

Abstract
Numerous studies have established a role for mineralocorticoids in the development of renal fibrosis. Originally, the research focus for mineralocorticoid-induced fibrosis was on the collecting duct, where “classical” mineralocorticoid receptors (MR) involved with electrolyte transport are present. Epithelial cells in this segment can, under selected circumstances, also respond to MR activation by initiating pro-fibrotic pathways. More recently, “non-classical” MR have been described in kidney cells not associated with electrolyte transport including mesangial cells and podocytes within the glomerulus. Activation of MR in these cells appears to lead to glomerular sclerosis. Mechanistically, aldosterone induces excess production of reactive oxygen species (ROS) and oxidative stress in glomerular cells through activation of NADPH oxidase. In mesangial cells, aldosterone also has pro-apoptotic, mitogenic, and pro-fibrogenic effects, all of which potentially promote active remodeling and expansion of the mesangium. While mitochondrial dysfunction seems to mediate the aldosterone-induced mesangial apoptosis, the ROS dependent EGFR transactivation is likely responsible for aldosterone-induced mesangial mitosis and proliferation. In podocytes, mitochondrial dysfunction elicited by oxidative stress is an early event associated with aldosterone-induced podocyte injury. Both the p38MAPK signaling and the redox sensitive glycogen synthase kinase (GSK) 3β pathways are centrally implicated in aldosterone-induced podocyte death. Aldosterone-induced GSK3β over-activity could potentially cause hyperphosphorylation and over-activation of putative GSK3β substrates, including structural components of the mitochondrial permeability transition (MPT) pore, all of which lead to cell injury and death. Clinically, proteinuria significantly decreases when aldosterone inhibitors are included in the treatment of many glomerular diseases further supporting the view that mineralocorticoids are important players in glomerular pathology.
Keywords: aldosterone, mineralocorticoid receptor, glomerulus, mesangial cell, podocyte, apoptosis, proliferation, glycogen synthase kinase 3β, mitochnodrial dysfunction, reactive oxygen species
Emergence and evolution of aldosterone-induced pathophysiology
Hans Selye [1] first noted a connection between aldosterone and the development of tissue inflammation over sixty years ago, shortly after the discovery and chemical characterization of the mineralocorticoid. Over the ensuing years, renal physiologists tended to set that salient observation aside in favor of studies describing renal sodium, potassium, and hydrogen ion transport [2]. As techniques advanced, investigators described and characterized specific mineralocorticoid receptors (MR), which appeared to be constitutively expressed in the distal portions of the mammalian nephron [3–5]. The MR, also known as the aldosterone receptor or nuclear receptor subfamily 3, group C, member 2 [6], (NR3C2) is a cytosolic receptor with equal affinity for both mineralocorticoids and glucocorticoids [7]. Additional factors independent of steroid-receptor binding, including the presence of the isoenzymes 11β-Hydroxysteroid Dehydrogenase (11β-HSD) types 1 and 2, appear to account for the much of the selectivity seen biologically [8, 9]. In most tissues where it is present including liver, fat cells, and vascular smooth muscle, 11β-HSD1 is considered to be a bi-directional enzyme [10–12] (a forward NADP dependent dehydrogenase and a reverse NADPH dependent reductase) with directionality largely a function of the redox state of the cofactor in a specific cell). However, 11β-HSD1 contained in proximal tubules of the kidney seems to be an exception, since the enzyme there only functions as a forward running NADP dependent dehydrogenase with no reductase activity seen in intact tissue, individual cultured cells, or in cell homogenates in the presence of excess NADPH [13, 14]. 11β-HSD2 usually co-localizes in cells containing MR (renal distal tubules and collecting ducts) and functions physiologically exclusively as a NAD dependent dehydrogenase [15, 16]. In humans, MR is encoded by the NR3C2 gene located on chromosome 4q31.1–31.2. When aldosterone bound to its receptor, a well-choreographed series of intracellular events occurred beginning with translocation of the receptor-ligand to the nucleus, the synthesis of selected new proteins, and finally changes in apical tubular epithelial cell membrane allowing for sodium reabsorption and potassium and hydrogen ion secretion [2, 17]. A second “non-classical” type of MR has been described mostly in non-electrolyte transporting cells [18–20]. This receptor binds mineralocorticoids but the biologic response is rapid occurring in seconds to minutes rather than hours and nuclear binding or protein synthesis doesn’t appear to be part of this process. Activation of protein kinase C and release of intracellular calcium follow MR binding in this signaling pathway [20]. These “non-classical” MR do not appear to respond to classic MR antagonists like spironolactone and are located in cell membranes. Wehling and his colleagues have suggested that an alternative G-protein coupled estrogen receptor, GPR30, may be a possible candidate for the “non-classical” MR receptor since it can bind aldosterone at physiologic concentrations [21]. Some confusion remains on this topic since classical MR antagonists do have an effect against aldosterone in some non-electrolyte transporting cells, especially cells in the glomerulus [22–25], and aldosterone may directly bind to other non-MR cell proteins and induce a biological effect [26].
MR in glomerular cells: pathogenic role of aldosterone in glomerular disease
MR have recently been described in the glomerulus, in mesangial cells [27, 28] and podocytes [29, 30], cells not normally associated with electrolyte transport. Whether these glomerular MR are expressed constitutively or are induced and what their biologic functions are, remains to be established. Prior studies conducted in normal renal tissues did not show evidence of MR in glomerular cells [31, 32] but the conflicting results may be related to unique characteristics of the antibodies used and/or the conditions specific to the animal model studied. In experiments done in our laboratory, conditionally immortalized mouse podocytes in culture were treated with adriamycin to induce injury (0.25μg/ml) or an equal volume of saline for 48 hours. Employing an anti-MR antibody kindly provided by Dr. Celeso Gomez-Sanchez, a Western immunoblot analysis (Figure 1) showed that MR expression was barely detectible in podocytes under basal conditions but expression was markedly up-regulated at 48 hours in injured cells (unpublished). This observation seems to explain the apparent absence of MR in “normal” glomeruli and reports of its presence following injury.
Figure 1. De novo expression of mineralocorticoid receptors (MR) in glomerular podocytes.
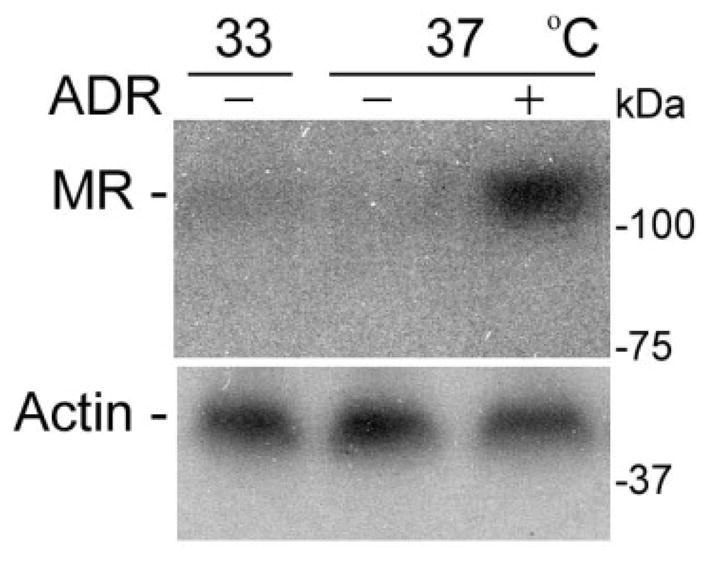
Conditionally immortalized mouse podocytes were cultured under permissive condition at 33°C or induced to differentiate at 37°C. Podocytes were treated with adriamycin (ADR - 0.25μg/ml) or saline for 48 hours before the cells were prepared for Western immunoblot analysis for MR using an anti-MR antibody kindly provided by Dr. Celso Gomez-Sanchez. MR expression was barely detected in podocytes under basal conditions but was markedly amplified after 48 hours of ADR-induced injury.
There is some suggestion that MR activation is associated with the progression of renal disease. Aldosterone can function as a growth factor in cultured collecting duct epithelial cells [33], a cell line more traditionally associated with classical MR and electrolyte transport. Aldosterone has also been shown to induce proliferation in cultured human mesangial cells [34], an effect inhibited by the MR antagonist eplerenone but not the glucocorticoid receptor antagonist RU-486 [27, 34]. Mesangial proliferation has also been noted in previously adrenalectomized mice infused for one week with aldosterone (8μg/kg/day) but not with the glucocorticoid corticosterone [24]. Moreover, MR were described in podocytes from glomeruli of previously uninephrectomized rats [29], as well as rat models of metabolic syndrome [30], and cultured podocytes [30]. Exposure to aldosterone in these various models was associated with signs of podocyte injury, most notably a decrease in the expression of podocin and nephrin and activation of reactive oxygen species [29]. The aldosterone-induced patterns of podocyte injury were all blocked with MR antagonists. Thus, MR activation, when it occurs in the injured glomerulus, appears to be clearly a pathological and not a physiological event.
The glomerular changes above mentioned are consistent with the previously described connection between aldosterone and inflammation; specifically the role aldosterone may play in advancing renal injury. Greene and colleagues were among the first to link aldosterone to disease progression in an animal model of pre-existing renal injury [35]. Others expanded those findings showing that aldosterone exposure enhanced the expression of a number of pro-inflammatory and pro-fibrotic cytokines in the kidney including PAI-1, NFκB, and CTGF [22, 36, 37]. Two fundamental questions rose from these studies: first, although it exacerbates pre-existing injury, could aldosterone also induce pro-inflammatory and pro-fibrotic pathways in the kidney in the absence of prior injury and/or hypertension and second, which renal cells might be the initial targets?
Experiments done in our laboratories combined with studies from other investigators demonstrate that mineralocorticoids like aldosterone or DOCA can, under selected laboratory conditions such as prior adrenalectomy with aldosterone replacement [24], cell culture [22, 24], or prior unilateral nephrectomy [38], induce fibrotic changes in renal tissues without evidence of prior tissue injury or systemic hypertension [24, 36]. Endogenously generated compounds such as progesterone [39] as well as 11-dehydro-glucocorticoid end products generated by renal 11β-HSD (both 11β-HSD1 contained in proximal tubules and 11β-HSD2 in distal tubules and the collecting duct) naturally inhibit the electrolyte transport pathways as well as the pro-fibrotic effects induced by aldosterone [24, 40–42]. Thus, under “normal physiologic conditions”, both of aldosterone’s biologic effects are limited. Only when these naturally occurring endogenous aldosterone antagonists are not present or significantly diminished because of decreased renal 11β-HSD isoform activity after injury or disease [43] does one begin to see the full effects of the mineralocorticoid in the kidney [24] and elsewhere [44]. At first glance, renal epithelial cells that contain constitutive MR and conduct electrolyte transport in the distal nephron are also sensitive to pro-fibrotic effects of aldosterone [36]. However, there is increasing evidence for aldosterone activated MR causing pathologic changes in glomerular mesangial cells and podocytes as discussed earlier.
Molecular mechanisms underlying the pathogenic roles of aldosterone in glomerular disease
Reactive oxygen species (ROS) play a key role in the progression of renal injury. Aldosterone increases ROS production possibly through the activation of NADPH oxidase. Activated MR mediates the translocation of the cytosolic components of p47 phagocytic oxidase (phox) and p67phox to the cell membrane [45]. Subsequently, ROS overproduction elicits oxidative stress and triggers redox sensitive cell signaling cascades that mediate mitochondrial dysfunction, cellular apoptosis, inflammatory response and fibrogenesis. These aldosterone induced inflammatory, fibrotic, and apoptotic pathways appear specifically activated in the response to injury in both glomerular mesangial cells and podocytes as described below.
A. Mechanism of aldosterone induced mesangial injury
In cultured human mesangial cells, aldosterone exposure induced both apoptotic and mitogenic effects (Figure 2). Aldosterone promoted mesangial cell apoptosis in a dose- and time-dependent manner. Spironolactone, an MR antagonist, inhibited aldosterone-induced mesangial cell apoptosis, inferring that an activated MR mediated the pro-apoptotic effect. Similarly, antioxidants and free radical scavengers partially attenuated pro-apoaptotic effects of aldosterone, consistent with the involement of ROS. Aldosterone also enhanced dephosphorylation of BAD, a protein, which when dephosphorylated, is linked to apoptosis and mitochondrial dysfunction in mesangial cells. Moreover, aldosterone-infused rats showed enhanced urinary albumin excretion rate and marked mesangial changes including both mesangial proliferation and mesangial apoptosis [46]. How aldosterone induces mesangial mitosis and proliferation remains largely unknown. There is evidence that aldosterone-induced mesangial cell proliferation may be mediated by epithelial growth factor receptor (EGFR) transactivation, because pre-treatment with the EGFR antagonist AG1478 blocked mesangial cell proliferation following aldostoreone exposure [27]. Moreover, the aldosterone-induced transactivation of EGFR is contingent on the oxidative stress. Pre-treatment with the antioxidant N-acetyl-L-cysteine, catalase, superoxide dismutase (SOD), mitochondrial respiratory chain complex I inhibitor rotenone, or NADPH oxidase inhibitors apocynin, and diphenylene iodonium significantly attenuated the aldosterone elicited EGFR transactivation and mesangial cell proliferation. Furthermore, the mitogenic effect of aldosterone was found to be mediated by the Ras/c-Raf/MEK/ERK pathway, an “off/on switch” comprised of extracellular-signal-regulated kinases, and PI3K/Akt/mTOR/p70S6K1, an EGF activated signaling pathway downstream from the EGF receptor [27] (Figure 2).
Figure 2. Aldosterone is centrally implicated in the pathogenesis of mesangial injuries in glomerular disease.

Mechanistically, aldosterone likely via the non-classic mineralocorticoid receptor (MR) induces reactive oxygen species (ROS) overproduction and oxidative stress in glomerular cells possibly by activating nicotinamide adenine dinucleotide phosphate (NADPH). In glomerular mesangial cells, aldosterone has both proapoptotic and mitogenic effects in addition to a profibrogenic activity and thereby potentially promotes active remodeling and expansion of glomerular mesangium. While mitochondria dysfunction seems to mediate the aldosterone induced mesangial cell death, the ROS dependent epithelial growth factor receptor (EGFR) transactivation together with the ensuing PI3K/Akt/p70(S6K) and Ras/MEK/ERK pathways is likely responsible for aldosterone induced mesangial mitosis and proliferation. The profibrogenic activity of aldosterone might involve a glucocorticoid receptor (GR) dependent as well as an MR/serum-and glucocorticoid-induced protein kinase (SGK)1 responsive mechanism, which amplifies connective tissue growth factor (CTGF) expression and results in overproduction of extracellular matrix. Collectively, all these mechanisms eventually lead to mesangial injury characterized by mesangial cell lysis and proliferation as well as mesangial matrix expansion and remodeling.
Aldosterone also demonstrated a pro-fibrotic effect in mesangial cells in addition to its pro-apoptotic and pro-mitotic effects. Mineralocorticoids promote extracellular matrix production in a variety of cell types containing both classical and non-classical MR, including cardiac myocytes, vascular smooth muscle cells, renal tubular cells, and mesangial cells [47]. In cultured rat mesangial cells, aldosterone (10−7~10−6M) rapidly (0~24 hours) induces mRNA and protein expression of CTGF, an early response pro-fibrotic growth factor, in a time- and concentration-dependent manner [48]. Surprisingly, MR antagonists, spironolactone (10−6M), canrenoate (10−5M), and eplerenone (10−5M), did not override this rapid CTGF induction [48], possibly suggesting an involvement of “non-classical” MR or an MR independent mechanism for this rapid effect. However, RU-486, a selective inhibitor of glucocorticoid receptors (GR), prevented aldosterone (10−7M)-induced CTGF expression, indicating that the aldosterone-mediated regulation of CTGF likely is conveyed through GR. GR nuclear translocation after acute aldosterone exposure further corroborated this observation [48]. In support of this point of view, Terada et al [22] demonstrated also in rat mesangial cells that aldosterone was able to induce CTGF expression at a lower concentration (10−8M) and that the MR antagonist eplerenone at 10−6M failed to completely block the effect. In contrast, any late effects (after 24 to 48 hours) of aldosterone exposure on CTGF and extracellular matrix overproduction is dependent on MR activation and may involve the pro-fibrotic Smad2-associated TGF-β1 pathway in mesangial cells [49]. The co-involvement of both MR and GR in a biologic response has been previously described in mineralocorticoid mediated sodium transport [50]. These late effects may also be influenced by factors over and above MR activation and nuclear transcriptional regulation, since other types of indirect regulation may also contribute to the response (Figure 2). Indeed, the increased expression of cardiac CTGF by aldosterone can be abolished in the heart tissue of SGK1 knockout mice [51]. This is important since SGK, is an enzyme, which is specifically induced by mineralocorticoids following MR binding and an up-regulation in its expression is associated with an increase in the long term expression of CTGF and activation of the TGF-β1 pathway.
B. Mechanism of aldosterone induced podocyte injury
An aldosterone infusion study provided direct evidence supporting the pathologic role of aldosterone in podocyte injury [25]. Following 14 days of aldosterone infusion (90 ng/day), mice excreted abundant urinary F2-isoprostane, a specific marker of renal oxidative stress. This dose of aldosterone likely produced high but probably biologically achievable blood levels although no values were reported in this study. Kidney sections from the aldosterone-infused mice also showed increased ROS generation in renal glomeruli. This was associated with evident podocyte injury in aldosterone-infused mice as indicated by diminished glomerular expression of nephrin and podocin, a 12-fold increase in urinary protein excretion, and ultrastuctural lesions in podocyte foot processes. Mechanistically, mitochondrial dysfunction seems to play an important role in aldosterone induced podocyte injury as evident by reduced mitochondrial membrane potential, reduced ATP levels, and a reduced mitochondrial DNA copy number were noted in aldosterone-treated podocytes found in the glomeruli of aldosterone-infused mice. Decreased expression of mitochondrial transcription factor A and PPARγ are likely to be responsible for aldosterone induced mitochondria dysfunction in podocytes and a PPARγ agonist or overexpression of mitochondrial transcription factor A significantly decreased the mitochondrial dysfunction and podocyte injury induced by aldosterone [52].
Podocyte depletion is a fundamental pathogenic mechanism that drives the development and progression of proteinuria and progressive glomerular sclerosis. Accumulating evidence is consistent with the view that aldosterone is a pro-apoptotic factor in podocytes as it is in other cells (Figure 3). In cultured rat podocytes, apoptosis could be induced in a dose and time dependent fashion [53]. The p38 MAPK signaling pathway seems to be responsible, at least in part, for the pro-apoptotic effect of aldosterone because inhibition of p38 MAPK by a small molecule inhibitor suppressed podocyte apoptosis. Moreover, the aldosterone induced podocyte death was also associated with an over-activity of glycogen synthase kinase (GSK)3β, which is a well-conserved, ubiquitously expressed serine/threonine protein kinase involved in multiple pathophysiological processes extending well beyond glycogen metabolism to cell death, embryo development and tissue injury, repair and regeneration. GSK3β is a redox sensitive signaling transducer situated at the nexus of multiple pathways influencing apoptotic cell death, cytoskeletal remodeling, control over development, insulin signaling, canonical wingless signaling, the NFκB pathway and more. More recently, GSK3β was found to regulate mitochondrial permeability transition and mitochondrial dysfunction in both excitable cells and non-excitable cells like kidney cells [54–56]. Thus, it is conceivable that ROS overproduction and oxidative stress elicited by aldosterone in podocytes induces GSK3β over-activity and subsequently enhance the phosphorylation and activation of GSK3β targeted substrates including structural components of mitochondrial permeability transition pore, like cyclophilin F and VDAC [54]. This effect will ultimately reduce the threshold of MPT in podocytes, promote mitochondrial dysfunction, and potentiate podocyte death (Figure 3).
Figure 3. Schematic diagram depicting the mechanisms underlying the pathogenic role of aldosterone in podocyte injury.

In glomerular podocytes, mitochnodrial dysfunction elicited by oxidative stress is an early event associated with aldosterone-induced podocytopathy likely via the non-classic mineralocorticoid receptor (MR). Both the p38MAPK signaling and the redox sensitive glycogen synthase kinase (GSK) 3β pathways are centrally implicated in aldosterone-induced podocyte injury. On one hand, activation of p38MAPK could induce podocyte apoptotic death via triggering the caspase death pathway. On the other hand, aldosterone-induced overactivity of the redox sensitive GSK3β could potentially cause hyperphosphorylation and overactivation of putative GSK3β substrates, including structural components of the mitochondrial permeability transition (MPT) pore. This accounts for the sensitized MPT, mitochondria dysfunction and potentiated podocyte death, resulting in podocytopenia. In addition, GSK3β overactivity is also a culprit for the disruption of both actin and microtubule cytoskeleton integrity and lead to podocyte shrinkage and foot process effacement, eventually culminating in proteinuria and progressive glomerular sclerosis.
Other abbreviations: MAPK, Mitogen-activated protein kinase; NADPH, Nicotinamide adenine dinucleotide phosphate ROS, reactive oxygen species.
Therapeutic targeting of aldosterone
A. Currently available aldosterone antagonists
There are 5 aldosterone antagonists that are commercially available, including spironolactone, eplerenone, canrenone, prorenone and mexrenone, which share a similar core molecular structure and antagonize the action of aldosterone at the level of MR. In current clinical practice, spironolatone and eplerenone are the two most common aldosterone antagonists that are being used. They have been commonly used in clinical settings as potassium sparing agents especially when added to other diuretics. They also have been used to attenuate cardiac fibrosis associated with aldosterone in patients with chronic congestive heart failure [57]. For research purposes, aldosterone antagonists are often used to differentiate between MR and GR actions. Spironolactone and its metabolite canrenoate bind with high affinity to the MR, but also may interact with other steroid receptors especially androgen receptors [58]. Interaction with androgen receptors has been used to account for the feminization, gynecomastia, impotence, low sex drive and reduction of size of male genitalia observed when these drugs are used therapeutically. However, there may be an alternative explanation for these side effects. It appears that MR are present in testicular cells and blocking MR activation in those cells suppresses testosterone production [59]. Eplerenone, although less potent as antagonist at the MR, is more specific and does not appear to interact with other steroid receptors as much. Compared to spironolactone, eplerenone is said to have a lower incidence of sexual side effects but given the new information on MR in the testes, its side effects may turn out to be not that different [57]. Other potential side effects of aldosterone antagonists including hyperkalaemia, hypotension, dizziness, altered renal function, and increased creatinine concentration are common to all these agents. Canrenone, a major active metabolite of spironolactone, has been used as a diuretic in Europe. However, prorenone and mexrenone are novel aldosterone antagonists and are currently under intensive pre-clinical investigations.
Glucocorticoid metabolites (11-dehydrocorticosterone and 11-dehydrocortisol, which is cortisone) generated endogenously by the forward reactions of the isoenzymes 11β-HSD1 and 11β-HSD2 also potently suppress both electrolyte transport [40, 42] and the pro-inflammatory/pro-fibrotic actions of aldosterone [24, 44]. These otherwise “inactive” metabolites block the actions of aldosterone in the kidney and in cultured renal epithelial cells where the metabolites cannot be enzymatically transformed back to the parent glucocorticoid, corticosterone or cortisol. 11-dehydrocorticosterone and cortisone appear to exert their effect on the transfer of the activated MR to the cell nucleus [41] and are less likely to function as competitive inhibitors for MR like spironolactone and related agents. Derivatives of these metabolites may eventually become available as pharmacological aldosterone antagonists.
In addition to the above steroidal MR antagonists, a next-generation non-steroidal MR antagonist, Finerenone or BAY94-8862, appears to have better selectivity for the MR when compared to spironolactone and eplerenone [60]. In a recent phase 2 clinical trial, this novel antagonist decreased levels of B-type natriuretic peptide as much as spironolactone in patients with chronic heart failure and kidney disease and did so without increasing serum potassium concentrations [61].
B. Aldosterone synthase inhibitors
Inhibition of aldosterone synthase is currently being investigated as a novel approach for the treatment of hypertension, heart failure, and renal disorders [62]. Inactivation of the enzymatic activity of aldosterone synthase reduced aldosterone concentrations in plasma and tissues and obliterated MR-dependent and independent effects in cardiac vascular and renal target organs [62]. In patients with primary aldosteronism, inhibition of aldosterone synthase reduced plasma and urinary aldosterone concentrations by 70~80%, rapidly corrected hypokalaemia, decreased blood pressure, and mildly increased plasma renin activity. The current, orally delivered, LCl699 has been found to be less specific to aldosterone synthase [62]. The second-generation aldosterone synthase inhibitors with higher selectivity to aldosterone synthase are under development. Nevertheless, the magnitude of the aldosterone synthase inhibition that is necessary to neutralize aldosterone in a biologically significant way is still unknown. Moreover, the accumulation of the mineralocorticoid 11-deoxycorticosterone during aldosterone synthase inhibition may act as a substitute for aldosterone.
Clinical implications
While there is ample evidence for improvement in pathology and/or function after treatment with aldosterone antagonists in various animal models of kidney disease [63, 64], data in human renal disease are limited. Most human clinical trials have been conducted in patients with renal disease already being treated with either an angiotensin II receptor blocker (ARB) or angiotensin converting enzyme (ACE) inhibitor. Aldosterone inhibition is most often an additional treatment not a separate treatment arm of its own. Despite that limitation, there is evidence favoring an additional benefit from using aldosterone inhibitors in patients with various forms of renal disease. Bianchi and colleagues [65] treated 83 patients with chronic kidney disease adding spironolactone 25 mg daily to either an ACE inhibitor or ARB. When compared to controls treated only with ACE inhibitor or ARB, patients given spironolactone demonstrated a marked decrease in proteinuria (2.1 versus 0.89 grams/gram creatinine) and a decreased monthly rate of decline in estimated glomerular filtration rate after one year. Another study, involving 221 patients with chronic kidney disease, demonstrated a similar fall in proteinuria after 16 weeks when spironolactone was added to ACE inhibitor or ARB [66]. There was no obvious effect on renal function reported in this last study however. In another controlled clinical trial, Mehdi and associates [67] treated 81 patients with evidence of diabetic nephropathy already maintained on lisinopril with the addition of either losartan or spironolactone over a period of 48 weeks. The group treated with spironolactone demonstrated a 34% drop in proteinuria compared to only 16.8% with the addition of losartan. There was no additional beneficial effect of spironolactone on renal function observed during this study. In a small observational study involving children with Alport’s syndrome, a genetically inherited disorder affecting the structure of the glomerular basement membrane and clinically characterized by hematuria, proteinuria and kidney dysfunction, Giani et al [68] described a similar decline in proteinuria as well as urinary TGF-β1 levels when spironolactone was added to therapy with an ACE inhibitor after 6 months of treatment. Thus, clinical trials seem to support the laboratory findings of aldosterone activated MR being a player in glomerular disease. A decrease in proteinuria is being considered as a surrogate maker for disease progression in these clinical studies, which may or may not be the case. All the trials thus far have been over too short a time period to clearly show an effect on renal function. Nevertheless, the addition of an MR antagonist may prove to be a useful additional aid in the treatment of renal diseases including those of glomerular origin.
Acknowledgments
This work was made possible in part by the funding from the U.S. National Institutes of Health grant R01DK092485 (to R.G.).
ABBREVIATIONS
- 11β-HSD
11β-hydroxysteroid dehydrogenase
- BAD
Bcl-2-associated death promoter
- CTGF
connective tissue growth factor
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GR
glucocorticoid receptors
- GSK3β
glycogen synthase kinase 3β
- MPT
mitochondrial permeability transition
- MR
mineralocorticoid receptors
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- NFκB
nuclear factor κB
- p38 MAPK
p38 mitogen activated protein kinase
- PAI-1
plasminogen activator inhibitor-1
- PI3K/Akt/mTOR/p70S6K1
phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin/p70S6K1- a pathway down stream from EGF activation
- PPARγ
peroxisome proliferator-activated receptor γ
- Ras/c-Raf/MEK/ERK
“off on switch” extracellular-signal-regulated kinases
- ROS
reactive oxygen species
- SGK-1
serum and glucocorticoid-induced kinase-1
- SOD
superoxide dismutase
- TGF-β1
transforming growth factor β1
- VDAC
voltage dependent anion channel
References
- 1.Selye H. Anticortisol action of aldosterone. Science. 1955;121:368–369. doi: 10.1126/science.121.3141.368. [DOI] [PubMed] [Google Scholar]
- 2.Porter GA, Edelman IS. The Action of Aldosterone and Related Corticosteroids On Sodium Transport Across the Toad Bladder. J Clin Invest. 1964;43:611–620. doi: 10.1172/JCI104946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funder JW, Feldman D, Edelman IS. The roles of plasma binding and receptor specificity in the mineralocorticoid action of aldosterone. Endocrinology. 1973;92:994–1004. doi: 10.1210/endo-92-4-994. [DOI] [PubMed] [Google Scholar]
- 4.Farman N, Kusch M, Edelman IS. Aldosterone receptor occupancy and sodium transport in the urinary bladder of Bufo marinus. Am J Physiol. 1978;235:C90–96. doi: 10.1152/ajpcell.1978.235.3.C90. [DOI] [PubMed] [Google Scholar]
- 5.Rossier BC, Wilce PA, Edelman IS. Spironolactone antagonism of aldosterone action on Na+ transport and RNA metabolism in toad bladder epithelium. J Membr Biol. 1977;32:177–194. doi: 10.1007/BF01905216. [DOI] [PubMed] [Google Scholar]
- 6.Le Menuet D, Viengchareun S, Muffat-Joly M, Zennaro MC, Lombes M. Expression and function of the human mineralocorticoid receptor: lessons from transgenic mouse models. Mol Cell Endocrinol. 2004;217:127–136. doi: 10.1016/j.mce.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 7.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- 8.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticord Action: Target Tissue Specificity Is Enzyme, Not Receptor, Mediated. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 9.Funder J, Myles K. Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: in vivo binding studies. Endocrinology. 1996;137:5264–5268. doi: 10.1210/endo.137.12.8940344. [DOI] [PubMed] [Google Scholar]
- 10.Brem AS, Bina RB, King T, Morris DJ. Bidirectional activity of 11 beta-hydroxysteroid dehydrogenase in vascular smooth muscle cells. Steroids. 1995;60:406–410. doi: 10.1016/0039-128x(94)00074-m. [DOI] [PubMed] [Google Scholar]
- 11.Brem AS, Bina RB, King TC, Morris DJ. Localization of 2 11beta-OH steroid dehydrogenase isoforms in aortic endothelial cells. Hypertension. 1998;31:459–462. doi: 10.1161/01.hyp.31.1.459. [DOI] [PubMed] [Google Scholar]
- 12.Hughes KA, Manolopoulos KN, Iqbal J, Cruden NL, Stimson RH, Reynolds RM, Newby DE, Andrew R, Karpe F, Walker BR. Recycling between cortisol and cortisone in human splanchnic, subcutaneous adipose, and skeletal muscle tissues in vivo. Diabetes. 2012;61:1357–1364. doi: 10.2337/db11-1345. doi:1310.2337/db1311-1345. Epub 2012 Apr 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brem AS, Bina RB, King T, Chobanian MC, Morris DJ. Influence of dietary sodium on the renal isoforms of 11 beta- hydroxysteroid dehydrogenase [In Process Citation] Proc Soc Exp Biol Med. 1997;214:340–345. doi: 10.3181/00379727-214-44101. [DOI] [PubMed] [Google Scholar]
- 14.Brem AS, Bina RB, Fitzpatrick C, King T, Tang SS, Ingelfinger JR. Glucocorticoid metabolism in proximal tubules modulates angiotensin II- induced electrolyte transport. Proc Soc Exp Biol Med. 1999;221:111–117. doi: 10.1046/j.1525-1373.1999.d01-63.x. [DOI] [PubMed] [Google Scholar]
- 15.Rusvai E, Naray-Fejes-Toth A. A new isoform of 11 beta-hydroxysteroid dehydrogenase in aldosterone target cells. Journal of Biological Chemistry. 1993;268:10717–10720. [PubMed] [Google Scholar]
- 16.Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 11β-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol. 1994;105:R11–R17. doi: 10.1016/0303-7207(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 17.Porter GA, Bogoroch R, Edelman IS. On the Mechanism of Action of Aldosterone On Sodium Transport: the Role of Rna Synthesis. Proc Natl Acad Sci U S A. 1964;52:1326–1333. doi: 10.1073/pnas.52.6.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christ M, Wehling M. Rapid actions of aldosterone: lymphocytes, vascular smooth muscle and endothelial cells. Steroids. 1999;64:35–41. doi: 10.1016/s0039-128x(98)00103-2. [DOI] [PubMed] [Google Scholar]
- 19.Boldyreff B, Wehling M. Non-genomic actions of aldosterone: mechanisms and consequences in kidney cells. Nephrol Dial Transplant. 2003;18:1693–1695. doi: 10.1093/ndt/gfg265. [DOI] [PubMed] [Google Scholar]
- 20.Mihailidou AS, Funder JW. Nongenomic effects of mineralocorticoid receptor activation in the cardiovascular system. Steroids. 2005;70:347–351. doi: 10.1016/j.steroids.2005.02.004. Epub 2005 Mar 2024. [DOI] [PubMed] [Google Scholar]
- 21.Wendler A, Albrecht C, Wehling M. Nongenomic actions of aldosterone and progesterone revisited. Steroids. 2012;77:1002–1006. doi: 10.1016/j.steroids.2011.12.023. doi:1010.1016/j.steroids.2011.1012.1023. Epub 2012 Jan 1020. [DOI] [PubMed] [Google Scholar]
- 22.Terada Y, Kuwana H, Kobayashi T, Okado T, Suzuki N, Yoshimoto T, Hirata Y, Sasaki S. Aldosterone-stimulated SGK1 activity mediates profibrotic signaling in the mesangium. J Am Soc Nephrol. 2008;19:298–309. doi: 10.1681/ASN.2007050531. Epub 2008 Jan 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldigier JC, Kanjanbuch T, Ma LJ, Brown NJ, Fogo AB. Regression of existing glomerulosclerosis by inhibition of aldosterone. J Am Soc Nephrol. 2005;16:3306–3314. doi: 10.1681/ASN.2004090804. Epub 2005 Sep 3328. [DOI] [PubMed] [Google Scholar]
- 24.Brem AS, Morris DJ, Ge Y, Dworkin LD, Tolbert E, Gong R. Direct Fibrogenic Effects of Aldosterone on Normotensive Kidney: An Effect Modified by 11{beta}-HSD Activity. Am J Physiol Renal Physiol. 2010;298:F1178–F1187. doi: 10.1152/ajprenal.00532.2009. [DOI] [PubMed] [Google Scholar]
- 25.Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B, Yang T, Zhang A. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: a therapeutic target of PPARgamma. Am J Pathol. 2011;178:2020–2031. doi: 10.1016/j.ajpath.2011.01.029. doi:2010.1016/j.ajpath.2011.2001.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alzamora R, Brown LR, Harvey BJ. Direct binding and activation of protein kinase C isoforms by aldosterone and 17beta-estradiol. Mol Endocrinol. 2007;21:2637–2650. doi: 10.1210/me.2006-0559. Epub 2007 Jul 2631. [DOI] [PubMed] [Google Scholar]
- 27.Huang S, Zhang A, Ding G, Chen R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol. 2009;296:F1323–1333. doi: 10.1152/ajprenal.90428.2008. Epub 2009 Apr 1321. [DOI] [PubMed] [Google Scholar]
- 28.Terada Y, Ueda S, Hamada K, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Kagawa T, Horino T, Takao T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin Exp Nephrol. 2012;16:81–88. doi: 10.1007/s10157-10011-10498-x. Epub 12011 Nov 10151. [DOI] [PubMed] [Google Scholar]
- 29.Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–364. doi: 10.1161/01.HYP.0000255636.11931.a2. Epub 2007 Jan 2002. [DOI] [PubMed] [Google Scholar]
- 30.Nagase M, Yoshida S, Shibata S, Nagase T, Gotoda T, Ando K, Fujita T. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–3446. doi: 10.1681/ASN.2006080944. Epub 2006 Nov 3432. [DOI] [PubMed] [Google Scholar]
- 31.Gomez-Sanchez CE, de Rodriguez AF, Romero DG, Estess J, Warden MP, Gomez-Sanchez MT, Gomez-Sanchez EP. Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology. 2006;147:1343–1348. doi: 10.1210/en.2005-0860. Epub 2005 Nov 1317. [DOI] [PubMed] [Google Scholar]
- 32.Sasano H, Fukushima K, Sasaki I, Matsuno S, Nagura H, Krozowski ZS. Immunolocalization of mineralocorticoid receptor in human kidney, pancreas, salivary, mammary and sweat glands: a light and electron microscopic immunohistochemical study. J Endocrinol. 1992;132:305–310. doi: 10.1677/joe.0.1320305. [DOI] [PubMed] [Google Scholar]
- 33.Thomas W, Dooley R, Harvey BJ. Aldosterone as a renal growth factor. Steroids. 2010;75:550–554. doi: 10.1016/j.steroids.2009.09.008. doi:510.1016/j.steroids.2009.1009.1008. Epub 2009 Sep 1024. [DOI] [PubMed] [Google Scholar]
- 34.Terada Y, Kobayashi T, Kuwana H, Tanaka H, Inoshita S, Kuwahara M, Sasaki S. Aldosterone stimulates proliferation of mesangial cells by activating mitogen-activated protein kinase 1/2, cyclin D1, and cyclin A. J Am Soc Nephrol. 2005;16:2296–2305. doi: 10.1681/ASN.2005020129. Epub 2005 Jun 2223. [DOI] [PubMed] [Google Scholar]
- 35.Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest. 1996;98:1063–1068. doi: 10.1172/JCI118867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leroy V, De Seigneux S, Agassiz V, Hasler U, Rafestin-Oblin ME, Vinciguerra M, Martin PY, Feraille E. Aldosterone activates NF-kappaB in the collecting duct. J Am Soc Nephrol. 2009;20:131–144. doi: 10.1681/ASN.2008020232. Epub 2008 Nov 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lea WB, Kwak ES, Luther JM, Fowler SM, Wang Z, Ma J, Fogo AB, Brown NJ. Aldosterone antagonism or synthase inhibition reduces end-organ damage induced by treatment with angiotensin and high salt. Kidney Int. 2009;75:936–944. doi: 10.1038/ki.2009.9. Epub 2009 Feb 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lam EY, Funder JW, Nikolic-Paterson DJ, Fuller PJ, Young MJ. Mineralocorticoid receptor blockade but not steroid withdrawal reverses renal fibrosis in deoxycorticosterone/salt rats. Endocrinology. 2006;147:3623–3629. doi: 10.1210/en.2005-1527. Epub 2006 Apr 3620. [DOI] [PubMed] [Google Scholar]
- 39.Rafestin-Oblin ME, Couette B, Barlet-Bas C, Cheval L, Viger A, Doucet A. Renal action of progesterone and 18-substituted derivatives. Am J Physiol. 1991;260:F828–832. doi: 10.1152/ajprenal.1991.260.6.F828. [DOI] [PubMed] [Google Scholar]
- 40.Alberti KGMM, Sharp GWG. Identification of four types of steroid by their interaction with mineralocorticoid receptors in the toad bladder. J Endocrinology. 1970;48:563–574. doi: 10.1677/joe.0.0480563. [DOI] [PubMed] [Google Scholar]
- 41.Odermatt A, Arnold P, Frey FJ. The intracellular localization of the mineralocorticoid receptor is regulated by 11beta-hydroxysteroid dehydrogenase type 2. J Biol Chem. 2001;276:28484–28492. doi: 10.1074/jbc.M100374200. Epub 22001 May 28411. [DOI] [PubMed] [Google Scholar]
- 42.Brem AS, Matheson KL, Barnes JL, Morris DJ. 11-Dehydrocorticosterone, a glucocorticoid metabolite, inhibits aldosterone action in toad bladder. American Journal of Physiology. 1991;261:F873–879. doi: 10.1152/ajprenal.1991.261.5.F873. [DOI] [PubMed] [Google Scholar]
- 43.Vierhapper H, Derfler K, Nowotny P, Hollenstein U, Waldhausl W. Impaired conversion of cortisol to cortisone in chronic renal insufficiency--a cause of hypertension or an epiphenomenon? Acta Endocrinologica. 1991;125:160–164. doi: 10.1530/acta.0.1250160. [DOI] [PubMed] [Google Scholar]
- 44.Brem AS, Morris DJ, Li X, Ge Y, Shaw S, Gong R. Adrenalectomy amplifies aldosterone induced injury in cardiovascular tissue: an effect attenuated by adrenally derived steroids. Steroids. 2013;78:347–355. doi: 10.1016/j.steroids.2012.12.007. doi:310.1016/j.steroids.2012.1012.1007. Epub 2012 Dec 1031. [DOI] [PubMed] [Google Scholar]
- 45.Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP, Kimura S, Yukimura T, Kiyomoto H, Kohno M, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. 2005;16:2906–2912. doi: 10.1681/ASN.2005040390. Epub 2005 Aug 2931. [DOI] [PubMed] [Google Scholar]
- 46.Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, Chander PN, Ding G, Singhal PC. Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol. 2008;295:F73–81. doi: 10.1152/ajprenal.00435.02007. Epub 02008 May 00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. 2013;9:459–469. doi: 10.1038/nrneph.2013.110. doi:410.1038/nrneph.2013.1110. Epub 2013 Jun 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gauer S, Segitz V, Goppelt-Struebe M. Aldosterone induces CTGF in mesangial cells by activation of the glucocorticoid receptor. Nephrol Dial Transplant. 2007;22:3154–3159. doi: 10.1093/ndt/gfm410. Epub 2007 Jun 3130. [DOI] [PubMed] [Google Scholar]
- 49.Lai L, Chen J, Hao CM, Lin S, Gu Y. Aldosterone promotes fibronectin production through a Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem Biophys Res Commun. 2006;348:70–75. doi: 10.1016/j.bbrc.2006.07.057. Epub 2006 Jul 2021. [DOI] [PubMed] [Google Scholar]
- 50.Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger JD, Rossier BC. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16:878–891. doi: 10.1681/ASN.2004121110. Epub 2005 Mar 2002. [DOI] [PubMed] [Google Scholar]
- 51.Vallon V, Wyatt AW, Klingel K, Huang DY, Hussain A, Berchtold S, Friedrich B, Grahammer F, Belaiba RS, Gorlach A, et al. SGK1-dependent cardiac CTGF formation and fibrosis following DOCA treatment. J Mol Med. 2006;84:396–404. doi: 10.1007/s00109-005-0027-z. Epub 2006 Apr 2008. [DOI] [PubMed] [Google Scholar]
- 52.Su M, Dhoopun AR, Yuan Y, Huang S, Zhu C, Ding G, Liu B, Yang T, Zhang A. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol Renal Physiol. 2013;305:F520–531. doi: 10.1152/ajprenal.00570.2012. doi:510.1152/ajprenal.00570.02012. Epub 02013 Jun 00512. [DOI] [PubMed] [Google Scholar]
- 53.Chen C, Liang W, Jia J, van Goor H, Singhal PC, Ding G. Aldosterone induces apoptosis in rat podocytes: role of PI3-K/Akt and p38MAPK signaling pathways. Nephron Exp Nephrol. 2009;113:e26–34. doi: 10.1159/000228080. Epub 000222009 Jul 000228089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, Ge Y, Bao H, Dworkin L, Peng A, Gong R. Redox-sensitive glycogen synthase kinase 3beta-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic Biol Med. 2013;65:849–58. doi: 10.1016/j.freeradbiomed.2013.1008.1169. Epub 2013 Aug 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao H, Ge Y, Zhuang S, Dworkin LD, Liu Z, Gong R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012;81:662–673. doi: 10.1038/ki.2011.443. doi:610.1038/ki.2011.1443. Epub 2012 Jan 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bao H, Ge Y, Wang Z, Zhuang S, Dworkin L, Peng A, Gong R. Delayed Administration of a Single Dose of Lithium Promotes Recovery from AKI. J Am Soc Nephrol. 2014;9:9. doi: 10.1681/ASN.2013040350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moore TD, Nawarskas JJ, Anderson JR. Eplerenone: a selective aldosterone receptor antagonist for hypertension and heart failure. Heart Dis. 2003;5:354–363. doi: 10.1097/01.hdx.0000089783.30450.cb. [DOI] [PubMed] [Google Scholar]
- 58.Armanini D, Karbowiak I, Goi A, Mantero F, Funder JW. In-vivo metabolites of spironolactone and potassium canrenoate: determination of potential anti-androgenic activity by a mouse kidney cytosol receptor assay. Clin Endocrinol (Oxf) 1985;23:341–347. doi: 10.1111/j.1365-2265.1985.tb01090.x. [DOI] [PubMed] [Google Scholar]
- 59.Ge RS, Dong Q, Sottas CM, Latif SA, Morris DJ, Hardy MP. Stimulation of testosterone production in rat Leydig cells by aldosterone is mineralocorticoid receptor mediated. Mol Cell Endocrinol. 2005;243:35–42. doi: 10.1016/j.mce.2005.08.004. Epub 2005 Sep 2026. [DOI] [PubMed] [Google Scholar]
- 60.Barfacker L, Kuhl A, Hillisch A, Grosser R, Figueroa-Perez S, Heckroth H, Nitsche A, Erguden JK, Gielen-Haertwig H, Schlemmer KH, et al. Discovery of BAY 94-8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. Chem Med Chem. 2012;7:1385–1403. doi: 10.1002/cmdc.201200081. [DOI] [PubMed] [Google Scholar]
- 61.Pitt B, Kober L, Ponikowski P, Gheorghiade M, Filippatos G, Krum H, Nowack C, Kolkhof P, Kim SY, Zannad F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J. 2013;34:2453–2463. doi: 10.1093/eurheartj/eht187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Azizi M, Amar L, Menard J. Aldosterone synthase inhibition in humans. Nephrol Dial Transplant. 2013;28:36–43. doi: 10.1093/ndt/gfs1388. Epub 2012 Oct 1098. [DOI] [PubMed] [Google Scholar]
- 63.Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-Induced Nephropathy in Obese Spontaneously Hypertensive Rats Via Paradoxical Activation of the Mineralocorticoid Receptor. Role of Oxidative Stress. Hypertension. 2007;17:17. doi: 10.1161/HYPERTENSIONAHA.107.091058. [DOI] [PubMed] [Google Scholar]
- 64.Han KH, Kang YS, Han SY, Jee YH, Lee MH, Han JY, Kim HK, Kim YS, Cha DR. Spironolactone ameliorates renal injury and connective tissue growth factor expression in type II diabetic rats. Kidney Int. 2006;70:111–120. doi: 10.1038/sj.ki.5000438. Epub 2006 May 2024. [DOI] [PubMed] [Google Scholar]
- 65.Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006;70:2116–2123. doi: 10.1038/sj.ki.5001854. Epub 2006 Oct 2111. [DOI] [PubMed] [Google Scholar]
- 66.Wang W, Li L, Zhou Z, Gao J, Sun Y. Effect of spironolactone combined with angiotensin-converting enzyme inhibitors and/or angiotensin II receptor blockers on chronic glomerular disease. Exp Ther Med. 2013;6:1527–1531. doi: 10.3892/etm.2013.1335. Epub 2013 Oct 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20:2641–2650. doi: 10.1681/ASN.2009070737. doi:2610.1681/ASN.2009070737. Epub 2009072009 Nov 2009070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giani M, Mastrangelo A, Villa R, Turolo S, Marra G, Tirelli AS, Hopfer H, Edefonti A. Alport syndrome: the effects of spironolactone on proteinuria and urinary TGF-beta1. Pediatr Nephrol. 2013;28:1837–1842. doi: 10.1007/s00467-013-2490-z. doi:1810.1007/s00467-00013-02490-z. Epub 02013 Jun 00411. [DOI] [PubMed] [Google Scholar]