

The structure of the dodecamer of the aminopeptidase APDkam598 from the archaeon D. kamchatkensis was determined using X-ray diffraction data to a resolution of 3.0 Å and single-particle electron microscopy.
Keywords: aminopeptidase, APDkam598, Desulfurococcus kamchatkensis, thermostability
Abstract
The crystal structure of the aminopeptidase APDkam589 from the thermophilic crenarchaeon Desulfurococcus kamchatkensis was determined at a resolution of 3.0 Å. In the crystal, the monomer of APDkam589 and its symmetry-related monomers are densely packed to form a 12-subunit complex. Single-particle electron-microscopy analysis confirms that APDkam589 is present as a compact dodecamer in solution. The APDkam589 molecule is built similarly to the molecules of the PhTET peptidases, which have the highest sequence identity to APDkam589 among known structures and were isolated from the more thermostable archaeon Pyrococcus horikoshii. A comparison of the interactions of the subunits in APDkam589 with those in PhTET1, PhTET2 and PhTET3 reveals that APDkam589 has a much lower total number of salt bridges, which correlates with the lower thermostability of APDkam589. The monomer of APDkam589 has six Trp residues, five of which are on the external surface of the dodecamer. A superposition of the structure of APDkam589 with those having a high sequence similarity to APDkam589 reveals that, although the positions of Trp45, Trp252 and Trp358 are not conserved in the sequences, the spatial locations of the Trp residues in these models are similar.
1. Introduction
The gene Dkam_0589 encoding APDkam598 was isolated from the genome of the anaerobic hyperthermophilic crenarchaeon Desulfurococcus kamchatkensis (Ravin et al., 2009 ▶), which inhabits terrestrial hot springs in Kamchatka, Russia. This organism is able to grow at temperatures between 65 and 87°C, with the optimal temperature being 85°C, which makes it of interest in studying the mechanisms of thermal adaptation.
According to the MEROPS database (Rawlings et al., 2004 ▶), APDkam598 belongs to the M42 family of metallo-aminopeptidases. Most enzymes of this family form dodecamers of a tetrahedral (TET) shape and are called TET peptidases. They are involved in the final stage of proteolysis, i.e. the degradation of short peptides (6–12 residues in length) into free amino acids. It has been shown that APDkam598 catalyzes the degradation of tripeptides (Slutskaya et al., 2012 ▶) and cleaves uncharged amino-acid residues from the N-terminus. An investigation of the activity of the enzyme showed that it is maximal in combination with Mn2+, Mg2+ and Zn2+ (Slutskaya et al., 2012 ▶).
Among known structures, the aminopeptidases PhTET1 (PDB entries 2wyr and 2cf4; 41% sequence identity to APDkam598; Schoehn et al., 2006 ▶; M. A. Durá & F. M. D. Vellieux, unpublished work), PhTET2 (PDB entries 1xfo and 1y0y; 48% sequence identity to APDkam598; Russo & Baumann, 2004 ▶; Borissenko & Groll, 2005 ▶) and PhTET3 (PDB entry 2wzn; 41% sequence identity to APDkam598; Durá et al., 2009 ▶) isolated from the same organism, the archaeon Pyrococcus horikoshii, share the highest sequence similarity with APDkam598. These enzymes cleave oligopeptides and display a broad substrate specificity with a preference for a particular type of amino acid. PhTET1 shows high specificity towards acidic amino acids, PhTET2 towards neutral and aliphatic amino acids and PhTET3 towards basic amino acids. It was found that PhTET2 and PhTET3 assemble as 12-subunit complexes and that PhTET1 can assemble into either 12-subunit or 24-subunit complexes (Schoehn et al., 2006 ▶). Along with the question of how these large molecular machines function, many other questions are of great interest. Thus, the stability of these complexes at high temperatures and the factors that determine this thermostability are subjects that have attracted attention (Durá et al., 2009 ▶). Another interesting line of research is the investigation of the self-oligomerization of the 12-subunit complex (Appolaire et al., 2013 ▶). Recently, it has been reported that PhTET2 and PhTET3 assemble to form heteromeric complexes (Appolaire et al., 2014 ▶).
In this paper, we present the crystal structure of APDkam598, which is another archaeal TET peptidase. We found that the APDkam598 molecule forms a 12-subunit complex both in the crystal and in solution and is built similarly to the molecules of PhTET1, PhTET2 and PhTET3. We analyzed the interactions between APDkam598 subunits and compare the APDkam598 structure with the structures of PhTET1, PhTET2 and PhTET3.
An interesting feature of the APDkam598 molecule is that the APDkam598 monomer contains as many as six Trp residues, all of which are located on its surface and all except one of which are located on the surface of the dodecamer. Trp residues are relatively scarce in proteins (Grohmann et al., 2003 ▶) and rarely occur on the protein surface (Samanta et al., 2000 ▶). A superposition of the models of APDkam589 and of structures possessing high sequence similarity to APDkam589 indicates that although the positions of Trp residues are not conserved in their sequences, the spatial locations of a few Trp residues in these models are similar. These findings are interesting and may suggest that Trp residues in APDkam598 not only stabilize the structure. It may be hypothesized that these Trp residues can participate, for example, in protein–protein interactions.
2. Materials and methods
2.1. Expression and purification
The gene Dkam_0589 was cloned into pET-15b vector as described in Slutskaya et al. (2012 ▶). Plasmid pET_APDkam589 was transformed into Escherichia coli BL21-CodonPlus (DE3)-RIPL cells (Stratagene, USA). The E. coli cells were cultivated at 37°C in 2×YT medium supplemented with ampicillin to an OD600 of 0.8 and were induced with 1 mM IPTG for 5 h. The culture was then harvested by centrifugation and frozen at −70°C.
The pellet was resuspended in 25 mM Tris–HCl buffer pH 7.5 containing 400 mM NaCl, 5 mM imidazole, 0.2%(v/v) Triton X-100, 5%(v/v) glycerol (buffer A). The cells were disrupted with an Ultrasonic Processor (Cole Parmer, USA) and centrifuged at 16 000g for 30 min. The supernatant was loaded onto a 5 ml Superflow Ni–NTA column (Qiagen, USA) equilibrated with the same buffer. The recombinant APDkam589 was eluted with a linear gradient from 20 to 500 mM imidazole in buffer A without Triton X-100 and was precipitated with 0.9 M ammonium sulfate. The pellet was resuspended in 20 mM Tris–HCl buffer pH 7.5 containing 100 mM NaCl, 5%(v/v) glycerol, 2 mM β-mercaptoethanol and transferred onto a Superdex G200 column (GE Healthcare, UK) equilibrated with the same buffer. High-molecular-weight fractions were collected, concentrated with a 30 kDa cutoff centrifugal filter device (Millipore, USA) to a concentration of 10–12 mg ml−1 and used in crystallization experiments.
2.2. Crystallization
Crystals were grown by a modified counter-diffusion technique in a capillary (Tanaka et al., 2004 ▶; Takahashi et al., 2010 ▶; Confocal Science, Japan). The reservoir solution consisted of 0.1 M HEPES pH 7.0, 0.8 M ammonium sulfate, 2% glycerol. The crystals grew over a period of several months at 20°C. The best crystal chosen for the X-ray data collection had a shape similar to a diamond and an approximate size of 70 × 70 µm. The crystal was cryoprotected with 0.1 M HEPES pH 7.0, 0.8 M ammonium sulfate, 20% glycerol.
2.3. X-ray data collection and treatment
X-ray diffraction data were collected on beamline BL41XU at the SPring-8 synchrotron using a Rayonix MX225HE CCD detector. A crystal was flash-cooled in a stream of cold nitrogen gas at 100 K. Data collection was performed using the HKL-2000 software package (Otwinowski & Minor, 1997 ▶). Experimental details are summarized in Table 1 ▶. X-ray diffraction images were indexed, integrated and subsequently scaled using the HKL-2000 package. Data reduction was performed using the CCP4 package (Winn et al., 2011 ▶). The data-collection statistics are given in Table 1 ▶.
Table 1. Experimental setup and statistics of data collection and processing.
Values in parentheses are for the highest resolution shell.
| Space group | F23 |
| Radiation source | BL41XU, SPring-8 |
| Unit-cell parameter () | a = 234.32 |
| Temperature (K) | 100 |
| Wavelength () | 1.0 |
| Crystal-to-detector distance (mm) | 320 |
| Oscillation range () | 0.5 |
| Exposure time per frame (s) | 2.5 |
| Mosaicity range () | 0.270.34 |
| No. of frames | 80 |
| Resolution limit () | 47.833.01 (3.163.00) |
| Total reflections | 107637 |
| B value estimated from Wilson plot (2) | 59.7 |
| Monomers per asymmetric unit | 2 |
| Independent reflections | 21317 (2018) |
| Average I/(I) | 19.4 (1.7) |
| Completeness (%) | 95.0 (67.2) |
| R merge (%) | 8.0 (81.9) |
2.4. Structure determination and refinement
The structure of APDkam589 was solved using BALBES (Long et al., 2008 ▶). The best solution found by BALBES was obtained using the structure of the aminopeptidase from P. horikoshii (PDB entry 1xfo; 48% sequence similarity) as a starting model for molecular replacement and refinement. The model was further refined using phenix.refine v.1.8 (Afonine et al., 2012 ▶). For each atom, the coordinates and the atomic B value were refined. Along with a set of standard stereochemical restraints, secondary-structure restraints and Ramachandran restraints were applied during the refinement. The examination of density maps and the manual rebuilding of the model were performed using Coot (Emsley & Cowtan, 2004 ▶). The geometry of the final model was inspected by the MolProbity server (http://molprobity.biochem.duke.edu; Chen et al., 2010 ▶). A summary of the refinement statistics and the model-quality indicators is given in Table 2 ▶. Figs. 1 ▶ and 3–7 were prepared using PyMOL (http://www.pymol.org).
Table 2. Refinement statistics.
| R work/R free | 0.21/0.26 |
| Size of the test set used for calculation of R free (%) | 5.16 |
| Wilson B value (2) | 53.8 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths () | 0.013 |
| Bond angles () | 1.460 |
| Average B, all atoms (2) | 36.0 |
| Ramachandran plot | |
| Most favourable (%) | 93.4 |
| Allowed (%) | 5.1 |
| Outliers (%) | 1.5 |
Figure 1.

The overall structure of APDkam589. (a) The structure of the monomer. The catalytic and dimerization domains are shown in blue and orange, respectively. The residues of the active site are shown as red sticks. The three strands localized on the surface are coloured green. The strands sandwiched between the helices are coloured lemon. (b) The APDkam589 dodecamer formed in the crystal by the assembled symmetry-related monomers. The monomers are shown in different colours. (c) The structure of the dimer. The monomers that form the dimer are shown in blue and red; the other monomers of the dodecamer are shown in grey. (d) Three monomers forming the trimer are shown in blue, red and orange; the other monomers of the dodecamer are shown in grey.
In the APDkam589 crystal, the asymmetric unit contained two almost identical monomers and 26 water molecules. There was no interpretable electron density for the four N-terminal residues of the molecule, 26 purification-tag residues and residues 126–131. The atomic coordinates and experimental structure factors were deposited in the Protein Data Bank and are accessible as entry 4wwv.
2.5. Electron microscopy
The purified protein was applied onto carbon-coated copper grids and subjected to negative glow discharging at −20 mA for 45 s in an air atmosphere. Excess protein was then blotted off with filter paper and the grids were washed once with buffer for 30 s. Samples were stained with 1% uranyl acetate twice for 30 s. The grids were then examined on a Jeol JEM2100 electron microscope at 200 kV. Images were recorded using an Ultrascan 1000XP CCD camera (Gatan, USA) at 40 000× magnification and 1.5–1.8 µm under focus. 3018 single particles were selected from the EM images using Boxer (Ludtke et al., 1999 ▶). They were then filtered and subjected to reference-free classification using IMAGIC (van Heel et al., 1996 ▶; Harauz & van Heel, 1986 ▶). The images of single protein particles were then categorized into 50 classes based on their orientations. Some particles and the class averages are presented in Fig. 2 ▶(b). Both the original particles (Fig. 2 ▶ b, row 1) and the averaged particles (Fig. 2 ▶ b, row 2) exhibited a compact structure with a diameter of about 150 Å. Some images showed a threefold symmetry, while others were twofold-symmetric. Since it has been demonstrated previously that these proteases possess a tetragonal symmetry (Russo & Baumann, 2004 ▶; Schoehn et al., 2006 ▶; Durá et al., 2009 ▶), we applied this type of symmetry to the final reconstruction, thus increasing the number of projections 12-fold. The resulting three-dimensional reconstruction has a resolution of 15 Å, as estimated by the method of Fourier shell correlation (Fig. 2c ▶; Harauz & van Heel, 1986 ▶).
Figure 2.

Single-particle EM analysis and three-dimensional reconstruction of the APDkam589 molecule. (a) Images of a protein particle negatively stained with uranyl acetate. The scale bar is 100 nm in length. (b) Some particles selected from raw images (row 1), the corresponding class averages (row 2), the corresponding reprojections of the three-dimensional structure (row 3) showing views that match the class averages and the surface representations of the three-dimensional structure shown in the same orientations as the reprojections in row 3 (row 4). The scale bar is 10 nm in length. (c) Fourier shell correlation plot calculated between three-dimensional structures each containing half of the data. (d) The surface representation of the final three-dimensional reconstruction and the APDkam589 dodecamer, which was docked into the electron density. Three different views are shown.
3. Results and discussion
3.1. Overall structure of APDkam589
The monomer of APDkam589 has a structure typical of that of TET aminopeptidases. It consists of two domains (Fig. 1 ▶ a): a globular catalytic domain (residues 1–74 and 170–369) and a smaller butterfly-shaped dimerization domain (residues 75–169). The catalytic domain contains eight β-strands sandwiched by two groups of α-helices (two α-helices from one side and five α-helices from the other side) and three β-strands on the surface of the monomer. The dimerization domain contains six β-strands whose spatial organization slightly resembles a β-barrel and two short α-helices. Both domains contain also several loops. The active site is located in the cleft between the two domains; all residues of the active site belong to the catalytic domain.
By applying the crystallographic symmetry of the F23 space group to an APDkam589 monomer, a 12-unit complex is obtained. The monomer of APDkam589 and the symmetry-related monomers in the crystal are densely packed to form a dodecamer with a tetrahedral shape (Fig. 1 ▶ b). Previously, it had been suggested that the building block of the dodecamer is most likely to be a dimer (Schoehn et al., 2006 ▶). The edges of the tetrahedron are formed by dimers (Fig. 1 ▶ c), while the vertices are formed by trimers (Fig. 1 ▶ d). In the inner part of the tetrahedron, there is a large cavity with all of the active sites, which are accessible from outside via four large openings in the middle of each facet (Fig. 1 ▶ b). The diameter of each opening is about 18 Å.
3.2. Three-dimensional structure of APDkam589 as determined by single-particle EM analysis
In the final EM three-dimensional reconstruction of the structure (Figs. 2 ▶ c and 2 ▶ d) at a resolution of 15 Å, a central cavity about 40 Å in diameter and channels of two types, a wider one in the middle of each facet and a narrower one on the vertices, are visible. The model of the APDkam589 dodecamer, which is formed in the crystal by the symmetry-related monomers, was docked using Chimera (Pettersen et al., 2004 ▶) into the electron density of the final EM reconstruction (Fig. 2 ▶ d). The shape and the dimensions of the EM image coincide well with those of the X-ray model (Fig. 2 ▶ d). Thus, the EM analysis confirms that APDkam589 is a dodecamer in solution.
3.3. Comparison of the structures of APDkam589 and PhTET peptidases
APDkam589 shares the highest sequence identity (48%) with the PhTET2 peptidase from P. horikoshii. The overall structures of the monomers of APDkam589 and PhTET2 are very similar (Fig. 3 ▶ a); the r.m.s.d. of the aligned Cα atoms is 0.93 Å (330 residues were aligned). However, there are some differences. The sequence of APDkam589 contains 17 more amino-acid residues than that of PhTET2. Loop 58–63 is two residues longer, and loop 266–272 is four resides longer than the corresponding loops in PhTET2. The monomer of APDkam589 is eight residues longer at the C-terminus. In addition, loop 106–114 has a different conformation to the corresponding loop in the PhTET2 monomer (Fig. 3 ▶ a). Note that the overall structure of the APDkam589 monomer is also similar to the structures of the PhTET2 and PhTET3 monomers. The residues of the active sites of APDkam589 and PhTET1, PhTET2 and PhTET3 are conserved (Fig. 3 ▶ b), and the geometry of the active sites of these structures is almost identical.
Figure 3.

Comparison of the structures and sequences of APDkam589 and the PhTET peptidases. (a) Superposition of the monomers of APDkam589 (shown in green) and the PhTET2 peptidase (PDB entry 1xfo; shown in magenta) performed using Coot. Three loops that are the most different structural elements in these two models are indicated by arrows. (b) Multiple sequence alignment of APDkam589, PhTET1, PhTET2 and PhTET3. The alignment was performed using Clustal Omega v.1.2.1 (Sievers et al., 2011 ▶). The residues of the active site are indicated by asterisks. These residues are conserved in the four enzymes.
The APDkam589 dodecamer is built up similarly to those found in the crystals of PhTET1, PhTET2 and PhTET3. However, there are a few notable differences in the regions of the large and small openings of the dodecamer. The large openings on the facets of the APDkam589 dodecamer seem to be slightly wider than those of the PhTET1, PhTET2 and PhTET3 dodecamers. The small openings, which are called ‘pores’ and are located at the interfaces of three monomers, are blocked in the APDkam589 structure by the Tyr226 residues (Fig. 4 ▶) and, deeper inside, by the Lys222 residues of the three monomers. In PhTET1, these openings are wider and open to the narrow channel, which leads into the interior of the tetrahedron and has been suggested to serve as the exit for free amino acids (Franzetti et al., 2002 ▶). In the PhTET2 and PhTET3 dodecamers these openings are also blocked as in the APDkam589 structure. However, the residues that block this opening and simultaneously form the interface of the three monomers are different. These residues (Arg220 and Phe224 in PhTET2) are conserved in PhTET2 and PhTET3 but not in APDkam589 (Lys222 and Tyr226 in APDkam589). It has been shown that Arg220 in PhTET2 is important for the formation of the dodecamer (Appolaire et al., 2013 ▶). In APDkam589, Lys222 is present instead of Arg.
Figure 4.

A small opening in the APDkam589 tetrahedron, which is formed by three monomers. Residues Tyr226 and Lys222 (shown in sticks) block the opening.
3.4. Analysis of contacts between monomers of APDkam589 and a comparison of the contacts in similar structures
In the APDkam589 dodecamer there are two kinds of interfaces between monomers: between two monomers in a dimer (Fig. 1 ▶ c) and between monomers of different dimers, i.e. between monomers in the trimer (Fig. 1 ▶ d). In all, there are six and 12 interfaces of the first and second kinds, respectively. The interface between the monomers in the dimer is more extensive. This interface in APDkam589 contains four salt bridges and 21 hydrogen bonds which are not salt bridges. The interface in the trimer contains many fewer hydrogen bonds. For APDkam589, it contains one salt bridge and 11 hydrogen bonds.
Table 3 ▶ shows that the total number of salt bridges in all interfaces of APDkam589 is much lower than those in the homologous TET peptidases from the archaeon P. horikoshii, which is a more thermophilic organism. It has been reported that the optimal temperature for growing P. horikoshii is 95°C (Gonzalez et al., 1998 ▶). The greater number of salt bridges in the interfaces between monomers in the models of PhTET1, PhTET2 and PhTET3 compared with the APDkam589 model correlates with the higher thermostability of P. horikoshii compared with D. kamchatkensis. Table 3 ▶ shows that this tendency is observed only for salt bridges, not for hydrogen bonds. This again confirms that the presence of salt bridges is a crucial structural factor for the thermostability of the enzyme molecule (Reed et al., 2013 ▶).
Table 3. The numbers of hydrogen bonds and salt bridges between monomers in the dodecamers of APDkam589 and TET peptidases from P. horikoshii .
The calculations were performed using the PISA server (http://www.ebi.ac.uk/pdbe/pisa).
| APDkam589 | PhTET1 | PhTET1 | PhTET2 | PhTET3 | |
|---|---|---|---|---|---|
| PDB entry | 4wwv | 2wyr | 2cf4 | 1xfo | 2wzn |
| Sequence identity to APDkam589 (%) | 37 | 37 | 48 | 41 | |
| Between monomers in the dimer | |||||
| No. of hydrogen bonds | 21 | 32 | 28 | 20 | 24 |
| No. of salt bridges | 4 | 2 | 8 | 16 | 8 |
| Between monomers in the trimer | |||||
| No. of hydrogen bonds | 11 | 12 | 8 | 2 | 10 |
| No. of salt bridges | 1 | 9 | 5 | 4 | 2 |
| Total No. of salt bridges between different monomers in the dodecamer | |||||
| No. of hydrogen bonds | 258 | 336 | 264 | 144 | 264 |
| No. of salt bridges | 36 | 120 | 108 | 144 | 72 |
3.5. Comparison of the positions of Trp residues in APDkam589 and homologous structures: a possible role of Trp residues
The monomer of APDkam589 contains as many as six Trp residues, the scarcest amino acid in proteins. Among 13 672 sequences from the IMG database (Markowitz et al., 2014 ▶) that have more than 30% identity to the sequence of APDkam589, only 15 contain six Trp residues, the same as in APDkam589, and only two sequences contain a greater number of Trp residues (seven and eight). In particular, the monomers of PhTET1, PhTET2 and PhTET3 contain two, four and two Trp residues, correspondingly.
All Trp residues of APDkam589 are located on the surface of the monomer (Fig. 5 ▶ a) and the dimer, and all Trp residues except for Trp49 are on the external surface of the dodecamer (Fig. 5 ▶ b). In general, Trp, which is the largest amino-acid residue, is relatively rarely located on the protein surface (Samanta et al., 2000 ▶). When the large aromatic rings of Trp occur on the surface of enzymes, they can serve in binding to either other proteins or ligands with specific structures. Thus, the Trp residues on the surface of cellulase are involved in binding to cellulose and polysaccharides (Sakon et al., 1997 ▶). In APDkam589, the Trp residues participate in interactions between secondary-structure elements. For instance, Trp5 is involved in interactions between helices 4–18 and 337–353 through a stacking interaction with Tyr341 (Fig. 6 ▶ a), and Trp175 is involved in interactions between loop 174–177 and helix 4–18 through a cation–π interaction and hydrogen bonding with Arg6 (Fig. 6 ▶ b). However, because the Trp residues are on the surface of the molecule, it can be supposed that their function is not only stabilization of the tertiary structure.
Figure 5.

Location of Trp residues in the APDkam589 structure. (a) The APDkam589 monomer and Trp residues, which all are located on its surface. (b) The APDkam589 dodecamer and the location of Trp residues. All Trp residues except one are located on the surface of the dodecamer.
Figure 6.
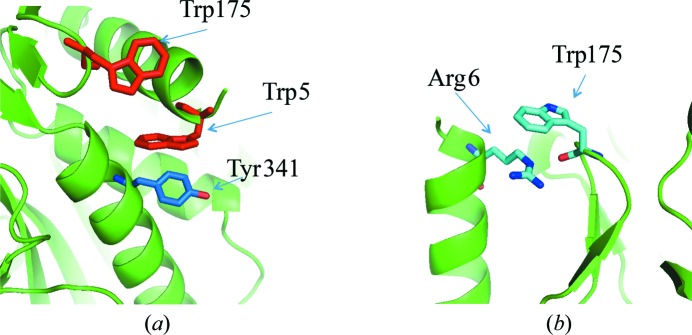
Trp residues that participate in interactions between secondary-structure elements. (a) Helix 4–18, helix 337–353, residues Trp5, Tyr341 and Trp175 (shown as sticks). (b) Loop 174–177, helix 4–18, residues Trp175 and Arg6 (shown as sticks).
Superposition of the models of APDkam589 with several structures [PhTET2 (PDB entry 1xfo, 48% sequence similarity to APDkam589; Russo & Baumann, 2004 ▶), endoglucanase from Thermotoga maritima (PDB entry 3isx, 39% sequence similarity to APDkam589; Joint Center for Structural Genomics, unpublished work), endoglucanase from T. maritima (PDB entry 2fvg, 37% sequence similarity to APDkam589; Joint Center for Structural Genomics, unpublished work) and protein S2589 from Shigella flexneri (PDB entry 1ylo, 31% sequence similarity to APDkam589; Midwest Center for Structural Genomics, unpublished work)] that possess a high sequence similarity to APDkam589 and have Trp residues on the surface revealed an interesting phenomenon. Although Trp45, Trp252 and Trp358 are not conserved in the sequences of these structures, their spatial positions are similar (Fig. 7 ▶). This observation leads us to suggest that the Trp residues in APDkam589 are not only involved in the interactions between secondary-structure elements. It may be speculated that these Trp residues can participate, for example, in protein–protein interactions. It was recently found that PhTET2 and PhTET3, which have the highest sequence similarity to APDkam589 among the known structures, can assemble to form hetero-oligomeric complexes (Appolaire et al., 2014 ▶). Further investigations are needed to verify the possible role of Trp residues in the functions of APDkam589.
Figure 7.

Superposition of the models of APDkam589 and structures that share a high sequence similarity with APDkam589 and have Trp residues on the surface. (a) Models of APDkam589 (shown in green) and PhTET2 (PDB entry 1xfo; shown in magenta). Trp358 of APDkam589 and Trp276 of PDB entry 1xfo are shown as sticks. (b) Models of APDkam589 (green) and endoglucanase from T. maritima (PDB entry 3isx; magenta). Trp45 of APDkam589 and Trp47 of 3isx are shown as sticks. (c) Models of APDkam589 (green) and endoglucanase from T. maritima (PDB entry 2fvg; brown). Trp252 of APDkam589 and Trp234 of PDB entry 2fvg are shown as sticks. (d, e) Models of APDkam589 (shown in green) and a possible aminopeptidase from S. flexneri (PDB entry 1ylo; magenta). Trp45 and Trp358 of APDkam589 and Trp191 and Trp368 of PDB entry 1ylo are shown in sticks.
Supplementary Material
PDB reference: APDkam598, 4wwv
Acknowledgments
We thank Natalia Mikhailova and Sergei Spirin for help and fruitful discussions. The EM study was performed at the Microscopy User Facilities Center of the Moscow State University and was supported by the Ministry of Education and Science of the Russian Federation. The part of the work involving purification, crystallization, structure determination and analysis of the structure was supported by the Russian Scientific Foundation (project No. 14-24-00172). The part of the work concerned with refinement of the structure was supported by the Russian Foundation for Basic Reseach (grant 13-04-00118). The part of the work concerned with X-ray data collection and treatment was supported by the Russian Federal Space Agency (Roscosmos).
References
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Appolaire, A., Durá, M. A., Ferruit, M., Andrieu, J.-P., Godfroy, A., Gribaldo, S. & Franzetti, B. (2014). Mol. Microbiol. 94, 803–814. [DOI] [PubMed]
- Appolaire, A., Rosenbaum, E., Durá, M. A., Colombo, M., Marty, V., Savoye, M. N., Godfroy, A., Schoehn, G., Girard, E., Gabel, F. & Franzetti, B. (2013). J. Biol. Chem. 288, 22542–22554. [DOI] [PMC free article] [PubMed]
- Borissenko, L. & Groll, M. (2005). J. Mol. Biol. 346, 1207–1219. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Durá, M. A., Rosenbaum, E., Larabi, A., Gabel, F., Vellieux, F. M. D. & Franzetti, B. (2009). Mol. Microbiol. 72, 26–40. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Franzetti, B., Schoehn, G., Hernandez, J.-F., Jaquinod, M., Ruigrok, R. W. H. & Zaccai, G. (2002). EMBO J. 21, 2132–2138. [DOI] [PMC free article] [PubMed]
- Gonzalez, J. M., Masuchi, Y., Robb, F. T., Ammerman, J. W., Maeder, D. L., Yanagibayashi, M., Tamaoka, J. & Kato, C. (1998). Extremophiles, 2, 123–130. [DOI] [PubMed]
- Grohmann, U., Fallarino, F. & Puccetti, P. (2003). Trends Immunol. 24, 242–248. [DOI] [PubMed]
- Harauz, G. & van Heel, M. (1986). Optik, 73, 146–156.
- Heel, M. van, Harauz, G., Orlova, E. V., Schmidt, R. & Schatz, M. (1996). J. Struct. Biol. 116, 17–24. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Ludtke, S. J., Baldwin, P. R. & Chiu, W. (1999). J. Struct. Biol. 128, 82–97. [DOI] [PubMed]
- Markowitz, V. M. et al. (2014). Nucleic Acids Res. 42, D560–D567. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Ravin, N. V., Mardanov, A. V., Beletsky, A. V., Kublanov, I. V., Kolganova, T. V., Lebedinsky, A. V., Chernyh, N. A., Bonch-Osmolovskaya, E. A. & Skryabin, K. G. (2009). J. Bacteriol. 191, 2371–2379. [DOI] [PMC free article] [PubMed]
- Rawlings, N. D., Tolle, D. P. & Barrett, A. J. (2004). Nucleic Acids Res. 32, D160–D164. [DOI] [PMC free article] [PubMed]
- Reed, C. J., Lewis, H., Trejo, E., Winston, V. & Evilia, C. (2013). Archaea, 2013, 373275. [DOI] [PMC free article] [PubMed]
- Russo, S. & Baumann, U. (2004). J. Biol. Chem. 279, 51275–51281. [DOI] [PubMed]
- Sakon, J., Irwin, D., Wilson, D. B. & Karplus, P. A. (1997). Nature Struct. Mol. Biol. 4, 810–818. [DOI] [PubMed]
- Samanta, U., Pal, D. & Chakrabarti, P. (2000). Proteins, 38, 288–300. [PubMed]
- Schoehn, G., Vellieux, F. M. D., Durá, M. A., Receveur-Brechot, V., Fabry, C. M. S., Ruigrok, R. W. H., Ebel, C., Roussel, A. & Franzetti, B. (2006). J. Biol. Chem. 281, 36327–36337. [DOI] [PubMed]
- Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., Thompson, J. D. & Higgins, D. G. (2011). Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed]
- Slutskaya, E. S., Bezsudnova, E. Y., Mardanov, A. V., Gumerov, V. M., Rakitina, T. V., Popov, V. O. & Lipkin, V. M. (2012). Dokl. Biochem. Biophys. 442, 30–32. [DOI] [PubMed]
- Takahashi, S., Tsurumura, T., Aritake, K., Furubayashi, N., Sato, M., Yamanaka, M., Hirota, E., Sano, S., Kobayashi, T., Tanaka, T., Inaka, K., Tanaka, H. & Urade, Y. (2010). Acta Cryst. F66, 846–850. [DOI] [PMC free article] [PubMed]
- Tanaka, H., Inaka, K., Sugiyama, S., Takahashi, S., Sano, S., Sato, M. & Yoshitomi, S. (2004). J. Synchrotron Rad. 11, 45–48. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: APDkam598, 4wwv