):231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- 95.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Arnst) 2006;5(2):145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 96.Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G. Neuro-protection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev Neurosci. 2010;32(5-6):480–487. doi: 10.1159/000323178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16(9):783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- 98.Mazzeo AT, Brophy GM, Gilman CB, et al. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26(12):2195–2206. doi: 10.1089/neu.2009.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith RA, Hartley RC, Murphy MP. Mitochondria targeted small molecule therapeutics and probes. Antioxid Redox Signal. 2011;15(12):3021–3038. doi: 10.1089/ars.2011.3969. [DOI] [PubMed] [Google Scholar]
- 100.Smith RA, Murphy MP. Animal and human studies with the mitochondria targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 101.Cho J, Won K, Wu D, et al. Potent mitochondria targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18(3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 102.Wipf P, Xiao J, Jiang J, et al. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc. 2005;127(36):12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 103.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 104.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14(4):615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 105.Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, Mcintosh TK. Experimental brain injury ind uces expression of interleukin 1 beta mRNA in the rat brain. Brain Res Mol Brain Res. 1995;30(1):125–130. doi: 10.1016/0169-328x(94)00287-o. [DOI] [PubMed] [Google Scholar]
- 106.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scherbel U, Raghupathi R, Nakamura M, et al. Differential acute and chronic responses of tumor necrosis factor deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96(15):8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chio CC, Chang CH, Wang CC, et al. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013;14:33. doi: 10.1186/1471-2202-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baratz R, Tweedie D, Rubovitch V, et al. Tumor necrosis factor-α synthesis inhibitor. 3,6'-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J Neurochem. 2011;118(6):1032–1042. doi: 10.1111/j.1471-4159.2011.07377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Su X, Wang H, Zhao J, Pan H, Mao L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expres sion and TLR4/NF-kenefB pathway after traumatic brain injury in the rat. Mediators Inflamm. 2011;2011:807–142. doi: 10.1155/2011/807142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang D, Li H, Li T, et al. TLR4 inhibitor resatorvid provides neuroprotection in experimental traumatic brain injury: implication in the treatment of human brain injury. Neurochem Int. 2014;75:11–18. doi: 10.1016/j.neuint.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 112.Sanchez-Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48(6):1393–1399. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 113.Clausen F, Hα̈nell A, Björk M, et al. Neutralization of interleukin-1 beta modifies the infla mm atory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30(3):385–396. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- 114.Tehranian R, Andell-Jonsson S, Beni SM, et al. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19(8):939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- 115.Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab. 2014;34(5):845–851. doi: 10.1038/jcbfm.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758) J Neurotrauma. 2008;25(8):1011–1017. doi: 10.1089/neu.2008.0554. [DOI] [PubMed] [Google Scholar]
- 117.Schneider EB, Efron DT, MacKenzie EJ, Rivara FP, Nathens AB, Jurkovich GJ. Premorbid statin use is associated with improved survival and functional outcomes in older head injured individuals. J Trauma. 2011;71(4):815–819. doi: 10.1097/TA.0b013e3182319de5. [DOI] [PubMed] [Google Scholar]
- 118.Giunti D, Parodi B, Cordano C, Uccelli A, Kerlero de Rosbo N. Can we switch microglia's phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology. 2014;141(3):328–339. doi: 10.1111/imm.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sikoglu EM, Heffernan ME, Tam K, et al. Enhancement in cognitive function recovery by granulocyte colony stimulating factor in a rodent model of traumatic brain injury. Behav Brain Res. 2014;259:354–356. doi: 10.1016/j.bbr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 120.Acosta SA, Tajiri N, Shinozuka K, et al. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS ONE. 2014;9(3):e90953. doi: 10.1371/journal.pone.0090953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dela-Peña I, Sanberg PR, Acosta S, Tajiri N, Lin SZ, Borlongan CV. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: aging as a comorbidity factor. J Neurosurg Sci. 2014;58(3):145–149. [PMC free article] [PubMed] [Google Scholar]
- 122.Povlishock JT. Pathobiology of traumatically induced axonal injury in animals and man. Ann Emerg Med. 1993;22(6):980–986. doi: 10.1016/s0196-0644(05)82738-6. [DOI] [PubMed] [Google Scholar]
- 123.Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12(4):555–564. doi: 10.1089/neu.1995.12.555. [DOI] [PubMed] [Google Scholar]
- 124.Smith DH, Hicks R, Povlishock JT. Therapy development for diffuse axonal injury. J Neurotrauma. 2013;30(5):307–323. doi: 10.1089/neu.2012.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Okonkwo DO, Büki A, Siman R, Povlishock JT. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport. 1999;10(2):353–358. doi: 10.1097/00001756-199902050-00026. [DOI] [PubMed] [Google Scholar]
- 126.Tang-Schomer MO, Patel AR, Baas PW, Smith DH. Mechanical breakingofmicrotubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEBJ. 2010;24(5):1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhilai Z, Hui Z, Anmin J, Shaoxiong M, Bo Y, Yinhai C. A combination of taxol infusion and human umbilical cord mesenchymal stem cells transplantation for the treatment of rat spinal cord injury. Brain Res. 2012;1481:79–89. doi: 10.1016/j.brainres.2012.08.051. [DOI] [PubMed] [Google Scholar]
- 128.Mbye LH, Keles E, Tao L, et al. Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2012;32(3):515–524. doi: 10.1038/jcbfm.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Büki A, Koizumi H, Povlishock JT. Moderate posttraumatic hypothermia decreases early calpain mediated proteolysis and con comitant cytoskeletal compromise in traumatic axonal injury. Exp Neurol. 1999;159(1):319–328. doi: 10.1006/exnr.1999.7139. [DOI] [PubMed] [Google Scholar]
- 130.Zafonte RD, Baglella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT) JAMA. 2012;308(19):1993–2000. doi: 10.1001/jama.2012.13256. [DOI] [PubMed] [Google Scholar]
- 131.Patel SP, Sullivan PG, Pandya JD, et al. N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Exp Neurol. 2014;257:95–105. doi: 10.1016/j.expneurol.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clifton GL, Valadka A, Zygun D, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 2011;10(2):l31–139. doi: 10.1016/S1474-4422(10)70300-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Su E, Bell MJ, Kochanek PM, et al. Increased CSF concentrations of myelin basic protein after TBI in infants and children: absence of significant effect of therapeutic hypothermia. Neurocrit Care. 2012;17(3):401–407. doi: 10.1007/s12028-012-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Miyauchi T, Wei EP, Povlishock JT. Evidence for the therapeutic efficacy of either mild hypothermia or oxygen radical scavengers after repetitive mild traumatic brain injury. J Neurotrauma. 2014;31(8):773–781. doi: 10.1089/neu.2013.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reeves TM, Phillips LL, Lee NN, Povlishock JT. Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res. 2007;1154:225–236. doi: 10.1016/j.brainres.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cantu RC, Gean AD. Second-impact syndrome and a small sub-dural hematoma: an uncommon catastrophic result of repetitive head injury with a characteristic imaging appearance. J Neuro trauma. 2010;27(9):1557–1564. doi: 10.1089/neu.2010.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bouma CJ, Muizelaar JP, Stringer WA, Choi SC, Fatouros P, Young HF. Ultra-early evaluation of regional cerebral blood fow in severely head-injured patients using xenon.enhanced computerized tomography. J Neurosurg. 1992;77(3):360–368. doi: 10.3171/jns.1992.77.3.0360. [DOI] [PubMed] [Google Scholar]
- 138.Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regluate cerebral blood fow in health and disease. Nature. 2014;508(7494):55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Terpolilli NA, Kim SW, Thal SC, Kuebler WM, Plesnila N. Inhaled nitric oxide reruces secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2013;33(2):311–318. doi: 10.1038/jcbfm.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Armstead WM, Kiessling JW, KolKe WA, Vavilala MS. SNP improves cerebral hemodynamics during normotension but fails to prevent sex dependent impaired cerebral autoregulation during hypotension after brain injury. Brain Res. 2010;1330:142–150. doi: 10.1016/j.brainres.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sinz EH, Kochanek PM, Dixon CE, et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104(5):647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Salonia R, Empey PE, Poloyac SM, et al. Endothelin-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes in children after severe traumatic brain injury. J Neurotrauma. 2010;27(10):1819–1825. doi: 10.1089/neu.2010.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Macdonald RL, Higashida RT, Keller E, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2) Lancet Neurol. 2011;10(7):618–625. doi: 10.1016/S1474-4422(11)70108-9. [DOI] [PubMed] [Google Scholar]
- 144.Giannopoulos S, Katsanos AH, Tsivgoulis G, Marshall RS. Statins and cerebral hemodynamics. J Cereb Blood Flow Metab. 2012;32(11):1973–1976. doi: 10.1038/jcbfm.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jansen JO, Lord JM, Thickett DR, Midwinter MJ, McAuley DF, Gao F. Clinical review: statins and trauma–a systematic review. Crit Care. 2013;17(3):227. doi: 10.1186/cc12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Elkind MS, Sacco RI, MacArthur RB, et al. The Neuroprotection with Statin Therapy for Acute Recovery Trial (NeuSTART): an adaptive design phase I dose-escalation study of high dose lovastatin in acute ischemic stroke. Int J Stroke. 2008;3(3):210–218. doi: 10.1111/j.1747-4949.2008.00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shaik JS, Ahmad M, Li W, et al. Soluble epoxide hydrolase inhibitor trans-4-14-(3-adamantan-1-yl-ureido)-cyclohexyloxyl-benzoic acid is neuroprotective in rat model of ischemic stroke. Am J Physiol Heart Circ Physiol. 2013;305(11):H1605–H1613. doi: 10.1152/ajpheart.00471.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary local ischemia. J Cereb Blood Flow Metab. 2006;26(12):1551–1561. doi: 10.1038/sj.jcbfm.9600309. [DOI] [PubMed] [Google Scholar]
- 149.Fordsmann JC, Ko RW, Choi HB, et al. Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci. 2013;33(6):2562–2570. doi: 10.1523/JNEUROSCI.2308-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hartings JA, Bullock MR, Okonkwo DO, et al. Co-Operative Study on Brain Injury Depolarisations. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. 2011;10(12):1058–1064. doi: 10.1016/S1474-4422(11)70243-5. [DOI] [PubMed] [Google Scholar]
- 151.Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217(4562):855–857. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- 152.Hovda DA, Sutton RL, Feeney DM. Amphetamine-induced recovery of visual cliff performance after bilateral visual cortex ablation in cats: measurements of depth perception thresholds. Behav Neurosci. 1989;103(3):574–584. doi: 10.1037//0735-7044.103.3.574. [DOI] [PubMed] [Google Scholar]
- 153.Feeney OM. Pharmacologic modulation of recovery after brain injury: a reconsideration of diaschisis. J Neurol Rehabil. 1991;5:113–128. [Google Scholar]
- 154.Kline AE, Chen MJ, Tso-Olivas DY, Feeney OM. Methylphenidate treatment following ablation induced hemiplegia in rat: experience during drug action alters effects on recovery of function. Pharmacol Biochem Behav. 1994;48(3):773–779. doi: 10.1016/0091-3057(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 155.Wagner AK, Ren D, Conley YP, et al. Sex and genetic associations with cerebrospinal fluid dopamine and metabolite production after severe traumatic brain injury. J Neurosurg. 2007;106(4):538–547. doi: 10.3171/jns.2007.106.4.538. [DOI] [PubMed] [Google Scholar]
- 156.Wagner AK, Seanlon JM, Niyonkuru C, et al. Genetic variation in the dopamine transporter gene and the D2 receptor gene influences striatal DAT binding in adults with severe TBI. J Cereb Blood Flow Metab. 2014 doi: 10.1038/jcbfm.2014.87. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bales JW, Kline AE, Wagner AK, Dixon CT. Targeting dopamine acutely in traumatic brain injury. Open Drug Discov J. 2010;2:119–128. doi: 10.2174/1877381801002010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dixon CE, Bales J, Kline AE, Wagner AK. Dopamine mechanisms of injury and recovery after TBI. In: Kobaissy FH, editor. Brain Neuro trauma: Molecular, Neuropsychological, and Rehabilitation Aspects in Brain Injury Models. CRC Press; Boca Raton, FL: 2015. [Google Scholar]
- 159.Kline AE, Yan HQ, Bao J, Marion DW, Dixon CE. Chronic methylphenidate treatment enhances water maze performance following traumatic brain injury in rats. Neurosci Lett. 2000;280(3):163–166. doi: 10.1016/s0304-3940(00)00797-7. [DOI] [PubMed] [Google Scholar]
- 160.Wagner AK, Drewencki LL, Chen X, et al. Chronic methylphenidate treatment enhances striatal dopamine neurotransmission after experimental traumatic brain injury. J Neurochem. 2009;108(4):986–997. doi: 10.1111/j.1471-4159.2008.05840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wagner AK, Kline AE, Ren D, et al. Gender associations with chronic methylphenidate treatment and behavioral performance following experimental traumatic brain injury. Behav Brain Res. 2007;181(2):200–209. doi: 10.1016/j.bbr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Meythaler JM, Brunner RC, Johnson A, Novack TA. Amancadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil. 2002;17(4):300–313. doi: 10.1097/00001199-200208000-00004. [DOI] [PubMed] [Google Scholar]
- 163.Dixon CE, Kraus MF, Kline AE, et al. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Rescor Neurol Neurosci. 1999;14(4):285–294. [PubMed] [Google Scholar]
- 164.Wang T, Huang XJ, Van KC, Went GT, Nguyen JT, Lyeth BG. Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats. J Neurotrauma. 2014;31(4):370–377. doi: 10.1089/neu.2013.2917. [DOI] [PubMed] [Google Scholar]
- 165.Grelak RP, Clark R, Stump JM, Vernier VG. Amantadine-dopamine interaction: possible mode of action in Parkinsonism. Science. 1970;169(3941):203–204. doi: 10.1126/science.169.3941.203. [DOI] [PubMed] [Google Scholar]
- 166.Gianutsos G, Chute S, Dunn JP. Pharmacological changes in dopaminergic systems induced by long-term administration of amantadine. Eur J Pharmacol. 1985;110(3):357–361. doi: 10.1016/0014-2999(85)90564-3. [DOI] [PubMed] [Google Scholar]
- 167.Allen RM. Role of amantadine in the management of neuroleptic-induced extrapyramidal syndromes: overview and pharmacology. Clio Neuropharmacol. 1983;6(Suppl 1):S64–S73. doi: 10.1097/00002826-198300061-00009. [DOI] [PubMed] [Google Scholar]
- 168.Harun R, Wagner AK. The neurobiological basis of pharmacological approaches for patients with traumatic brain injury. In: Levin, Shun, Chan, editors. Traumatic Brain Injury: A Review of the Research and Future Directions. Oxford University Press; New York, NY: 2013. [Google Scholar]
- 169.Kline AE, Massucci JL, Ma X, Zafonte RD, Dixon CE. Bromocriptine reduces lipid peroxidation and enhances spacial learning and hippocampal neuron surviva1 in a rodent model of focal brain trauma. J Neurotrauma. 2004;21(12):1712–1722. doi: 10.1089/neu.2004.21.1712. [DOI] [PubMed] [Google Scholar]
- 170.Brannan T, Martinez-Tica J, Di Rocco A, Yahr MD. Low and high dose bromocriptine have different effects on striatal dopamine release: an in vivo study. J Neural Transm Park Dis Dement Sect. 1993;6(2):81–87. doi: 10.1007/BF02261001. [DOI] [PubMed] [Google Scholar]
- 171.Zhu J, Hamm RJ, Reeves TM, Povlishock JT, Phillips LL. Postinjury administration of L-deprenyl improves cognitive function and enhances neuroplasticicy after traumatic brain injury. EXp Neurol. 2000;166(1):136–152. doi: 10.1006/exnr.2000.7484. [DOI] [PubMed] [Google Scholar]
- 172.Kraus MF, Maki PM. Effect of amantadine hydrochloride on symptoms offrontal lobe dysfunction in brain injury: case studies and review. J Neuropsychiatry Clio Neurosci. 1997;9(2):222–230. doi: 10.1176/jnp.9.2.222. [DOI] [PubMed] [Google Scholar]
- 173.McDowell S, Whyte J, D'Esposito M. Diferential effect of a dopaminergic agonist on prefrontal function in traumatic brain injury patients. Brain. 1998;121:1155–1164. doi: 10.1093/brain/121.6.1155. Pt 6. [DOI] [PubMed] [Google Scholar]
- 174.Verrier JD, Jackson TC, Bansal R, et al. The brain in vivo expresses the 2',3'-cAMP-adenosine pathway. J Neurochem. 2012;122(1):115–125. doi: 10.1111/j.1471-4159.2012.07705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kochanek PM, Verrier JD, Wagner AK, Jackson EK. The many roles of adenosine in traumatic brain injury. In: Boisen D, Masino S, editors. Adenosine: A Key Link Between Metabolism and Central Nervous System Activity. Springer; New York, NY: 2012. pp. 307–322. [Google Scholar]
- 176.Kochanek PM, Vagni VA, Janesko KL, et al. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26(4):565–575. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- 177.Wagner AK, Miller MA, Scanlon J, Ren D, Kochanek PM, Conley VP. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 2010;90(3):259–272. doi: 10.1016/j.eplepsyres.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Haselkorn ML, Shellington DK, Jackson EK, et al. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J Neurorrauma. 2010;27(5):901–910. doi: 10.1089/neu.2009.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kim M, Ham A, Kim KY-M, Brown KM, Lee HT. The volatile anesthetic isoflurane increases endothelial adenosine generation via microparticle ecto-5'-nucleotidase (CD73) release. PLoS ONE. 2014;9(6):e99950. doi: 10.1371/journal.pone.0099950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tas PWL, Eisemann C, Roewer N. The volatile anesthetic isoflurane suppresses spontaneous calcium oscillations in vitro in rat hippocampal neurons by activation of adenosine A1 receptors. Neurosci Lett. 2003;338(3):229–232. doi: 10.1016/s0304-3940(02)01420-9. [DOI] [PubMed] [Google Scholar]
- 181.Sachse KT, Jackson EK, Wisniewski SR, et al. Increases in cerebrospinal fluid caffeine concentration are associated with favorable outcome after severe traumatic brain injury in humans. J Cereb Blood Flow Metab. 2008;28(2):395–401. doi: 10.1038/sj.jcbfm.9600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Boisen D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol. 2008;84(3):249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Huber A, Padrun V, Deglon N, Aebischer P, Möhler H, Boisen D. Grafts of adenosine releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci US A. 2001;98(13):7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Baroncelli L, Braschi C, Spolidoro M, Begenisic T, Sale A, Maffei L. Nurturing bran plasticity: impact of environmental enrichment. Cell Death Offer. 2010;17(7):1092–1103. doi: 10.1038/cdd.2009.193. [DOI] [PubMed] [Google Scholar]
- 185.Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res. 2014;114(10):1601–1610. doi: 10.1161/CIRCRESAHA.114.303822. [DOI] [PubMed] [Google Scholar]
- 186.Dezfulian C, Shiva S, Alekseyenko A, et al. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. 2009;120(10):897–905. doi: 10.1161/CIRCULATIONAHA.109.853267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schwarzschild MA, Ascherio A, Beal MF, et al. Parkinson Study Group SURE PD Investigators, Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: a randomized clinical trial. JAMA Neurol. 2014;71(2):141–150. doi: 10.1001/jamaneurol.2013.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chip S, Zelmer A, Ogunshola OO, et al. The RNA binding protein RBM3 is involved in hypothermia induced neuroprotection. Neurobiol Dis. 2011;43(2):388–396. doi: 10.1016/j.nbd.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 189.Jackson TC, Verrier JD, Kochanek PM. 2013;4:e451. doi: 10.1038/cddis.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mahmood A, Lu D, Yi L, Chen JL, Chopp M. Intracranial bone marrow transplantation after traumatic brain injury improving functional outcome in adult rats. J Neurosurg. 2001;94(4):589–595. doi: 10.3171/jns.2001.94.4.0589. [DOI] [PubMed] [Google Scholar]
- 191.Lu D, Li Y, Wang I, Chen J, Mahmood A, Chopp M. Intraarterial administration of marrow stromal cells in a rat model of traumatic brain injury. J Neurotrauma. 2001;18(8):813–819. doi: 10.1089/089771501316919175. [DOI] [PubMed] [Google Scholar]
- 192.Mahmood A, Lu D, Qu C, Goussev A, Chopp M. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J Neurosurg. 2006;104(2):272–277. doi: 10.3171/jns.2006.104.2.272. [DOI] [PubMed] [Google Scholar]
- 193.Lu D, Mahmood A, Wang L, Li Y, Lu M, Chopp M. Adult bone-marrow stromal cells administered intravenously to rats after traumatic brain injury migrate into brain and improve neurological outcome. NeuroReport. 2001;12(3):559–563. doi: 10.1097/00001756-200103050-00025. [DOI] [PubMed] [Google Scholar]
- 194.Riess P, Zhang C, Saatman KE, et al. Transplanted neural stem cells survive, differentiate, and improve neurological motor function after experimental traumatic brain injury. Neurosurgery. 2002;51(4):1043–1052. doi: 10.1097/00006123-200210000-00035. [DOI] [PubMed] [Google Scholar]
- 195.Hoane MR, Becerra GD, Shank JE, et al. Transplantation of neuronal and glial precursors dramatically improves sensorimotor function but not cognitive function in the traumatically injured brain. J Neurotrauma. 2004;21(2):163–174. doi: 10.1089/089771504322778622. [DOI] [PubMed] [Google Scholar]
- 196.Liu SJ, Zou Y, Belegu V, et al. Co-grafting of neural stem cells with olfactory en sheathing cells promotes neuronal restoration in traumatic brain injury with an anti in flammatory mechanism. J Neuroinflammation. 2014;11:66. doi: 10.1186/1742-2094-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mahmood A, Wu H, Qu C, Xiong Y, Chopp M. Effects of treating traumatic brain injury with collagen scaffolds and human bone marrow stromal cells on sprouting of corticospinal tract axons into the denervated side of the spinal cord. J Neurosurg. 2013;118(2):381–389. doi: 10.3171/2012.11.JNS12753. [DOI] [PubMed] [Google Scholar]
- 198.Tajiri N, Acosta SA, Shahaduzzaman M, et al. Intravenous trans plants of human adi pose derived stem cell protect the brain from traumatic brain injury ind uced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34(1):313–326. doi: 10.1523/JNEUROSCI.2425-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sun D, Bullock MR, McGinn MJ, et al. Basic fibroblast growth factor enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol. 2009;216(1):56–65. doi: 10.1016/j.expneurol.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. En hanced hippocampal neurogenesis by intraventricular S100B in fusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22(6):645–655. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- 201.Bregy A, Nixon R, Lotocki G, et al. Posttraumatic hypothermia increases doublecortin expressing neurons in the dentate gyrus after traumatic brain injury in the rat. Exp Neurol. 2012;233(2):821–828. doi: 10.1016/j.expneurol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Carlson SW, Madathil SK, Sama DM, Gao X, Chen J, Saatman KE. Conditional overexpression of insulin like growth factor 1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J Neuropathol Exp Neurol. 2014;73(8):734–746. doi: 10.1097/NEN.0000000000000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11(5):339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WO. Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J Neurotrauma. 2014;31(5):476–486. doi: 10.1089/neu.2013.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br J Pharmacol. 2011;164(4):1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Margulies S, Hicks R. Combination Therapies for Traumatic Brain Injury Workshop Leaders. Combination therapies fo r traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26(6):925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N acetyl cysteine in traumatic brain injury. PLoS ONE. 2014;9(4):e90617. doi: 10.1371/journal.pone.0090617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N acetylcysteine: a double blind, placebo controlled study. PLoS ONE. 2013;8(1):e54163. doi: 10.1371/journal.pone.0054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N acetylcysteine. Biochim Biophys Acta. 2013;1830(8):4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 211.Abdel-Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ. Minocycline synergizes with N acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS ONE. 2010;5(8):e12490. doi: 10.1371/journal.pone.0012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Haber M, Abdel-Baki SG, Grin'kina NM, et al. Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Exp Neurol. 2013;249:169–177. doi: 10.1016/j.expneurol.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 213.Empey PE, Alexander HL, Ocque AJ, et al. Probenecid increases n-acetylcys teine brain penetration following experimental pediat ric traumatic brain injury. J Neurotrauma. 2012;29:229–230. [Google Scholar]
- 214.Tang H, Hua F, Wang J, et al. Progesterone and vitamin D: im provement after traumatic brain injury in middle aged rats. Horm Behav. 2013;64(3):527–538. doi: 10.1016/j.yhbeh.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shear DA, Tortella FC. A military centered approach to neuroprotection for traumatic brain injury. Front Neural. 2013;4:73. doi: 10.3389/fneur.2013.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chesnut RM. Secondary brain insults after head injury: clinical perspectives. New Horiz. 1995;3(3):366–375. [PubMed] [Google Scholar]
- 217.Jungner M, Grande PO, Mattiasson G, Bentzer P. Effects on brain edema of crystalloid and albumin fluid resuscitation after brain trauma and hemorrhage in the rat. Anesthesiology. 2010;112(5):1194–1203. doi: 10.1097/ALN.0b013e3181d94d6e. [DOI] [PubMed] [Google Scholar]
- 218.Myburgh J, Cooper DJ, Finfer S, et al. SAFE Study Investigators: Australian and New Zealand Intensive Care Society Clinical Trials Group; Australian Red Cross Blood Service; George Institute for International Health. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357(9):874–884. doi: 10.1056/NEJMoa067514. [DOI] [PubMed] [Google Scholar]
- 219.Chodobski A, Zink BJ. Szmydynger Chodobska J. Blood brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2(4):492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Brockman EC, Baytr H, Blasiole B, et al. Polyni troxylated pegy lated hemoglobin attenuates fluid requirements and brain edema in combined traumatic brain injury plus hemorrhagic shock in mice. J Cereb Blood Flow Metab. 2013;33(9):1457–1464. doi: 10.1038/jcbfm.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shellington DK, Du L, Wu X, et al. Polyn itroxylated pegylated hemoglobin: a novel neuroprotective hemoglobin for acute volume limited fluid resuscitation after combined traumatic brain injury and hemorrhagic hypotension in mice. Crit Care Med. 2011;39(3):494–505. doi: 10.1097/CCM.0b013e318206b1fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Brands J, Kliner D, Lipowsky HH, Kameneva MV, Villanueva FS, Pacella JJ. New insights into the microvascular mechanisms of drag reducing polymers: effect on the cell free layer. PLoS ONE. 2013;8(10):e77252. doi: 10.1371/journal.pone.0077252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Pacella JJ, Kameneva MV, Brands J, et al. Modulation of precapillary arteriolar pressure with drag reducing polymers: a novel method for enhancing microvascular perfusion. Microcir culation. 2012;19(7):580–585. doi: 10.1111/j.1549-8719.2012.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Macias CA, Kameneva MV, Tenhunen JJ, Puyana JC, Fink MP. Survival in a rat model of lethal hemorrhagic shock is prolonged fo llowing resuscitation with a small volume of a solution con taining a drag reducing polymer derived from aloe vera. Shock. 2004;22(2):151–156. doi: 10.1097/01.shk.0000131489.83194.1a. [DOI] [PubMed] [Google Scholar]
- 225.Khuman J, Zhang J, Park J, Carroll JD, Donahue C, Whalen MJ. Low level laser light therapy improves cognitive deficits and inhibits microglial activation after controlled cortical impact in mice. J Neurotrauma. 2012;29(2):408–417. doi: 10.1089/neu.2010.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Singleton RH, Yan HQ, Fellows-Mayle W, Dixon CE. Resveratrol attenuates behavioral impairments and reduces cortical and hippocampal loss in a rat controlled cortical impact model of traumatic brain injury. J Neurotrauma. 2010;27(6):1091–1099. doi: 10.1089/neu.2010.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wu A, Ying Z, Gomez-Pinilla F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J Neurotrauma. 2011;28(10):2113–2122. doi: 10.1089/neu.2011.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dash PK, Johnson D, Clark J, et al. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neuro cognitive outcome. PLoS ONE. 2011;6(9):e24648. doi: 10.1371/journal.pone.0024648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Valle EJ, Allen CJ, Van Haren RM, et al. Do all trauma patients benefit from tranexamic acid? J Trauma Acute Care Surg. 2014;76(6):1373–1378. doi: 10.1097/TA.0000000000000242. [DOI] [PubMed] [Google Scholar]
- 230.Kabadi SV, Stoica BA, Loane DJ, Luo T, Faden AI. CR8, a novel inh ibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental trau matic brain injury. J Cereb Blood Flow Metab. 2014;34(3):502–513. doi: 10.1038/jcbfm.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Maruta J, Heaton KJ, Kryskow EM, Maule AL, Ghajar J. Dynamic visuomotor synchronization: quantification of predictive timing. Behav Res Methods. 2013;45(1):289–300. doi: 10.3758/s13428-012-0248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]