

Abstract
Purpose: Tissue-engineered esophagus (TEE) may serve as a therapeutic replacement for absent foregut. Most prior esophagus studies have favored microdesigned biomaterials and yielded epithelial growth alone. None have generated human TEE with mesenchymal components. We hypothesized that sufficient progenitor cells might only require basic support for successful generation of murine and human TEE.
Materials and Methods: Esophageal organoid units (EOUs) were isolated from murine or human esophagi and implanted on a polyglycolic acid/poly-l-lactic acid collagen-coated scaffold in adult allogeneic or immune-deficient mice. Alternatively, EOU were cultured for 10 days in vitro prior to implantation.
Results: TEE recapitulated all key components of native esophagus with an epithelium and subjacent muscularis. Differentiated suprabasal and proliferative basal layers of esophageal epithelium, muscle, and nerve were identified. Lineage tracing demonstrated that multiple EOU could contribute to the epithelium and mesenchyme of a single TEE. Cultured murine EOU grew as an expanding sphere of proliferative basal cells on a neuromuscular network that demonstrated spontaneous peristalsis in culture. Subsequently, cultured EOU generated TEE.
Conclusions: TEE forms after transplantation of mouse and human organ-specific stem/progenitor cells in vivo on a relatively simple biodegradable scaffold. This is a first step toward future human therapies.
Introduction
Previous approaches to develop esophageal substitutes have resulted in partial tissues that do not contain all the key components required for a durable long-term conduit. Initially, investigators attempted to stent esophageal defects with nonabsorbable material, but these were often extruded by the host animal.1 Later techniques include wrapping absorbable cell-seeded scaffolds around nonabsorbable scaffold tubes and implanting them in vivo; however, this required the eventual removal of the nonabsorbable scaffold tube.2,3 Previously, the experimental model to address full-thickness esophageal tissue loss has been seeding an inert scaffold with epithelial cells. This approach has the benefit of simplicity and a singular cell source but cannot generate a replacement organ with all cell layers and functions. To meet such needs both epithelium and mesenchyme will be required. In other laboratories, scaffolds have been seeded with epithelial cells (oral mucosal or esophageal) and/or mesenchymal cells (fibroblasts or smooth muscle) from different donors.4 However, these chimeric approaches are cumbersome, costly, and do not readily translate.5,6 Furthermore, they do not provide other cell types known to be crucial to esophageal function, such as enteric nerves. An ideal source of tissue-engineered esophagus (TEE) would grow on an absorbable, biocompatible scaffold,1 be directly derived from the patient's own cells, and contain all cell types to retain the function of native esophagus.
We have previously reported an approach to tissue engineering the gastrointestinal tract in which multicellular clusters, or organoid units (OUs), are transplanted, resulting in the generation of regions from esophagus to colon in the Lewis rat.7–11 TEE was studied in a replacement model in the rat by experimental substitution by onlay patch or interposition to the rat's native esophagus.7 However, to investigate the regenerative mechanisms underpinning the tissue generation, we recently transitioned to the mouse model, which necessitated redesigning the scaffold for the smaller size of the recipient. Both tissue-engineered small intestine (TESI)12 and stomach13 were generated by this approach in the mouse. However, the esophagus has several key differences from the rest of the gastrointestinal tract. Unlike the known Lgr5 cells of the remaining intestine, the progenitor cells of the esophagus are unknown.14,15 The esophagus lacks a serosa, which is present throughout the rest of the intestine, and the embryologic origin is thought to be different. Large variations in the microenvironment are noted from mouth to anus, including pH, osmolality, absorption, and resident microbiota.16
Esophageal atresia occurs 1 in 2500 to 3000 live births,17 and esophageal adenocarcinoma is the fasting growing cancer in the United States with a rising incidence of 4–23 cases per million from 1975 to 2001.18 Esophageal replacement, including gastric transposition,19 colonic interposition,20 or jejunal interposition,21 is required in children with congenital esophageal anomalies when primary repair is impossible. Similar esophageal replacement strategies interposing various segments of the gastrointestinal tract are performed in adults after esophagectomy for neoplasia.22 Each of these techniques has disadvantages, such as anastomotic leak, stricture, reflux, neoplasia, and dysmotility, which may result in poor quality of life.17,22 Furthermore, reconstruction with stomach or intestine may not be possible in some patients due to prior surgery, infection, or anatomic variation.
TEE contains all of the key components of the native esophagus, including epithelium and mesenchyme. In contrast to current surgical replacements such as gastric tubes or colon interpositions, TEE is an attractive solution to esophageal lack or loss, particularly if generated from autologous cells. TEE could offer a near exact replacement with restoration of an appropriate, functional organ. Furthermore, because complex microdesigned biomaterials are not required, clinical translation, especially with regard to regulatory hurdles, may be simplified.
Materials and Methods
Animals
All experimental protocols were in accordance with and approved by the Institutional Animal Care and Use Committee. Animals were maintained in a temperature-regulated environment on a 12-h light–dark cycle and given access to chow and water ad libitum. All recipient mice were inspected daily for the duration of the study. Wild-type C57BL/6 mice were obtained from The Jackson Laboratory. NOD/SCID gamma mice were obtained from The Jackson Laboratory. ActinGFP23 and B6.Cg-Tg(CAG-mRFP1)1F1Hadj/J mice were maintained on a C57BL/6 background.
Scaffold construction
Tubular microporous biodegradable scaffolds were generated from nonwoven, highly porous, polyglycolic acid (PGA) felt (bulk density 50 mg/cm3, porosity >95%, 2 mm thickness Biofelt®; Concordia Medical, Warwick, RI). Scaffolds were formed on a glass mandril by sealing a 5×4 mm square piece of the PGA felt into a tube with 5% poly-l-lactic acid (PLLA) (Durect Corporation, Cupertino, CA) that was solubilized in chloroform (Sigma-Aldrich, St. Louis, MO). Two hundred microliters of PLLA was delivered onto the polymer secured to the mandril twice via pipet. After the polymers dried in a dessicator, they were sterilized with 100% ethanol. Finally, the polymer tubes were coated with collagen type I solution (0.4 mg/mL; Sigma-Aldrich) for 20 min at 4°C, rinsed with phosphate-buffered saline, and stored in a desiccator to avoid premature hydrolysis.
Esophageal organoid unit isolation
Nonfasted, <3-week-old, neonatal mice (both females and males) that were maintained on a C57BL/6 background were euthanized by exposure to carbon dioxide (CO2) in an inhalation chamber. The mouse esophagi were harvested and opened lengthwise with scissors. Human surgical samples were obtained as waste tissue after esophageal resection, collected at the time of surgery. All esophageal tissue was washed several times with 4°C Hank's balanced salt solution (Invitrogen, Carlsbad, CA) on ice, sedimenting between washes, to remove debris and mucous. Each lavaged esophagus was minced into <1 mm3 pieces with sterile scissors. Tissue fragments were enzymatically digested with 0.12 mg/mL dispase (Invitrogen) and 800 U/mL collagenase type 1 (Sigma-Aldrich) at 37°C on an orbital shaker for 20 min. Tissue fragments were further mechanically digested with a 10 mL pipet for trituration. Digestion was stopped with 4°C 4% sorbitol (Sigma-Aldrich), 10% fetal bovine serum (FBS) (Invitrogen) in high glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen). After centrifugation at 39 g for 10 min, the supernatant was poured off, the pellet resuspended in 4°C 10% FBS in DMEM, and then centrifuged at 100 g for 5 min. The resulting pellet contained the isolated OU, heterogeneous multicellular clusters of epithelium and mesenchyme. The esophageal organoid unit (EOU) generated from one 21-day-old mouse esophagus were sufficient to populate one scaffold to around the density of 105 EOU per cm2.
Implantation
EOU were seeded onto scaffolds, and adult syngeneic or NOD/SCID gamma mice (both females and males) served as recipients. NOD/SCID gamma mice were exposed to a single dose (350 cGy) of total body radiation immediately prior to implantation. Anesthesia was affected with inhaled 1–5% isoflurane. Through an upper midline vertical incision, the omentum was exposed. The seeded scaffold was wrapped completely in the omentum, secured with a 5-0 Monocryl suture (Ethicon, New Brunswick, NJ), and placed back into the abdominal cavity.24 The abdominal muscles and skin were closed with 4-0 Vicryl suture (Ethicon) in two layers. Postoperative pain was controlled with 2 mg/kg ketoprofen (Fort Dodge Animal Health, Fort Dodge, IA) subcutaneously administered immediately after implantation, and 2.5 mL ibuprofen (100 mg/5 mL; Major Pharmaceuticals, Livonia, MI) per 300 mL drinking water ad libitum for at least 3 days postoperatively. Drinking water was also supplemented with 3 mL of trimethoprim sulfamethoxazole (200 mg/40 mg/5 mL; Qualitest Pharmaceuticals, Inc., Huntsville, AL) per 300 mL for the duration of the postoperative period. The recipient animals were euthanized by exposure to CO2 in an inhalation chamber after 4 weeks for tissue harvest.
Lineage tracing
Interactions between epithelium and mesenchyme within TEE were further evaluated by lineage tracing. Instead of obtaining donor tissue from a single mouse strain, a combination of three breeds was pooled. In addition to standard C57BL/6, EOU from actinGFP and B6.Cg-Tg(CAG-mRFP1)1F1Hadj/J transgenic strains were also harvested. All three strains are maintained in our laboratory on a C57BL/6 background, and therefore can be transplanted into C57BL/6 hosts. Importantly, both transgenic strains expressed their respective fluorescent protein in a constitutive manner. This mixture of EOU was then pooled and loaded onto a single scaffold implanted into the omentum of a wild-type host and harvested at 4 weeks.
Culture
Isolated EOU were suspended in an equal volume of growth factor reduced Matrigel (BD Biosciences, Franklin Lakes, NJ) and evenly spread over the bottom of a six-well plate at a total of ∼2 mL per well. The suspension was solidified by incubation for 20 min at 37°C at which point medium consisting of DMEM with 10% FBS, 1× MEM nonessential amino acids (Life Technologies, Grand Island, NY) and 1× antibiotic-antimycotic (Life Technologies) was added. The EOU cultures were incubated at 37°C with 5% CO2 for 10 days with culture medium changed every other day. After 10 days the EOU were freed from the wells with a cell scraper and centrifuged at 39 g for 5 min. The pellet was then mounted in a liquid mold of low melting agarose (Gold Biotechnology, St. Louis, MO) for microscopic analyses. Alternatively, the cultured EOU were seeded onto a polymer scaffold and implanted, as above, to generate TEE.
Histology and immunofluorescence
EOU were fixed in 10% formalin overnight, resuspended in 3% agarose, and then embedded in paraffin. The TEE was fixed in 10% formalin and embedded in paraffin. Serial sections of all samples were cut at 5 μm thickness. Histology slides were stained with hematoxylin and eosin (H&E) per standard protocol. The remaining slides were prepared for immunofluorescence. These slides were dehydrated to water, and then an antigen retrieval step was performed by boiling the slides in a microwave for 12 min in 10 mM sodium citrate buffer pH=6.0. The slides were incubated with the primary antibody diluted in universal blocking solution with 2% goat serum overnight at 4°C. The primary antibodies included rabbit anti-cytokeratin 13 (CK13) (1:100; Abcam, Cambridge, MA), mouse anti-cytokeratin 4 (CK4) (1:100; Abcam), rabbit anti-keratin 14 (CK14) (1:100; NecMarkers, Fremont, CA), mouse anti-proliferating cell nuclear antigen (PCNA) (1:100; Vector Laboratories, Burlingame, CA), mouse Cy3-coupled anti-α-smooth muscle actin (SMA) (1:300; Sigma-Aldrich), mouse anti-desmin (1:50; Dako, Carpinteria, CA), rabbit anti-E-cadherin (1:100; Santa Cruz, Santa Cruz, CA), mouse anti-Tuj-1 (1:1000; Covance, Princeton, NJ), rabbit anti-RFP (1:100; Abcam), and mouse anti-GFP (1:100; Abcam). Corresponding Cy3, Cy5, or FITC-conjugated goat anti-rabbit or anti-mouse secondary antibodies were applied. Slides were mounted in Vectashield with DAPI (Vector Laboratories) as mounting medium. Adult murine esophagus and colon and human esophagus served as controls.
Results
TEE grows as an expanding sphere. EOU seeded onto biodegradable scaffolds and implanted into the omentum of syngeneic or irradiated NOD/SCID gamma mice grew in vivo over 4 weeks. At the time of tissue harvest, gross examination demonstrates a sphere of TEE (Fig. 1A) with a lumen filled with white-yellow mucous. One hundred percent of our harvested wild-type murine constructs, seven out of seven, grew TEE. On histologic examination, all implants regenerated an intact epithelium and mesenchyme. H&E staining of murine TEE demonstrates a keratinized stratified squamous epithelium at 4 weeks (Fig. 1C) similar to that of native esophagus (Fig. 1B). Keratin accumulates within the lumen over multiple layers of squamous cells, and adjacent smooth muscle cells organize into a rudimentary muscle layer below the epithelium. Similarly, human TEE (Fig. 3E, F) organizes around a central lumen and recapitulates a rudimentary squamous epithelium approaching the complexity of the donor human esophagus (Fig. 3A–C) from which it was generated. Often more than one area of developing epithelium with associated mesenchyme was present within each construct.
FIG. 1.
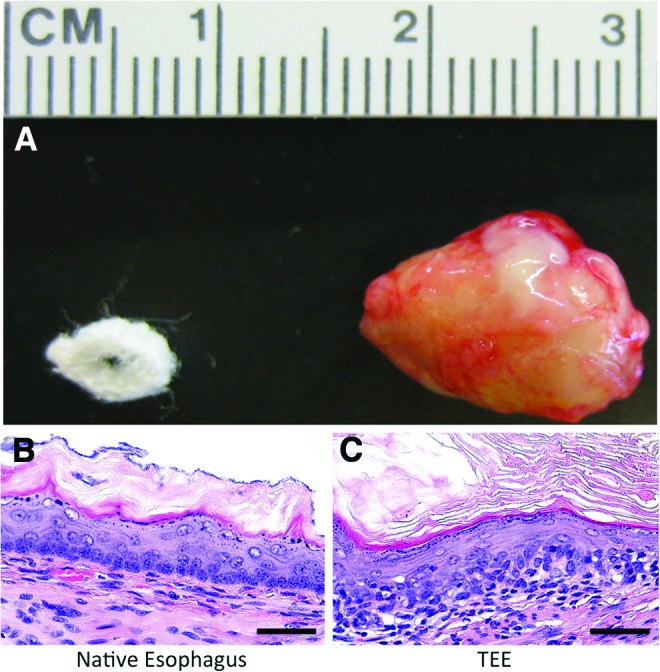
Tissue-engineered esophagus (TEE) grows as an expanding sphere. (A) Gross photograph of a biodegradable scaffold measuring 5×4×3 mm and 2 mm thick, and TEE harvested at 4 weeks measuring 14×10×7 mm. Hematoxylin and eosin staining demonstrates the keratinized stratified squamous epithelium of (B) native murine esophagus architecture well recapitulated in (C) murine TEE at 4 weeks including a subjacent muscularis (scale bars=50 μm). Color images available online at www.liebertpub.com/tea
FIG. 3.

Human TEE is composed of proliferating human esophageal epithelial cells. Native human esophagus (A–C) serves as a control in comparison to human TEE (D–E). Immunofluorescence staining for β-2-microglobulin (A, D) identifies human cells and confirms that all cells comprising human TEE are human, rather than of the murine host. Furthermore, E-cadherin (B, E) demonstrates that the flattened apical cells facing the dominant lumen of the human TEE are epithelial. CK14 (C, F) identifies basal esophageal cells in the human TEE, some of which also express PCNA (arrowheads), confirming their proliferative capacity. DAPI stains all nuclei blue. Scale bars=50 μm. Color images available online at www.liebertpub.com/tea
TEE demonstrates proper epithelial differentiation. To demonstrate normal esophageal epithelial differentiation, TEE specimens were analyzed for the presence of differentiated suprabasal cells and compared to native esophagus via immunofluorescence staining (Fig. 2). CK4 (Fig. 2A, E) and CK13 (Fig. 2B, F), known markers of differentiated suprabasal cells,25–27 stain cells throughout the upper layers of epithelium of all murine TEE specimens. To demonstrate that the epithelial proliferation in TEE was similar to that in native esophagus, immunohistochemistry was performed to detect expression of CK14, a known marker of proliferative basal cells,25–27 and PCNA. Proliferative basal cells with cytoplasmic staining for CK14 and nuclear staining for PCNA are identified in the correct location28 in both TEE (Fig. 2G, H) and native esophagus (Fig. 2C, D). This same pattern of proliferating cells is observed in human TEE (Fig. 3). Positive staining of human TEE for β-2-microglobulin (Fig. 3D) confirms that all cells arose from human donor origin, and consecutive sections are also found to be E-cadherin+ (Fig. 3B). This demonstrates that the flattened apical cells facing the dominant lumen are indeed epithelial. Finally, CK14 (Fig. 3F) identifies basal esophageal cells in the human TEE epithelium, some of which also express PCNA (Fig. 3D, arrowheads, inset) confirming their proliferative capacity.
FIG. 2.

TEE demonstrates epithelial differentiation and proliferation. (A–D) Native esophagus. (E–H) TEE harvested at 4 weeks. Positive staining for both cytokeratin 4 (A, E) and cytokeratin 13 (B, F) confirms the presence of differentiated suprabasal cells. Positive cytoplasmic staining for cytokeratin 14 (C, G) and positive nuclear staining for proliferating cell nuclear antigen (PCNA) (D, H) confirms the presence of proliferative basal cells. DAPI stains all nuclei blue. Scale bars=50 μm. Color images available online at www.liebertpub.com/tea
Subjacent to TEE epithelium a rudimentary muscularis is identified on H&E staining and confirmed with double immunofluorescence staining for SMA and desmin at 4 weeks (Fig. 4B), which resembles that of native esophagus (Fig. 4A). Additionally, cells staining positive for neuronal marker Tuj-1 (Fig. 4C) are also present at the same level, as would be expected with the innervated smooth muscle layers of native esophagus.
FIG. 4.
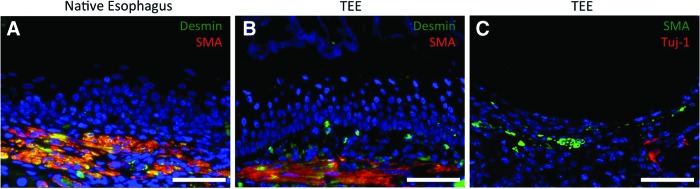
Murine TEE demonstrates a muscularis and nerves. The muscularis stains positive with alpha smooth muscle actin (SMA) and desmin in (A) native esophagus and (B) TEE harvested at 4 weeks. Nerve cells in TEE stain positive with Tuj-1. DAPI stains all nuclei blue. Scale bars=50 μm. Color images available online at www.liebertpub.com/tea
EOU are heterogenous multicellular clusters. H&E staining of the EOU just after isolation show mesenchyme in close contact with esophageal epithelium. The mesenchyme, as in native esophagus, is positive for both SMA and SMA/Desmin cells in the muscle layer while the epithelium is positive for CK4, 13, 14, and PCNA (data not shown).
By pooling EOU from different donors we sought to evaluate the conservation of initial intercellular relationships. While some TEE constructs were found to have epithelium and mesenchyme of the same donor origin, such as GFP+ epithelium and GFP+ mesenchyme (Fig. 5A) or RFP+ epithelium with RFP+ mesenchyme (Fig. 5B), others do not conserve this association. For example, in certain TEE the epithelium is of B6.Cg-Tg(CAG-mRFP1)1F1Hadj/J donor mouse origin while the underlying mesenchyme is of actinGFP origin (Fig. 5C). Still other murine TEE constructs demonstrate even greater heterogeneity in both compartments. In one example, there is a single continuous epithelial layer composed of GFP+ cells merging with RFP+ cells (Fig. 5D). This may indicate a fluid process with dynamic interactions between progenitors cells of different germ layers within developing TEE.
FIG. 5.

TEE demonstrates variable donor origin of epithelium and mesenchyme. (A–D) TEE was generated from pooled organoid units of actinGFP, B6.Cg-Tg(CAG-mRFP1)1F1Hadj/J, and wild-type donor mice. TEE lumen is oriented toward the top left corner of each image. Immunofluorescence costaining for green fluorescent protein (GFP) and red fluorescent protein (RFP) demonstrates conservation of original esophageal organoid unit (EOU) epithelial–mesenchymal relationships in some samples (A, B) where the epithelial and mesenchymal layers are of the same origin. Both the epithelial and mesenchymal layers in (A) stain positive for GFP, and in (B) both layers stain positive for RFP. However, other TEE samples (C, D) show variable donor origin of epithelium and mesenchyme. The TEE epithelium in (C) stains positive for RFP while its subjacent mesenchyme is GFP+. (D) Shows TEE with even greater heterogeneity of donor origin with an epithelial layer that is GFP+ on the left but RFP+ on the right and a mesenchyme of predominantly GPF+ with occasional RFP+ cells. DAPI stains all nuclei blue. Scale bars=50 μm. Color images available online at www.liebertpub.com/tea
EOU are capable of proliferating in vitro and do so without the support of exogenous growth factors. This robust system reliably produces expanding OU spheres of esophageal epithelium supported by a network of mesenchymal cells that carpet the dish and begin to envelop the growing organoid over the 10-day time course. High-resolution live microscopy of the EOU from an actinGFP murine donor imaged at 6 days demonstrates an expanding sphere with buds forming at the surface (Fig. 6A). Immunostaining of wild-type EOU at day 6 shows a dense ring of E-cadherin+ cells (Fig. 6B) consistent with an epithelial layer. Further analysis demonstrates that the cultured EOU is composed almost exclusively of PCNA+CK14+CK13−CK4− proliferative basal esophageal epithelial cells by the 6-day time point (Fig. 6C, D). The supportive layer of mesenchymal cells becomes more densely packed over the course of the culture, usually reaching confluence by day 4–6 and beginning to envelop the organoid by day 8–10. Live imaging microscopy of EOU culture of actinGFP murine cells reveals that both the expanding epithelial sphere and its mesenchymal support cells are GFP+ via native fluorescence without the need for immunostaining (Fig. 7A). Positive immunofluorescence staining for Tuj-1 and SMA in the same experiment identifies the surrounding mesenchyme as a complex network (Fig. 7B, C). This initially flat layer of cells is noted to be investing in and enveloping the three-dimensional sphere of esophageal epithelium. After 10 days in culture actinGFP EOU were loaded onto the polymer scaffold implanted into the omentum of a syngeneic, nonfluorescent, wild-type host mouse. The resultant TEE demonstrated the expected cellular structure, with a continuous layer of squamous cells facing a central lumen. Furthermore, these cells costain positive for GFP and E-cadherin E confirming that they represent an epithelium originating from the cultured actinGFP EOU (Fig. 7D).
FIG. 6.

Cultured EOUs are composed of proliferating basal esophageal epithelial cells. (A) EOU culture from an actinGFP murine donor imaged by live imaging microscopy after 6 days demonstrates an expanding sphere with buds forming at the surface. Native GFP fluorescence is appreciated without immunostaining. EOU culture after 6 days from C57 wild-type murine donor stains positive for epithelial marker E-cadherin (B). Positive cytoplasmic staining for cytokeratin 14 and nuclear staining for PCNA (C) confirms that the expanding esophageal sphere is predominantly composed of proliferative basal cells. Conversely, negative staining for both cytokeratin 4 and 13 (D) indicates that differentiated suprabasal cells are not present in culture after 6 days. DAPI stains all nuclei blue. Scale bars=25 μm. Color images available online at www.liebertpub.com/tea
FIG. 7.

EOU in culture grow as an expanding sphere supported by a robust network of nerve and SMA+ cells. (A) EOU culture from an actinGFP murine donor imaged by live imaging microscopy after 10 days demonstrates an expanding sphere supported by a network of mesenchymal support cells. Native GFP fluorescence is appreciated without immunostaining. (B, C) Positive immunofluorescence staining for Tuj-1 and SMA demonstrates a mesenchymal network of nerves and SMA+ cells supporting a murine EOU after 8 days in culture. (D) TEE generated from actinGFP EOU cultured in vitro for 10 days prior to implantation. E-cadherin staining demonstrates TEE epithelium that is GFP+ confirming donor origin. DAPI stains all nuclei blue. Scale bars=100 μm. Color images available online at www.liebertpub.com/tea
Additionally, we observe in vitro function of these mesenchymal components. During days 8–10 of EOU culture, the muscular fibers begin to contract spontaneously and induce neighboring cells to contract (Supplementary Video SV1; Supplementary Data are available online at www.liebertpub.com/tea). In wells where the mesenchyme completely envelops the esophageal epithelial sphere, this spontaneous contraction can be seen to propagate and results in squeezing of the EOU lumen (Supplementary Video SV2) in a fashion similar to peristalsis of native esophagus.
Discussion
Biomaterials approaches to tissue engineering various hollow structures such as the trachea, gastrointestinal tract, and components of the genitourinary system have evolved from nonabsorbable inert surfaces toward the inclusion of microdesign and biomimetics.
Among the imaginary countries in Gulliver's Travels, a satire written by Jonathan Swift in 1726, Lemuel Gulliver visits both Luggnagg and Lilliput.29 Luggnagg is remarkable for its immortal citizens while Lilliput is well known for recapitulating the world in miniature. While microdesign of transplanted scaffolds may possibly support the growth of engineered tissues, herein we demonstrate that a simple and versatile biodegradable polymer is sufficient for the growth of TEE as long as the correct progenitor population is provided. Luggnagg trumps Lilliput. TEE grows on a simple and versatile polyglycolic acid/poly-l-lactic acid (PGA/PLLA) collagen-coated scaffold, even from human donor cells. The resulting TEE contains all of the key components of the esophagus including mesenchymal structures such as nerve and muscle.
This study demonstrates the successful generation of mouse and human TEE for the first time to our knowledge. TEE was generated on a simple PGA/PLLA scaffold that is now proven effective across multiple tissue and species. Furthermore, it contains only materials likely to be acceptable to government regulatory bodies for rapid translation. Additionally, EOU contain adequate progenitor cells to survive in vitro culture with subsequent generation of TEE in vivo. Lineage tracing demonstrates a remarkable heterogeneity in the epithelial and mesenchymal relationships that are included in TEE, indicating that multiple combinations of cell populations allow subsequent formation of an engineered tissue. In our opinion this argues for supplying the necessary and sufficient progenitor cell population in regenerative medicine approaches and that microscale design of the delivery biomaterials may not be required.
The pursuit of viable clinical esophageal replacements that do not rely on native tissue substitutions has led to investigations in numerous animal models, including rat,5–7,28,30–32 dog,3,33–37 pig,38–40 rabbit,41 and sheep.2 In some experiments in which a defect was patched, it is unclear if the esophagus was actually engineered or if tissue ingrowth from healthy surrounding tissue bridged the defect. Few techniques have developed any mesenchyme in their attempts and none have identified nerve elements or generated human TEE. Most investigators have employed elaborate, costly scaffold systems and only concentrating on the generation of an epithelial layer. As a corollary, basic esophageal stem cell biology continues to be poorly understood, despite advancement in other fields of intestine research.
Murine TEE architecture resembles that of native esophagus and demonstrates an intact epithelium and mesenchyme (Fig. 1) and appropriate epithelial differentiation and proliferation. Immunohistochemistry confirms CK14−CK13+CK4+ cells comprising a differentiated suprabasal layer and a proliferative basal layer of PCNA+CK14+CK13−CK4− cells (Fig. 2). The mesenchyme shows a well-developed muscularis and also contains nerve (Fig. 2B, C), which has never been observed previously in TEE. Together these comprise a diverse mesenchymal support structure with the potential to develop a mature tissue-engineered enteric nervous system regulating the function of its tissue-engineered muscularis. Although present, the muscularis does not recapitulate the inner circular and outer longitudinal layers normally observed in the native esophagus. We have seen this in previous tissue-engineered regions of the intestine, with marked improvement after connection to luminal flow.
The ability to generate TEE from human esophageal cells in an immunosuppressed mouse host confirms the translational potential of tissue engineering in future human therapy. Previously the most complete efforts to engineer human esophagus had come from Hayashi et al. who cultured esophageal epithelial cells and smooth muscle cells in vitro with subsequent embedding of muscle cells onto collagen sheets and layering of epithelial cells on top.5 In distinction from such studies, our model produces full-thickness TEE (Fig. 1E) from a single cell source, EOU, and does not require in vitro manipulation of donor cells, a potential source of contamination and possible obstacle to clinical translation. Additionally, our TEE can be confirmed as human in origin by β-2-microglobulin (Fig. 3D) and demonstrates a proliferative basal layer of PCNA+CK14+ epithelial cells (Fig. 3F) as in native esophagus. These are key characteristics that will be required to generate a functional replacement organ for human translation.
The application of tissue engineering to esophagus not only serves as a potential source of tissue but also of knowledge, as a model of esophageal stem cell physiology does not yet exist. Our experience with rudimentary lineage tracing in TESI led us to believe that epithelial and mesenchymal cells in donor OU always maintained close intercellular contact during genesis of TESI.12 Despite the promise of recent observations that certain esophageal epithelial cell populations can switch behavior in response to injury42 there are no other robust models currently in place to study esophageal stem cell dynamics and potential niche interactions. There is a lack of knowledge regarding esophageal regeneration and epithelial maintenance, but TEE might not require maintenance of epithelial–mesenchymal relationships, thus expanding the pool of potential donor cells.
To date, all successful protocols for generating TESI have required both epithelium and mesenchyme in the transplanted OUs. Maintenance of the intestinal stem cell niche, including Paneth cells and mesenchymal cells such as intestinal subepithelial myofibroblasts, is believed to be critical to intestinal stem cell health.43 In support of this hypothesis we previously identified preserved epithelial–mesenchymal relationships during TESI formation.12 Based on our published observations in TESI,12 we expected to observe conservation of original epithelial–mesenchymal spatial relationships in TEE (Fig. 5A, B). Instead, we noted variable donor origin of epithelium and mesenchyme (Fig. 5C) and heterogeneity of donor origin within the same germ cell layer (Fig. 5D). This is contrary to data from analogous experiments in tissue-engineered intestine,12 and may indicate that esophageal progenitor cells are supported in multiple conformations. TEE formation is a dynamic process, and preservation of initial donor epithelial–mesenchymal cellular contact is not necessary for successful formation of TEE. This novel observation may enlarge the potential donor pool for engineered tissues and indicates that if pursuing the alternate strategy of microdesign of the transplanted biomaterials it may be difficult to predict which design is best as there seem to be multiple “correct” conformations.
While monoculture of esophageal epithelial cells is well described,44 it requires the addition of numerous expensive, exogenous growth factors, whereas cultured EOU grow in simple medium with 10% FBS. Furthermore, no in vitro system currently exists to study multicellular esophageal cellular interactions. Cultured murine EOU grow as expanding spheres of esophageal epithelium with active budding along the surface (Fig. 6A, B) supported by a network of mesenchymal cells. Predictably, the predominant cell type is the proliferative basal cell population (Fig. 6C); however, the discovery of this massive network of Tuj-1+ and SMA+ cells (Fig. 7B, C) supporting the epithelial spheres was not expected. In addition this neuromuscular network enveloped the epithelial sphere and began to demonstrate spontaneous contraction in culture without any external stimuli (Supplementary Video SV2). The automaticity and self-organization demonstrated functional mesenchymal components that may assist TEE to provide clinical functional replacement. Cultured EOU that survive and grow in vitro demonstrate the same diverse cell populations observed in TEE. In addition to the supportive network of mesenchymal cells (Fig. 7B, C), cultured EOU manifest an expanding epithelium (Fig. 7A, B) composed of proliferating basal esophageal cells (Fig. 7C). Subsequently, cultured EOU could generate TEE in vivo. GFP labeling confirmed donor origin (Fig. 7D). As a current in vitro model, EOU may facilitate the study of esophageal growth and neurogenesis to better understand both normal development and dysmotility disorders such as achalasia. Furthermore, in vitro manipulation and expansion of EOU prior to TEE generation could improve the yield and quality of resultant tissue for esophageal replacement. We hypothesize that, maintaining the intact cellular relationships between mesenchymal and epithelial layers provides the unidentified esophageal stem cells with the physical and biochemical signals to survive and proliferate, because we did not need to supply exogenous growth factors. This in vitro model may aid in defining mechanisms of esophageal disease such as esophageal neoplasia and dysmotility and perhaps allow better definition of an esophageal stem cell population.
The growth of TEE is efficient. One hundred percent of our harvested wild-type constructs, seven out of seven, grew TEE. This efficiency is similar to that observed in Lewis rats with a similar but larger and more dense PGA/PLLA scaffold in which 100% of the harvested constructs, eight out of eight, formed TEE from neonatal OU.7 Our efficiency with murine esophagus is similar (when accounting for the smaller sample size) to our results in murine TESI (89% of harvested implants, 39 out of 44)12 while tissue-engineered stomach appears to be less successful (50%, 15 out of 30, harvested implants).13 Although variable in percentage, the successful tissue engineering of multiple gastrointestinal segments on the same biomaterial highlights its versatility for several progenitor cell populations. This tissue engineering technique is simple and requires only one cell source, EOU, for all of the donor cells (epithelial, fibroblasts, smooth muscle, and nerve cells), unlike other more complex models2,3,5,6,28,30,36,37 that require cells from numerous sites or even different animals.
The main limitation of this murine TEE model is size and moderate surgical complexity. Small animal models limit the ability to perform experiments with autologous tissue. Although this technique in the mouse involves a syngeneic implantation, clinical application would demand autologous human EOU. This approach has been previously demonstrated in a preclinical Yorkshire swine model in which both TESI and tissue-engineered stomach grew from autologous OU.45 TEE will need to be generated in a large animal model to further investigate function and pretranslational qualification and quantification. In these models it will be possible to further investigate the function of the enteric nervous system identified in TEE and the in vitro culture of EOU.
This study demonstrates the successful generation of TEE in a mouse model, and is the first report of human TEE containing all the key components of the full-thickness tissue. TEE epithelial and mesenchymal layers are present and organized appropriately with proliferation and differentiation that is similar to that of native esophagus. In vitro culture of these multicellular esophageal organoids reveals an intricate network of functional neuromuscular support cells intimately associated with the growing epithelium. The ability to generate TEE from human esophageal cells in an immunosuppressed mouse host indicates possible translational potential, and further investigation into the mechanism of TEE formation is now possible. TEE grows on a simple biomaterial that could be acceptable to regulatory bodies. These are all necessary steps for future human therapies, and they argue for an approach to tissue engineering that focuses on identifying and transplanting the necessary and sufficient progenitor cell population.
Supplementary Material
Acknowledgment
This study was supported by grants from the California Institute for Regenerative Medicine (RN2-00946-1, TG2-01168).
Disclosure Statement
No competing financial interests exist.
References
- 1.Chen M.K., and Beierle E.A.Animal models for intestinal tissue engineering. Biomaterials 25,1675, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Saxena A.K., Baumgart H., Komann C., Ainoedhofer H., Soltysiak P., Kofler K., et al. Esophagus tissue engineering: in situ generation of rudimentary tubular vascularized esophageal conduit using the ovine model. J Pediatr Surg 45,859, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Nakase Y., Nakamura T., Kin S., Nakashima S., Yoshikawa T., Kuriu Y., et al. Intrathoracic esophageal replacement by in situ tissue-engineered esophagus. J Thorac Cardiovasc Surg 136,850, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Hodde J.Naturally occurring scaffolds for soft tissue repair and regeneration. Tissue Eng 8,295, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Hayashi K., Ando N., Ozawa S., Kitagawa Y., Miki H., Sato M., et al. A neo-esophagus reconstructed by cultured human esophageal epithelial cells, smooth muscle cells, fibroblasts, and collagen. ASAIO J 50,261, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Miki H., Ando N., Ozawa S., Sato M., Hayashi K., and Kitajima M.An artificial esophagus constructed of cultured human esophageal epithelial cells, fibroblasts, polyglycolic acid mesh, and collagen. ASAIO J 45,502, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Grikscheit T., Ochoa E.R., Srinivasan A., Gaissert H., and Vacanti J.P.Tissue-engineered esophagus: experimental substitution by onlay patch or interposition. J Thorac Cardiovasc Surg 126,537, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Grikscheit T., Srinivasan A., and Vacanti J.P.Tissue-engineered stomach: a preliminary report of a versatile in vivo model with therapeutic potential. J Pediatr Surg 38,1305, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Grikscheit T.C., Siddique A., Ochoa E.R., Srinivasan A., Alsberg E., Hodin R.A., et al. Tissue-engineered small intestine improves recovery after massive small bowel resection. Ann Surg 240,748, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grikscheit T.C., Ochoa E.R., Ramsanahie A., Alsberg E., Mooney D., Whang E.E., et al. Tissue-engineered large intestine resembles native colon with appropriate in vitro physiology and architecture. Ann Surg 238,35, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grikscheit T.C., Ogilvie J.B., Ochoa E.R., Alsberg E., Mooney D., and Vacanti J.P.Tissue-engineered colon exhibits function in vivo. Surgery 132,200, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Sala F.G., Matthews J.A., Speer A.L., Torashima Y., Barthel E.R., and Grikscheit T.C.A multicellular approach forms a significant amount of tissue-engineered small intestine in the mouse. Tissue Eng Part A 17,1841, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speer A.L., Sala F.G., Matthews J.A., and Grikscheit T.C.Murine tissue-engineered stomach demonstrates epithelial differentiation. J Surg Res 171,6, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Barker N., van Es J.H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449,1003, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Kushner J.A.Development. Esophageal stem cells, where art thou? Science 337,1051, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneeman B.O.Gastrointestinal physiology and functions. Br J Nutr 88(Suppl 2),S159, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Spitz L.Oesophageal atresia. Orphanet J Rare Dis 2,24, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pohl H., and Welch H.G.The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 97,142, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Spitz L., Kiely E., and Pierro A.Gastric transposition in children—a 21-year experience. J Pediatr Surg 39,276–281, 2004; discussion 276–281 [DOI] [PubMed] [Google Scholar]
- 20.Hamza A.F., Abdelhay S., Sherif H., Hasan T., Soliman H., Kabesh A., et al. Caustic esophageal strictures in children: 30 years' experience. J Pediatr Surg 38,828, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Bax N.M., and van der Zee D.C.Jejunal pedicle grafts for reconstruction of the esophagus in children. J Pediatr Surg 42,363, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Low D.E.Update on staging and surgical treatment options for esophageal cancer. J Gastrointest Surg 15,719, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Wright D.E., Cheshier S.H., Wagers A.J., Randall T.D., Christensen J.L., and Weissman I.L.Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood 97,2278, 2001 [DOI] [PubMed] [Google Scholar]
- 24.Barthel E.R., Speer A.L., Levin D.E., Sala F.G., Hou X., Torashima Y., et al. Tissue engineering of the intestine in a murine model. J Vis Exp e4279,2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalabis J., Oyama K., Okawa T., Nakagawa H., Michaylira C.Z., Stairs D.B., et al. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. J Clin Invest 118,3860, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Squier C.A., and Kremer M.J.Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr 7,2001 [DOI] [PubMed] [Google Scholar]
- 27.Green N., Huang Q., Khan L., Battaglia G., Corfe B., MacNeil S., et al. The development and characterization of an organotypic tissue-engineered human esophageal mucosal model. Tissue Eng Part A 16,1053, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Saxena A.K., Ainoedhofer H., and Hollwarth M.E.Esophagus tissue engineering: in vitro generation of esophageal epithelial cell sheets and viability on scaffold. J Pediatr Surg 44,896, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Swift J., and Swift JGST.Travels into several Remote Nations of the World. In four parts. By Lemuel Gulliver, first a surgeon, and then a captain of several ships. [By Swift Jonathan. With plates.]. London: Benj, Motte, 1726 [Google Scholar]
- 30.Saxena A.K., Kofler K., Ainodhofer H., and Hollwarth M.E.Esophagus tissue engineering: hybrid approach with esophageal epithelium and unidirectional smooth muscle tissue component generation in vitro. J Gastrointest Surg 13,1037, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Urita Y., Komuro H., Chen G., Shinya M., Kaneko S., Kaneko M., et al. Regeneration of the esophagus using gastric acellular matrix: an experimental study in a rat model. Pediatr Surg Int 23,21, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Lopes M.F., Cabrita A., Ilharco J., Pessa P., Paiva-Carvalho J., Pires A., et al. Esophageal replacement in rat using porcine intestinal submucosa as a patch or a tube-shaped graft. Dis Esophagus 19,254, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Ohki T., Yamato M., Murakami D., Takagi R., Yang J., Namiki H., et al. Treatment of oesophageal ulcerations using endoscopic transplantation of tissue-engineered autologous oral mucosal epithelial cell sheets in a canine model. Gut 55,1704, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isch J.A., Engum S.A., Ruble C.A., Davis M.M., and Grosfeld J.L.Patch esophagoplasty using AlloDerm as a tissue scaffold. J Pediatr Surg 36,266, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Badylak S., Meurling S., Chen M., Spievack A., and Simmons-Byrd A.Resorbable bioscaffold for esophageal repair in a dog model. J Pediatr Surg 35,1097, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Badylak S.F., Vorp D.A., Spievack A.R., Simmons-Byrd A., Hanke J., Freytes D.O., et al. Esophageal reconstruction with ECM and muscle tissue in a dog model. J Surg Res 128,87, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Wei R.Q., Tan B., Tan M.Y., Luo J.C., Deng L., Chen X.H., et al. Grafts of porcine small intestinal submucosa with cultured autologous oral mucosal epithelial cells for esophageal repair in a canine model. Exp Biol Med 234,453, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Komuro H., Nakamura T., Kaneko M., Nakanishi Y., and Shimizu Y.Application of collagen sponge scaffold to muscular defects of the esophagus: an experimental study in piglets. J Pediatr Surg 37,1409, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Kajitani M., Wadia Y., Hinds M.T., Teach J., Swartz K.R., and Gregory K.W.Successful repair of esophageal injury using an elastin based biomaterial patch. ASAIO J 47,342, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Marzaro M., Vigolo S., Oselladore B., Conconi M.T., Ribatti D., Giuliani S., et al. In vitro and in vivo proposal of an artificial esophagus. J Biomed Mater Res A 77,795, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Lynen Jansen P., Klinge U., Anurov M., Titkova S., Mertens P.R., and Jansen M.Surgical mesh as a scaffold for tissue regeneration in the esophagus. Eur Surg Res 36,104, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Doupé D.P., Alcolea M.P., Roshan A., Zhang G., Klein A.M., Simons B.D., et al. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 337,1091, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King S.L., and Dekaney C.M.Small intestinal stem cells. Curr Opin Gastroenterol 29,140, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Saxena A.K., Ainoedhofer H., and Höllwarth M.E.Culture of ovine esophageal epithelial cells and in vitro esophagus tissue engineering. Tissue Eng Part C Methods 16,109, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Sala F.G., Kunisaki S.M., Ochoa E.R., Vacanti J., and Grikscheit T.C.Tissue-engineered small intestine and stomach form from autologous tissue in a preclinical large animal model. J Surg Res 156,205, 2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.