

Abstract
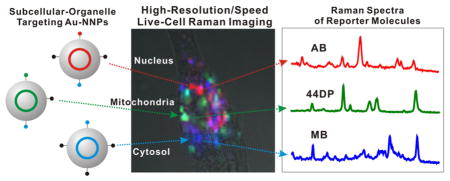
We report a method to achieve high speed and high resolution live cell Raman images using small spherical gold nanoparticles with highly narrow intra-nanogap structures responding to NIR excitation (785 nm) and high-speed confocal Raman microscopy. The three different Raman-active molecules placed in the narrow intra-nanogap showed a strong and uniform Raman intensity in solution even under transient exposure time (10 ms) and low input power of incident laser (200 μW), which lead to obtain high-resolution single cell image within 30 s without inducing significant cell damage. The high resolution Raman image showed the distributions of gold nanoparticles for their targeted sites such as cytoplasm, mitochondria, or nucleus. The high speed Raman-based live cell imaging allowed us to monitor rapidly changing cell morphologies during cell death induced by the addition of highly toxic KCN solution to cells. These results strongly suggest that the use of SERS-active nanoparticle can greatly improve the current temporal resolution and image quality of Raman-based cell images enough to obtain the detailed cell dynamics and/or the responses of cells to potential drug molecules.
Keywords: SERS, live cell Raman imaging, intra-nanogap gold nanoparticle, drug-screening
Since its first inception, Raman spectroscopy has been widely used as an analytical tool in many fields.1 Confocal Raman microscopy, which combines confocal microscopy with conventional Raman spectroscopy, has been extensively used to obtain three-dimensional information and can be used to achieve high resolution label-free imaging using the fingerprint peaks of biomolecules of interest such as DNA and protein.2–5 However, observing a living cell with Raman microscopy is quite challenging due to the intrinsically weak Raman signal intensity of endogenous cellular components.1,6,7 The Raman scattering cross-section is about 10−30 cm2/sr, which is much smaller than the absorption cross-section of typical fluorescent molecules (10−16 cm2/sr). Therefore, Raman-based cell imaging requires a relatively long exposure or integration time (typically few seconds per pixel), making it unrealistic to image a live cell whose cellular changes can occur on the scale of milliseconds. To address this issue, many researchers have developed high-speed Raman microscopy systems. Slit-scanning Raman microscopy,3 multifocal Raman microscopy,8 and direct Raman imaging systems have all been previously reported.9 In order to overcome a weak Raman intensity and the spectral overlapping from cell body, exogenous small molecule Raman tags with strong signals were introduced into the Raman-silent cellular region10,11 and plasmonic nanoparticles to enhance the Raman signals of biomolecules placed inside cells were also extensively investigated.12–15 In spite of these efforts, the technologies for high speed live-cell Raman imaging without sacrificing the image resolution have not been firmly established.14–17
Recently, we developed a high-speed confocal Raman microscopy system using commercially available galvano mirrors instead of mechanical stage movement.4,18 This custom-built high-speed Raman system was successfully applied to monitor the transient uptake of single-walled carbon nanotubes with a 1 min temporal resolution. In this work, 50 ms integration time per pixel (30 × 30 spectra from 17 μm × 17 μm field of view) were applied for single cell imaging.18 Although spatial and temporal tracking of two cell-intrinsic and nine nanotube-derived Raman bands were conducted simultaneously in RAW 264.7 macrophages, the previous study lacked targeting capability, and acquisition time was not fast enough to monitor the rapidly changing cell dynamics. (It should be noted that 50 ms integration time per pixel is still not desirable for high speed imaging purposes since 125 s has been required to obtain all Raman spectra from 50 × 50 for high resolution images).
In order to overcome current spatial and temporal resolution of the live cell Raman imaging techniques, we introduced the near-infrared (NIR)-sensitive SERS-active nanoparticles that can selectively target specific intracellular organelles. The use of highly SERS-sensitive nanoparticles can greatly decrease acquisition time per pixel and allow us to obtain the intracellular particle distributions for specific subcellular organelles. In addition, the use of NIR (785 nm) for imaging can further improve spatial resolution by minimizing the autofluorescence background from the cell body.
In this work, we report high resolution and high speed single live cell Raman imaging using a galvano mirror-equipped confocal Raman microscopy system4 and NIR-sensitive SERS-active nanoparticles,19 both leading to accomplish the high resolution (50 × 50 pixels) single live cell imaging within 30 s (10 ms/pixel). Furthermore, the use of SERS-active nanoparticles allowed us to monitor particle distributions within their targeted sites of cell and enabled us to monitor rapidly changing cell morphologies without inducing significant damages on cells.
For a microscopy system, a previously reported custom-built NIR confocal Raman microscopy system was used. Briefly, 785 nm wavelength from Ti:Sapphire laser (3900S, Spectra-Physics) was focused onto the sample, and backscattered light is collected by the same objective lens. After series of filters, Raman spectrum is acquired by a spectrograph (Holospec f/1.8i, Kaiser Optical Systems) and a thermoelectric-cooled, back-illuminated, and deep depleted CCD (PIXIS: 100BR_eXcelon, Princeton Instruments). High-speed XY scanning (0.1 ms response time) was performed by the galvanometer mirrors (CT-6210, Cambridge Technology) (Figure S1, Supporting Information).4,18
As a SERS-active nanoparticle for Raman imaging, we selected Raman dye-coded spherical-shape gold nanoparticles with uniform intra-nanogap (Au-NNP) prepared with DNA-modified AuNP.19 The small particle size (~42 nm), uniform Raman response in solution, and straightforward surface chemistries required for targeting are the major benefits of using Au-NNP.20 However, the previously reported Au-NNP was not sensitive to NIR (785 nm) excitations due to the lack of plasmonic absorptions at 785 nm wavelength.19,21 In this work, we used NIR-sensitive Raman reporters. A large number of molecules could be localized in the nanogap through electrostatic interactions between molecules and Au surface.22 Furthermore, we found that the selection of a specific spacer DNA sequence in the core DNA-AuNP was very critical in order to obtain a strong Raman response with an excitation in the NIR region of the spectrum. The Au-NNPs prepared from thymine spacer (T10)-oligonucleotide modified AuNP showed the strongest Raman response when excited using NIR-wavelength (Figure 1).
Figure 1.

(A) Synthetic scheme of Raman dye (44DP)-coded Au-NNPs using DNA-AuNPs as a core particle (four different kinds of spacer sequences such as adenine (A10), guanine (G10), cytosine (C10), and thymine (T10) in the thiolated DNA (3′-(CH2)3-spacer sequence-PEG9-AAACTCTTTGCGCAC-5′) were investigated). 4,4′-Dipyridyl (44DP) molecules were loaded on the surface of DNA-AuNP via electrostatic interactions, then the Au shell was formed. (B) The solution color and HR-TEM image of 44DP-coded Au-NNPs prepared from A10 spacer DNA-AuNP (1), G10 spacer DNA-AuNP (2), C10 spacer DNA-AuNP (3), and T10 spacer DNA-AuNP (4). (C,D) Raman spectra of 44DP-coded Au-NNP solution prepared from four different spacer DNA with an excitation of 633 (C) and 785 nm (D). (Each spectrum was obtained using conditions 0.5 nM particle concentration and 1.0 s exposure time).
First, we prepared oligonucleotide modified AuNP using thiolated DNA (3′-(CH2)3-spacer sequence (A10, G10, C10, or T10)-PEG9-AAACTCTTTGCGCAC-5′) and then incubated 1.0 mL of DNA-AuNP solution in the presence of 100 μL of 0.1 M 4,4′-dipyridyl (44DP) in distilled water (DW) for 1 week at room temperatures. After centrifugation (12,000 rpm, 15 min) of the mixtures to remove excess 44DP in the supernatant, it was dispersed in 1.0 mL of DW and formed the Au shell using NH2OH-HCl (500 μL, 10 mM) and HAuCl4 (500 μL, 5 mM) in the presence of 0.3 M phosphate buffered saline and 1% PVP (Mw 40,000) (Figure 1A). The TEM images and solution color (deep red wine) in Figure 1B showed the formation of spherical shape nanoparticle (diameter; ~42 nm). Interestingly, the Au-NNPs prepared from A10, G10, or C10 spacer showed clearly visible intra-nanogap structures (1.2 nm) as shown in Figure 1B-1,2,3, but the Au-NNP prepared from T10 spacer showed very narrow nanogap distance, which is hard to define the distance (Figure 1B-4). The UV–visible spectra of the Au-NNPs prepared with T10 spacer showed the absence of plasmonic absorptions between 640–900 nm, which distinguishes from Au-NNPs with visible intra-nanogap structures (Figure S2, Supporting Information). Next we used two different excitation wavelengths (i.e., 633 and 785 nm) to evaluate the Raman responses of prepared particles. When excited with a 633 nm laser to obtain the Raman response of Au-NNPs (Figure 1C), the Au-NNPs prepared with T10 spacer (green line) showed the strongest Raman intensity, which is 3.6 times higher than that of Au-NNPs from A10 spacer (black line). When excited with 785 nm (Figure 1D), the Au-NNPs prepared with T10 spacer showed a 7.7 times higher Raman intensity (based on the Raman shift at 1609 cm−1) than that of Au-NNPs from A10 spacer. The presence of narrower intra-nanogap or small nanogap space in the Au-NNPs prepared with T10 spacer as compared with A10, G10, or C10 spacer is strongly expected, which is well matched with the results in the recent paper.21 We investigated the intermediate structures involved in the formation of Au-NNP structures by adding controlled amount of NH2OH-HCl and HAuCl4 (Figure S3, Supporting Information). We found the number of small budding particles on DNA-AuNP to be critical in determining the intra-nanogap structures as indicated by the red arrow in Figure S3, Supporting Information. In the case of A10, G10, and C10 spacers, one small budding particle produced on the DNA-AuNP core particle was grown into a complete shell with a wide and uniform intra nanogap structure (1.2 nm) (Figure S3a,b,c, Supporting Information). However, in the case of T10 spacer it showed higher numbers of small budding particles on the core gold nanoparticle as shown in Figure S3d, Supporting Information, which lead to the formation of very narrow intra-nanogap structures.21 The high Raman cross-section of 4,4′-dipyridyl (44DP) molecules was also responsible for the strong Raman response at NIR-wavelength.22
For targeting and multiplexed Raman-based cell imaging, we selected two more Raman dyes (i.e., methylene blue (MB) and 4,4′-azobis (pyridine) (AB)) for NIR-sensitive Raman reporters.22 The Raman dyes (MB, AB) were successfully localized on the surface of core gold nanoparticle through physical adsorption, then Au shell (~11 nm thickness) was formed to generate NIR-sensitive Au-NNPs. After PEGylation of Raman dye-coded Au-NNPs with methoxy PEG-thiol (mPEG thiol, 5k), the Au-NNPs were subsequently modified with cell penetrating peptide (RGD; RGDRGDRGDRGDPGC), and/or mitochondria-targeting peptide (MLS; MLALLGWWWFFSRKKC), and/or nucleus-targeting peptide (NLS; CGGGPKKKRKVGG) via covalent linkages between gold and thiol group of the cysteine (bold in peptide sequence) (Figure 2) (See Supporting Information for synthetic details).23 The MB-coded, 44DP-coded, and AB-coded Au-NNP solutions (0.5 nM) showed highly strong and uniform Raman response as shown in Figure 2B,C,D, respectively. It should be noted that each Raman spectrum was obtained with an excitation of 785 nm wavelength (10 ms and 4.0 mW incident laser power density). The strong and uniform Raman responses with time in solution state analysis are strongly required properties of the Raman probe for cellular imaging applications.12,13
Figure 2.

(A) Scheme for the PEGylation and peptide modifications on the Raman dye-coded Au-NNPs: MB-coded Au-NNP was modified with mPEG thiol and RGD peptide (RGDRGDRGDRGDPGC) targeting the cytoplasm; 44DP-coded Au-NNP modified with mPEG thiol, RGD peptide, and MLS peptide (MLALLGWWWFFSRKKC) targeting the mitochondria; and AB-coded Au-NNP modified with mPEG thiol, RGD peptide, and NLS peptide (CGGGPKKKRKVGG). (B–D) Representative Raman spectra of MB-coded Au-NNPs (B), 44DP-coded Au-NNPs (C), and AB-coded Au-NNPs (D) obtained from 0.5 nM particle concentration with an incident laser power of 4.0 mW and 10 ms exposure time per spectra.
Next, we selected human oral cancer cells (HSC-3) for single cell imaging due to the well-established interactions between nanoparticle with targeting ligands and HSC-3 cell.23,24 HSC-3 cells were plated onto a custom-built quartz bottomed Petri dish (043210-KJ, Alfa Aesar). The cells were allowed to adhere to the quartz bottomed plates for at least 24 h before adding Au-NNPs. Raman dye (i.e., 44DP, MB, AB)-coded Au-NNPs (100 μL, 0.5 nM) were added into the human oral cancer cells (HSC-3) (1.0 mL of culture media), after incubating cells with Au-NNPs for 3, 6, and 12 h, and then obtained bright field cell image and confocal Raman image. After replacing the culture media, laser beam (200 μW, 785 nm) was focused to a micron spot size and raster scanned the cells. Then 50 × 50 spectra were acquired from 38 μm × 38 μm area with an integration time of 10 ms per pixel. With 1.0 ms CCD readout time, the total measurement time was approximately 27.5 s.
All Raman spectra were fit with a linear combination of basis spectra with non-negative restriction. Raman spectra from Au-NNPs solutions (Figure 2B,C,D) were normalized and used as basis spectra. SERS signals are several orders of magnitude stronger than intrinsic cellular Raman signals allowing us to easily exclude the cellular Raman signals from the decomposition process. To avoid data overfitting, the process was carefully monitored by restricting the fitting coefficient to nonnegative values. As a result, each Raman mapping resulted in 2500 coefficients for each basis spectrum. Blue, green, and red colors are assigned to MB-coded Au-NNPs for cytoplasm targeting, 44DP-coded Au-NNPs for mitochondria, and AB-coded Au-NNPs for nucleus, respectively. Then these Raman images are overlaid with corresponding bright field images as shown in Figure 3.
Figure 3.
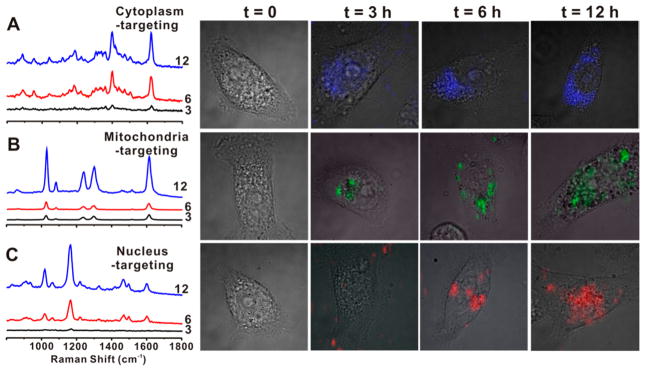
Time-dependent live cell Raman images (50 × 50 pixel) after incubating with subcellular targeting NNPs. (A) Representative Raman spectra obtained from inside cells incubated with MB-coded NNPs for cytoplasm targeting, bright field image (t = 0), and bright field images overlaid with Raman images (t = 3, 6, 12 h) (4.0 mW laser power; 100 ms exposure time per pixel). (B) Representative Raman spectra from cell incubated with 44DP-coded NNPs for mitochondria targeting, bright field image (t = 0), and bright field images overlaid with Raman images (t = 3, 6, 12 h) (200 μW laser power; 10 ms exposure time per pixel). (C) Representative Raman spectra from cell incubated with AB-coded NNPs for nucleus targeting, bright field image (t = 0), and bright field images overlaid with Raman images (t = 3, 6, 12 h) (200 μW laser power; 10 ms exposure time per pixel).
MB-coded Au-NNPs designed to be distributed in the cytoplasm showed time-dependent intracellular uptake behaviors through the interactions of RGD peptide and αvβ6 integrins on HSC-3 cell surface (Figure 3A).24 The Raman spectra in Figure 3A are representative Raman spectra obtained from inside the cell at each time point. With an increase of incubation time, strong spectra and extensive color distributions around cytoplasm were observed. The blue color indicates the distributions of MB-coded Au-NNP in single cell overlaid with bright field image. To obtain a single cell image in Figure 3A, we used 4.0 mW incident laser power density and 100 ms integration time per pixel, which allowed us to obtain a 40 × 40 pixel single cell image within 162.6 s because of relatively low Raman intensity and intracellular uptake efficiency of MB-coded Au-NNPs. On the contrary, the 44DP-coded Au-NNPs designed to target mitochondria showed much higher intra-cellular uptake efficiency as compared with MB-coded Au-NNPs, which lead to the strong and extensive green color distributions around the cytoplasm with an increased incubation time (Figure 3B). The distributions of Au-NNPs were well matched with typical distributions of mitochondria in HSC-3 cells. For single cell Raman imaging in Figure 3B, 200 μW of incident laser power and 10 ms of integration time per pixel were applied to 50 × 50 pixel. Therefore, the total imaging time was 27.5 s without inducing any cell damage. AB-coded Au-NNPs designed to target nucleus through the interactions between NLS peptide and importins α and β, karyopherins that are associated with the nuclear pore complex, also showed time-dependent intracellular uptake of Au-NNPs and successful localization of Au-NNPs at nucleus as shown in Figure 3C. Reportedly, the localization of AgNPs at the nucleus with the same targeting ligand showed significant cytotoxic effects;24,25 the Au-NNPs localized at the nucleus showed no such significant cytotoxicity. For this single cell Raman imaging, we used 200 μW incident laser power density and 10 ms integration time per pixel for 50 × 50 pixel, leading to a total imaging time per cell of 27.5 s.
Combining high-end optical and electrical components, the label-free Raman imaging for a live cell was possible by use of fingerprint peak of DNA (785 cm−1), protein (1004 cm−1), and lipid (1450 cm−1) in the cell as shown in Figures 4A and S4, Supporting Information.21 To obtain the high resolution (40 × 40 pixel) single cell Raman image displayed in Figure 4A, 60 mW of incident laser power was delivered to single HSC-3 cell, and long integration time (500 ms per pixel) was applied to obtain a high-quality single cell Raman image. It took about 13.5 min to acquire the single cell Raman image in Figure 4A. The time for cell imaging should be reduced greatly to monitor rapidly changing and complex biological events occurring inside cells. In this regards, the use of SERS-active nanoparticle can provide various advantages for live cell Raman imaging such as reduced imaging time, decreased excitation power, particle tracking capabilities, and enhanced Raman signals of biomolecules of interest inside the cell.
Figure 4.
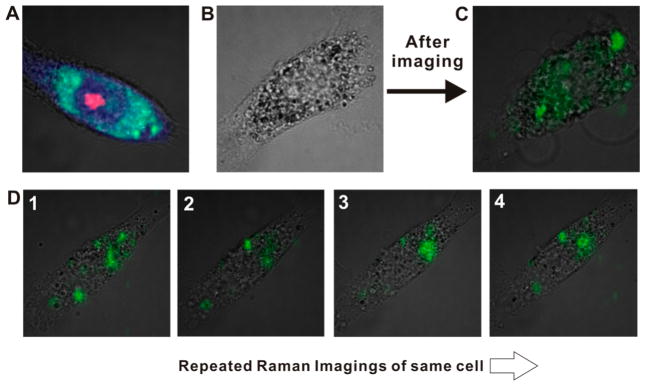
(A) Label-free Raman images overlaid with bright field images of single HSC-3 cell. Red, green, and blue colors represent the Raman intensities from 785, 1450, and 1004 cm−1, respectively. (B) Bright field images of HSC-3 cell after 12 h incubation with mitochondria-targeting Au-NNPs (before Raman mapping). (C) Overlaid Raman images with bright field image after Raman mapping (15 mW, 100 ms exposure time per pixel). (D) Repeated Raman analysis for HSC-3 cell incubated with 44DP-coded Au-NNPs obtained with a low laser power (200 μW) and exposure time (10 ms exposure time per pixel).
One potential issue, when using SERS-active gold nanoparticle for cell imaging, will be the photothermal damages on cells during the laser illumination for Raman signal acquisition. In spite of no plasmonic absorption property of Au-NNP at 785 nm as shown in Figure S5, Supporting Information, significant cell damages after single cell imaging were observed when applied incident laser power more than 4.0 mW. The bright field cell images in Figure 4B showed significant morphological changes in cell after illuminating the laser for Raman measurement, indicating significant cell damage occurred by the presence of nanoparticles. Importantly, we found the photothermal damages could be minimized with the use of low laser power density (less than 2.0 mW) without sacrificing Raman intensity from Au-NNP. The repeated Raman imaging for HSC-3 cell, which is incubated for 12 h with 44DP-coded Au-NNPs, with the power of 200 μW and 10 ms integration time per pixel showed no significant cell damages as demonstrated in Figure 4D (1–4, images were obtained 1–2 min time intervals). The bright field images of a single HSC-3 cell showed very little morphological changes. More importantly, almost identical Raman images in terms of Raman signal distributions and intensity were obtained, indicative of no significant photothermal damages in cell and photobleaching of the Raman dyes in Au-NNPs. Obtaining Raman image without inducing cell damage is essential for further application of SERS-based analytical methods such as cell-based drug screening (the laser power dependences on the cell morphology and viability were summarized in Figure S6, Supporting Information).
To demonstrate the potentials of high speed and high-resolution live-cell Raman imaging technology for drug screening capabilities, we monitored the rapidly changing cell morphologies and Raman signal distributions of 44DP-coded Au-NNPs in the mitochondria of HSC-3 cell by inducing cell death using strong cytotoxic reagents such as potassium cyanide (KCN) (Figure 5). After addition of 100 μL of KCN solution (1.0 M) into a HSC-3 cell, the single cell was imaged repeatedly at every 2.5 min with a 200 μW of laser power and 10 ms exposure per pixel (50 × 50 pixel). The overlaid images of bright field and Raman image at t = 0, it showed typical distributions of 44DP-coded Au-NNPs in the mitochondria as shown in Figure 5A. After 2.5 min of addition, it showed some changes of Raman signal distributions in the cell. After 5 min, the cell morphologies were significantly changed, and the changes of Raman signal distributions were also observed (green in Figure 5A). Eventually it was completely changed into circular cell structure, indicative of cell death (necrosis). In the case of 10 μL of KCN solution (1.0 M) addition, it also showed the changes of cell morphologies and Raman signal distributions with time (Figure S7, Supporting Information).
Figure 5.
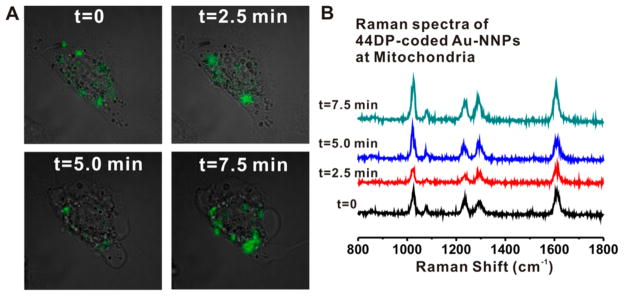
(A) Time-dependent Raman images overlaid with bright field image after additions of 100 μL of potassium cyanide solution (1.0 M) into HSC-3 cell. (B) Representative Raman spectra obtained from inside cells at designated time points (0, 2.5, 5.0, and 7.5 min). Imaged at every 2.5 min after additions of KCN with an analysis conditions (50 × 50 pixel, 200 μW laser power density, 10 ms/pixel, and 27.5 s (total imaging time)).
In summary, we found the use of specific spacer sequence (T10) of oligonucleotides on the core DNA-modified AuNP could lead to the formation of very narrow intra-nanogap structures in Au-NNPs even in the presence of a large number of Raman reporters on core DNA-AuNP. The narrow intra-nanogap and the presence of a large number of NIR-sensitive Raman reporters (i.e., 44DP) in the nanogap were responsible for the 7.7 times increased magnitudes of Raman responses at NIR wavelength and the strong and uniform Raman responses in solution. The bright SERS-intensity of Au-NNP in solutions and a custom-built high-speed confocal Raman microscopy enabled to accomplish high-resolution single live-cell Raman imaging within 30 s. The current methods also showed excellent subcellular organelle targeting capabilities as clearly evidenced by the colocalized Raman images with fluorescent probes (Figure S8, Supporting Information) and multiplexed imaging capabilities for informative cellular imaging (Figure S9, Supporting Information). It is expected that the synthetic methods for Au-NNP would be a useful concept for the future design of SERS-active nanostructures, and the high resolution live cell Raman imaging method within half minutes can open new opportunities for Raman-based high throughput and high contents drug screening platforms.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF-2013R1A1A1061387) and Jeon-Dae Memorial Fund. J.W.K., R.R.D., and P.T.C.S. acknowledge support from NIH 9P41EB015871-27 and the MIT SkolTech initiative. PTCS further acknowledges support from NIH 5R01EY017656-02, 5R01NS051320, 4R44EB012415-02, NSF CBET-0939511, the Singapore-MIT Alliance 2, the Hamamatsu Corp., and the Koch Institute for Integrative Cancer Research Bridge Project Initiative. The authors would like to thank Dr. Freddy T. Nguyen for proofreading the manuscript.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
Materials, experimental details including synthesis, and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Abramczyk H, Brozek-Pluska B. Chem Rev. 2013;113:5766–5781. doi: 10.1021/cr300147r. [DOI] [PubMed] [Google Scholar]
- 2.Fujita K, Ishitobi S, Hamada K, Smith NI, Taguchi A, Inouye Y, Kawata S. J Biomed Opt. 2009;14:024038–7. doi: 10.1117/1.3119242. [DOI] [PubMed] [Google Scholar]
- 3.Hamada K, Fujita K, Smith NI, Kobayashi M, Inouye Y, Kawata S. J Biomed Opt. 2008;13:044027–4. doi: 10.1117/1.2952192. [DOI] [PubMed] [Google Scholar]
- 4.Kang JW, Lue N, Kong CR, Barman I, Dingari NC, Goldfless SJ, Niles JC, Dasari RR, Feld MS. Biomed Opt Express. 2011;2:2484–2492. doi: 10.1364/BOE.2.002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puppels GJ, de Mul FFM, Otto C, Greve J, Robert-Nicoud M, Arndt-Jovin DJ, Jovin TM. Nature. 1990;347:301–303. doi: 10.1038/347301a0. [DOI] [PubMed] [Google Scholar]
- 6.Freudiger CW, Min W, Saar BG, Lu S, Holtom GR, He C, Tsai JC, Kang JX, Xie XS. Science. 2008;322:1857–1861. doi: 10.1126/science.1165758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puppels GJ, Grond M, Greve J. Appl Spectrosc. 1993;47:1256–1267. [Google Scholar]
- 8.Okuno M, Hamaguchi H-o. Opt Lett. 2010;35:4096–4098. doi: 10.1364/OL.35.004096. [DOI] [PubMed] [Google Scholar]
- 9.Ling J, Weitman SD, Miller MA, Moore RV, Bovik AC. Appl Opt. 2002;41:6006–6017. doi: 10.1364/ao.41.006006. [DOI] [PubMed] [Google Scholar]
- 10.Palonpon AF, Ando J, Yamakoshi H, Dodo K, Sodeoka M, Kawata S, Fujita K. Nat Protoc. 2013;8:677–692. doi: 10.1038/nprot.2013.030. [DOI] [PubMed] [Google Scholar]
- 11.Yamakoshi H, Dodo K, Okada M, Ando J, Palonpon A, Fujita K, Kawata S, Sodeoka M. J Am Chem Soc. 2011;133:6102–6105. doi: 10.1021/ja108404p. [DOI] [PubMed] [Google Scholar]
- 12.Huefner A, Kuan WL, Barker RA, Mahajan S. Nano Lett. 2013;13:2463–2470. doi: 10.1021/nl400448n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kneipp J, Kneipp H, McLaughlin M, Brown D, Kneipp K. Nano Lett. 2006;6:2225–2231. doi: 10.1021/nl061517x. [DOI] [PubMed] [Google Scholar]
- 14.Kang B, Austin LA, El-Sayed MA. ACS Nano. 2014;8:4883–4892. doi: 10.1021/nn500840x. [DOI] [PubMed] [Google Scholar]
- 15.Aioub M, Kang B, Mackey MA, El-Sayed MA. J Phys Chem Lett. 2014;5:2555–2561. doi: 10.1021/jz501091x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell SEJ, Sirimuthu NMS. J Am Chem Soc. 2006;128:15580–15581. doi: 10.1021/ja066263w. [DOI] [PubMed] [Google Scholar]
- 17.Lu G, De Keersmaecker H, Su L, Kenens B, Rocha S, Fron E, Chen C, Van Dorpe P, Mizuno H, Hofkens J, Hutchison JA, Uji-i H. Adv Mater. 2014;26:5124–5128. doi: 10.1002/adma.201401237. [DOI] [PubMed] [Google Scholar]
- 18.Kang JW, Nguyen FT, Lue N, Dasari RR, Heller DA. Nano Lett. 2012;12:6170–6174. doi: 10.1021/nl302991y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim DK, Jeon KS, Hwang JH, Kim H, Kwon S, Suh YD, Nam JM. Nat Nanotechnol. 2011;6:452–460. doi: 10.1038/nnano.2011.79. [DOI] [PubMed] [Google Scholar]
- 20.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 21.Oh JW, Lim DK, Kim GH, Suh YD, Nam JM. J Am Chem Soc. 2014;136:14052–14059. doi: 10.1021/ja504270d. [DOI] [PubMed] [Google Scholar]
- 22.Zavaleta CL, Smith BR, Walton I, Doering W, Davis G, Shojaei B, JNM, Gambhir SS. Proc Natl Acad Sci USA. 2009;106:13511–13516. doi: 10.1073/pnas.0813327106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang B, Mackey MA, El-Sayed MA. J Am Chem Soc. 2010;132:1517–1519. doi: 10.1021/ja9102698. [DOI] [PubMed] [Google Scholar]
- 24.Austin LA, Kang B, Yen CW, El-Sayed MA. J Am Chem Soc. 2011;133:17594–17597. doi: 10.1021/ja207807t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Austin LA, Kang B, Yen CW, El-Sayed MA. Bioconjugate Chem. 2011;22:2324–2331. doi: 10.1021/bc200386m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.