

Crystals of a complex between a fragment of TssM obtained by limited proteolysis and a nanobody have been obtained and diffracted to 1.9 Å resolution.
Keywords: type VI secretion system, TssM, nb25
Abstract
The type VI secretion system (T6SS) is a machine evolved by Gram-negative bacteria to deliver toxin effectors into target bacterial or eukaryotic cells. The T6SS is functionally and structurally similar to the contractile tail of the Myoviridae family of bacteriophages and can be viewed as a syringe anchored to the bacterial membrane by a transenvelope complex. The membrane complex is composed of three proteins: the TssM and TssL inner membrane components and the TssJ outer membrane lipoprotein. The TssM protein is central as it interacts with both TssL and TssJ, therefore linking the membranes. Using controlled trypsinolysis, a 32.4 kDa C-terminal fragment of enteroaggregative Escherichia coli TssM (TssM32Ct) was purified. A nanobody obtained from llama immunization, nb25, exhibited subnanomolar affinity for TssM32Ct. Crystals of the TssM32Ct–nb25 complex were obtained and diffracted to 1.9 Å resolution. The crystals belonged to space group P64, with unit-cell parameters a = b = 95.23, c = 172.95 Å. Molecular replacement with a model nanobody indicated the presence of a dimer of TssM32Ct–nb25 in the asymmetric unit.
1. Introduction
The type VI secretion system (T6SS) is one of the key players during the intense warfare for nutrients that bacteria encounter in their ecological niche (Russell et al., 2014 ▶). This large multi-protein complex is widespread in Gram-negative bacteria and is dedicated to the delivery of enzymatic effectors directly into bacterial or eukaryotic prey cells (Russell et al., 2014 ▶; Durand et al., 2014 ▶). Among the effectors that have already been identified and characterized, peptidoglycan hydrolases, DNases and phospholipases are the most common (Russell et al., 2014 ▶; Durand et al., 2014 ▶). Although T6SS phospholipase and DNase effectors might also affect the integrity of eukaryotic cells, eukaryotic specific effectors have been described such as the VgrG-borne actin cross-linking domain in Vibrio cholerae (Pukatzki et al., 2007 ▶; Satchell, 2009 ▶; Durand, Derrez et al., 2012 ▶).
The T6SS comprises a set of 13 conserved structural components that participate in the architecture and dynamics of the secretion system (Cascales & Cambillau, 2012 ▶; Coulthurst, 2013 ▶; Ho et al., 2014 ▶; Zoued et al., 2014 ▶). Architecturally, the T6SS can be seen as a syringe-like module tethered to the cell envelope through contacts with a membrane-associated complex composed of the TssJ, TssL and TssM proteins (Cascales & Cambillau, 2012 ▶; Zoued et al., 2014 ▶). In several instances such as in enteroaggregative Escherichia coli (EAEC), an additional component, TagL, mediates anchoring of this complex to the peptidoglycan layer (Aschtgen, Gavioli et al., 2010 ▶; Aschtgen, Thomas et al., 2010 ▶). TagL, TssL and TssM are all inner membrane proteins, whereas TssJ is an outer membrane lipoprotein (Aschtgen et al., 2008 ▶, 2012 ▶; Ma et al., 2009 ▶; Aschtgen, Thomas et al., 2010 ▶; Felisberto-Rodrigues et al., 2011 ▶; Durand, Zoued et al., 2012 ▶). In EAEC, TssM (accession No. EC042_4539; GenBank gi:284924260) is a large, 1129-amino-acid protein bearing a ∼750-residue periplasmic domain. In addition to making contacts with the TssL subunit in the inner membrane, the C-terminal region of the TssM protein interacts with the TssJ outer membrane lipoprotein (Felisberto-Rodrigues et al., 2011 ▶). Three-dimensional structures of the soluble domains of TagL, TssJ and TssL have recently been reported (Aschtgen, Gavioli et al., 2010 ▶; Felisberto-Rodrigues et al., 2011 ▶; Rao et al., 2011 ▶; Durand, Zoued et al., 2012 ▶; Robb et al., 2012 ▶). However, although TssM is of the utmost importance as it constitutes the scaffold of the T6SS membrane-associated complex, structural information on this component is still lacking. To facilitate the crystallization of TssM, we have raised camelid antibodies against the periplasmic domain of EAEC TssM (amino acids 386–1129; hereafter called TssMp). Camelid antibodies differ from conventional antibodies as they harbour unique variable heavy, antigen-binding domains called VHH domains or nanobodies (Hamers-Casterman et al., 1993 ▶; Muyldermans, 2013 ▶). These single-chain antibodies are easy to produce and have demonstrated their efficiency in improving the crystallization of awkward proteins (Pardon et al., 2014 ▶). In parallel, we have subjected TssMp to controlled enzymatic digestion to obtain stable structural domains that are more prone to crystallization. We report here that a combination of these two approaches is successful: we obtained crystals of a C-terminal fragment of TssMp in complex with a nanobody at a resolution sufficient for structure determination
2. Materials and methods
2.1. Production, controlled trypsinolysis and purification of TssM32Ct
The periplasmic domain of the enteroaggregative E. coli TssM protein (EAEC_042_4539; gi:284924260), TssMp (residues 386–1129), was produced and purified as described previously (Felisberto-Rodrigues et al., 2011 ▶). The purified recombinant TssMp was digested with trypsin [1000:1(m:m)] at room temperature for 24 h. The reaction was quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (PMSF) and insoluble TssMp fragments were discarded by centrifugation at 20 000g for 30 min. A proteolysis-resistant fragment of apparent size ∼32 kDa (hereafter called TssM32Ct) was further purified by consecutive ion-exchange (Mono Q 5/50 GL column; GE Healthcare) and size-exclusion (Superdex 75 16/600 HL column) chromatography using an ÄKTA system (GE Healthcare). The purified fragment was subjected to N-terminal Edman sequencing. A PVDF membrane was rinsed three times with a water/ethanol mixture (10:90) and inserted into the A cartridge of a Procise 494A sequencer. After five cycles of Edman degradation, the sequence DYGSL was identified, indicating that cleavage after Arg834 generated a C-terminal fragment of theoretical mass 32 398 Da, in agreement with the 32 kDa band observed on SDS–PAGE.
2.2. Generation of nanobodies against TssM
Four injections of 1 mg purified TssMp (in 20 mM Tris–HCl pH 8.0, 150 mM NaCl) were performed subcutaneously at two-week intervals followed by a fifth injection one month later in two llamas (Lama glama; Capralogics Inc., Hardwick, Masachusetts, USA). Lymphocytes were isolated from blood samples obtained one week after the last immunization. The cDNA was synthesized from purified total RNA by reverse transcription and was used as a template for PCR amplification to amplify the sequences corresponding to the variable domains of the heavy-chain antibodies. PCR fragments were then cloned into the phagemid vector pHEN4 (Pardon et al., 2014 ▶) to create a nanobody phage display library. Selection and screening of nanobodies were performed as described previously (Desmyter et al., 2013 ▶). Three rounds of panning resulted in the isolation of TssMp-specific binders. Nanobodies nb02 and nb25 were selected, sequenced and cloned into the pHEN6 expression vector downstream of the pelB signal peptide and fused to a C-terminal 6×His tag (Conrath et al., 2009 ▶).
2.3. Production and purification of nanobodies
E. coli WK6 cells carrying the pHEN6 derivatives were grown at 37°C in Terrific Broth medium containing 0.1% glucose and ampicillin to an optical density (600 nm) of ∼0.8. Expression of VHH was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubation for 16 h at 28°C. The periplasmic fraction containing the nanobodies was prepared by osmotic shock. The His-tail-containing fusion proteins were purified by immobilized metal-affinity chromatography on a 5 ml Ni–NTA column equilibrated in 20 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole. The fractions eluted in 250 mM imidazole were concentrated on an Amicon Ultra 10 kDa cutoff concentrator prior to loading onto a HiLoad 16/60 Superdex 75 gel-filtration column equilibrated in 20 mM Tris–HCl pH 8.0, 150 mM NaCl buffer.
2.4. Bio-layer interferometry (BLI)
Nanobody nb25 was first biotinylated using the EZ-Link NHS-PEG4-Biotin kit (Perbio Science, France). The reaction was stopped by removing excess biotin using a Zeba Spin Desalting column (Perbio Science, France). BLI studies were performed in black 96-well plates (Greiner) at 25°C using an OctetRed96 (ForteBio, USA). Streptavidin biosensor tips (ForteBio, USA) were hydrated with 0.2 ml kinetic buffer (KB; ForteBio, USA) for 20 min and then coupled to biotinylated nb25 (10 µg ml−1 in KB). The association of nb25 with various concentrations of TssM32Ct (0.13, 0.4, 1.22, 3.66, 11 and 33 nM) was recorded for a period of 600 s followed by monitoring the kinetics of dissociation in KB for 900 s.
2.5. Crystallization, data collection and molecular replacement
Crystallization trials were initiated with the TssM32Ct fragment and the nb25 nanobody. The purified His-tagged nanobody (10 mg ml−1) was mixed with TssM32Ct (1 mg ml−1) in a 1:1 ratio and the TssM32Ct–nb25 complex was purified by gel filtration on a Superdex 75 16/60 HL column using an ÄKTA purifier (GE Healthcare). The complex was concentrated to 11.5 mg ml−1 using a 10 kDa cutoff Centricon in 20 mM Tris–HCl pH 8.0, 150 mM NaCl and subjected to crystallization screening with a Mosquito robot (TTP Labtech) using various commercial kits. Drops were prepared by mixing different volumes (100, 200 and 300 nl) of the TssM32Ct–nb25 complex solution and 100 nl precipitant solution, and were equilibrated against a 150 µl reservoir volume in Greiner plates. Promising hits were obtained with 20%(m/v) PEG 4000, 0.2 M ammonium sulfate. These crystals were subjected to grid optimization using 10–30%(m/v) PEG 4000, 0.1–0.3 M ammonium sulfate. The best conditions consisted of 18–20% PEG 4000, 0.25 M ammonium sulfate.
TssM32Ct–nb25 crystals were briefly soaked in crystallization solution supplemented with 20%(v/v) glycerol before being flash-cooled in a nitrogen-gas stream at 100 K. An X-ray data set was collected from a single crystal of TssM32Ct–nb25 on an in-house rotating-anode generator (Brucker MicroStar) with a MAR345 image-plate detector. An additional data set was collected on the PROXIMA1 beamline at SOLEIL, Saint Aubin, France with a PILATUS 6M detector (Table 1 ▶). 1000 images were collected with an oscillation step of 0.20° and 0.4 s exposure time. The data were integrated, scaled and merged using the XDS package (Kabsch, 2010 ▶). The merged intensities were converted to structure-factor amplitudes using TRUNCATE from the CCP4 package (Winn et al., 2011 ▶). Molecular replacement was performed using nanobody structures with high sequence similarity to nb25. As starting models for MOLREP (Vagin & Teplyakov, 2010 ▶), we successively used three nanobodies with more than 70% sequence similarity to nb25 (PDB entries 3stb, 4nc2 and 4hem; Park et al., 2012 ▶; Murase et al., 2014 ▶; Desmyter et al., 2013 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the outer shell.
| Beamline | PROXIMA1, SOLEIL |
| Wavelength () | 0.931 |
| Detector | PILATUS 6M |
| Space group | P64 |
| Unit-cell parameters () | a = b = 95.2, c = 172.9 |
| Resolution range () | 50.01.92 (1.971.92) |
| R merge | 0.079 (1.08) |
| CC1/2 | 99.9 (84) |
| Total No. of reflections | 771552 (55929) |
| No. of unique reflections | 67557 (4957) |
| Completeness (%) | 100.0 (100.0) |
| Multiplicity | 11.4 (11.3) |
| I/(I) | 18.0 (2.0) |
| B factor from Wilson plot (2) | 37.1 |
| MOLREP contrast with 3stb † | 7.1, 6.5 [3.1] |
| MOLREP contrast with 4nc2 † | 6.9, 6.2 [3.2] |
| MOLREP contrast with 4hem † | 6.5, 5.9 [3.0] |
The contrast of the next solution is given in square brackets.
3. Results and discussion
TssM is a central component of the membrane-anchoring complex of the type VI secretion system. Topology studies of TssM showed that this protein is composed of a short N-terminal cytoplasmic domain followed by three transmembrane segments and a large periplasmic domain (Ma et al., 2009 ▶). The C-terminal fragment of this periplasmic domain contacts the TssJ outer membrane lipoprotein (Felisberto-Rodrigues et al., 2011 ▶) and therefore TssM bridges the inner and outer membranes. Although the TssM periplasmic domain (TssMp) readily purified and behaved as a monomer (Felisberto-Rodrigues et al., 2011 ▶), crystallization screening was unsuccessful. Here, we have subjected TssMp to limited proteolysis with the goal of selecting stable structural domains that are more prone to crystallization. Interestingly, SDS–PAGE analyses showed that a stable fragment with an apparent molecular mass of ∼32 kDa resisted digestion with trypsin [1000:1(m:m)] at room temperature for 24 h (Fig. 1 ▶). This fragment, TssM32Ct, starts at residue Asp835 as determined by Edman sequencing and corresponds to the C-terminal region of TssMp. To further facilitate the crystallization process, we selected TssMp-specific camelid antibodies after the immunization of llamas with TssMp. Among the four anti-TssMp nanobodies, nanobody nb25 binds to TssM32Ct as shown by size-exclusion chromatography (Fig. 2 ▶). The binding constant was determined by bio-layer interferometry (BLI) using an OctetRed96 apparatus (ForteBio). Analysis of BLI curves indicated that nb25 binds to TssM32Ct with a K d value of 0.88 ± 0.005 nM (Fig. 3 ▶).
Figure 1.

Isolation of the TssM32Ct fragment by controlled trypsinolysis. Fragments of purified TssMp (lane 1) subjected to trypsin digestion at room temperature for 2, 5, 10, 20, 40, 80 min and 20 h (lanes 2–8, respectively) were analysed by SDS–PAGE with Coomassie Blue staining. The molecular-weight markers are shown in lane 9 and are labelled in kDa. The full-length TssM periplasmic domain (TssMp) and the trypsinolysed fragment TssM32Ct are indicated.
Figure 2.

TssM32Ct–nb25 complex formation. (a) Amino-acid sequences of TssM32Ct and nb25. The cleavage site in TssM is indicated by a triangle. The CDRs of nb25 are coloured blue, green and red for CDRs 1, 2 and 3, respectively. The nb25 cysteines are coloured orange. (b) Gel-filtration analysis of TssM32Ct (red) and the TssM32Ct–nb25 complex (blue). The volume of elution is indicated above each peak. (c) Selected fractions were analyzed by SDS–PAGE. Molecular-weight markers are shown in lane 1 (labelled in kDa). Lanes 2 and 3 correspond to the insoluble fraction (pellet) and the soluble fraction (supernatant) obtained after the digestion of TssMp with trypsin, respectively, lanes 7–10 correspond to fractions at 50–63 ml and lane 13 corresponds to the fractions centred at 81.6 ml. The peak at 56.66 ml therefore corresponds to the TssM32Ct–nb25 complex, while the peak at 81.63 ml corresponds to excess nb25.
Figure 3.
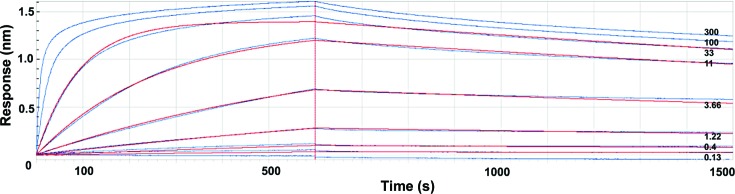
Bio-layer interferometry analysis of the TssM32Ct–nb25 interaction. The chip was covered with biotinylated nb25 nanobody and the responses after injection of various concentrations of TssM32Ct (0.13, 0.4, 1.22, 3.66, 11, 33, 100 and 300 nM) were recorded (blue curves). The response (in nanometres) is plotted versus the time (in seconds). The k on and k off analysis (red curves) excluded the outlier values obtained at 100 and 300 nM. The values of k on and k off are 3.26 × 105 and 2.4 × 10−4, respectively, and K d is 0.88 ± 0.005 nM.
Crystals of the TssM32Ct–nb25 complex grew after 20 d from a protein solution concentrated to 11.5 mg ml−1 in 20 mM Tris–HCl pH 8.0, 150 mM NaCl with precipitation conditions consisting of 20% PEG 4000, 0.25 M ammonium sulfate. The dimensions of the crystals were 0.04 × 0.04 × 0.06 mm (Fig. 4 ▶). The crystals belonged to space group P64, with unit-cell parameters a = b = 95.2, c = 172.9 Å. With a complex molecular weight of 47.1 kDa, a Matthews coefficient of 2.41 Å3 Da−1 (49% solvent) corresponds to the presence of two molecules in the asymmetric unit.
Figure 4.

Representative crystal of the TssM32Ct–nb25 complex.
A complete data set was collected using a PILATUS 6M detector on the PROXIMA1 beamline at SOLEIL, Saint-Aubin, France, yielding a resolution of 1.9 Å and excellent statistics (Table 1 ▶). Owing to the uncertainty in the asymmetric unit content, we performed molecular-replacement assays with MOLREP (Vagin & Teplyakov, 2010 ▶) using three different nanobodies sharing high sequence homology with nb25 (PDB entries 3stb, 4nc2 and 4hem). All three molecular-replacement attempts returned a common outcome with two nanobodies in the asymmetric unit (Table 1 ▶). The crystal packing (Fig. 5 ▶) is compatible with the presence of two TssM32Ct molecules bound to the two nanobodies. Further phasing experiments with SeMet labelling are under way to solve the structure of the complex. This information will be of great interest in order to better understand the T6SS architecture, as TssM32Ct corresponds to a conserved domain that mediates interaction with the TssJ outer membrane lipoprotein.
Figure 5.

Crystal packing of nb25 in the TssM32Ct–nb25 complex after molecular replacement. Two orientations rotated by 90° from each other are presented. The two nanobodies in the asymmetric unit are identified by white arrows and are rainbow-coloured (views were generated using Coot; Emsley & Cowtan, 2004 ▶).
Acknowledgments
We thank the SOLEIL synchrotron for data-collection time and the PROXIMA1 beamline staff for assistance. This work was supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01.
References
- Aschtgen, M.-S., Bernard, C. S., De Bentzmann, S., Lloubès, R. & Cascales, E. (2008). J. Bacteriol. 190, 7523–7531. [DOI] [PMC free article] [PubMed]
- Aschtgen, M.-S., Gavioli, M., Dessen, A., Lloubès, R. & Cascales, E. (2010). Mol. Microbiol. 75, 886–889. [DOI] [PubMed]
- Aschtgen, M.-S., Thomas, M. S. & Cascales, E. (2010). Virulence, 1, 535–540. [DOI] [PubMed]
- Aschtgen, M.-S., Zoued, A., Lloubès, R., Journet, L. & Cascales, E. (2012). MicrobiologyOpen, 1, 71–82. [DOI] [PMC free article] [PubMed]
- Cascales, E. & Cambillau, C. (2012). Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1102–1111. [DOI] [PMC free article] [PubMed]
- Conrath, K., Pereira, A. S., Martins, C. E., Timóteo, C. G., Tavares, P., Spinelli, S., Kinne, J., Flaudrops, C., Cambillau, C., Muyldermans, S., Moura, I., Moura, J. J. G., Tegoni, M. & Desmyter, A. (2009). Protein Sci. 18, 619–628. [DOI] [PMC free article] [PubMed]
- Coulthurst, S. J. (2013). Res. Microbiol. 164, 640–654. [DOI] [PubMed]
- Desmyter, A., Farenc, C., Mahony, J., Spinelli, S., Bebeacua, C., Blangy, S., Veesler, D., van Sinderen, D. & Cambillau, C. (2013). Proc. Natl Acad. Sci. USA, 110, E1371–E1379. [DOI] [PMC free article] [PubMed]
- Durand, E., Cambillau, C., Cascales, E. & Journet, L. (2014). Trends Microbiol. 22, 498–507. [DOI] [PubMed]
- Durand, E., Derrez, E., Audoly, G., Spinelli, S., Ortiz-Lombardia, M., Raoult, D., Cascales, E. & Cambillau, C. (2012). J. Biol. Chem. 287, 38190–38199. [DOI] [PMC free article] [PubMed]
- Durand, E., Zoued, A., Spinelli, S., Watson, P. J., Aschtgen, M. S., Journet, L., Cambillau, C. & Cascales, E. (2012). J. Biol. Chem. 287, 14157–14168. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Felisberto-Rodrigues, C., Durand, E., Aschtgen, M. S., Blangy, S., Ortiz-Lombardia, M., Douzi, B., Cambillau, C. & Cascales, E. (2011). PLoS Pathog. 7, e1002386. [DOI] [PMC free article] [PubMed]
- Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hammers, C., Songa, E. B., Bendahman, N. & Hammers, R. (1993). Nature (London), 363, 446–448. [DOI] [PubMed]
- Ho, B. T., Dong, T. G. & Mekalanos, J. J. (2014). Cell Host Microbe, 15, 9–21. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Ma, L.-S., Lin, J.-S. & Lai, E.-M. (2009). J. Bacteriol. 191, 4316–4329. [DOI] [PMC free article] [PubMed]
- Murase, T., Eugenio, L., Schorr, M., Hussack, G., Tanha, J., Kitova, E. N., Klassen, J. S. & Ng, K. K. S. (2014). J. Biol. Chem. 289, 2331–2343. [DOI] [PMC free article] [PubMed]
- Muyldermans, S. (2013). Annu. Rev. Biochem. 82, 775–797. [DOI] [PubMed]
- Pardon, E., Laeremans, T., Triest, S., Rasmussen, S. G., Wohlkönig, A., Ruf, A., Muyldermans, S., Hol, W. G. J., Kobilka, B. K. & Steyaert, J. (2014). Nature Protoc. 9, 674–693. [DOI] [PMC free article] [PubMed]
- Park, Y.-J., Pardon, E., Wu, M., Steyaert, J. & Hol, W. G. J. (2012). Nucleic Acids Res. 40, 1828–1840. [DOI] [PMC free article] [PubMed]
- Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D. & Mekalanos, J. J. (2007). Proc. Natl Acad. Sci. USA, 104, 15508–15513. [DOI] [PMC free article] [PubMed]
- Rao, V. A., Shepherd, S. M., English, G., Coulthurst, S. J. & Hunter, W. N. (2011). Acta Cryst. D67, 1065–1072. [DOI] [PMC free article] [PubMed]
- Robb, C. S., Nano, F. E. & Boraston, A. B. (2012). J. Mol. Biol. 419, 277–283. [DOI] [PubMed]
- Russell, A. B., Peterson, S. B. & Mougous, J. D. (2014). Nature Rev. Microbiol. 12, 137–148. [DOI] [PMC free article] [PubMed]
- Satchell, K. J. (2009). Toxins, 1, 123–133. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zoued, A., Brunet, Y. R., Durand, E., Aschtgen, M.-S., Logger, L., Douzi, B., Journet, L., Cambillau, C. & Cascales, E. (2014). Biochim. Biophys. Acta, 1843, 1664–1673. [DOI] [PubMed]