

Abstract
Together with vector control, chemotherapy is an essential tool for the control of visceral leishmaniasis (VL), but its efficacy is jeopardized by growing resistance and treatment failure against first-line drugs. To delay the emergence of resistance, the use of drug combinations of existing antileishmanial agents has been tested systematically in clinical trials for the treatment of visceral leishmaniasis (VL). In vitro, Leishmania donovani promastigotes are able to develop experimental resistance to several combinations of different antileishmanial drugs after 10 weeks of drug pressure. Using an untargeted liquid chromatography-mass spectrometry (LC-MS) metabolomics approach, we identified metabolic changes in lines that were experimentally resistant to drug combinations and their respective single-resistant lines. This highlighted both collective metabolic changes (found in all combination therapy-resistant [CTR] lines) and specific ones (found in certain CTR lines). We demonstrated that single-resistant and CTR parasite cell lines show distinct metabolic adaptations, which all converge on the same defensive mechanisms that were experimentally validated: protection against drug-induced and external oxidative stress and changes in membrane fluidity. The membrane fluidity changes were accompanied by changes in drug uptake only in the lines that were resistant against drug combinations with antimonials, and surprisingly, drug accumulation was higher in these lines. Together, these results highlight the importance and the central role of protection against oxidative stress in the different resistant lines. Ultimately, these phenotypic changes might interfere with the mode of action of all drugs that are currently used for the treatment of VL and should be taken into account in drug development.
INTRODUCTION
Visceral leishmaniasis (VL) is a protozoan disease caused by different species of Leishmania, including Leishmania donovani, and affects ≥0.2 to 0.4 million people in 98 countries. More than 90% of VL cases develop in India, Bangladesh, Nepal, Sudan, and Brazil (1). Since the disease is lethal if left untreated, treatment is one of the major tools in the fight against VL, although there is a limited arsenal of available drugs. Pentavalent antimonials (SbV) have been used since 1923 and are still the recommended first-line treatment, except on the Indian subcontinent, where they were abandoned because of resistance (2, 3). Antimonials (Sb) have a complex (and not yet fully understood) multifactorial mode of action involving (i) a direct inhibitory effect of SbIII on trypanothione reductase of the parasite (4) and (ii) an indirect effect of SbV by the stimulation of macrophages that kill their intracellular invaders (5). Several molecular adaptations have been encountered in Sb-resistant clinical isolates (including genomic structural variation and a wide range of metabolic changes in RNA-DNA synthesis, phospholipid, and energy metabolism), suggesting a complex multifactorial mechanism of resistance (6, 7). Amphotericin B (AmB) or one of its lipid-carrier formulations is another highly effective antileishmanial agent often used in second-line treatments (8, 9). AmB forms a complex with ergosterol, the major sterol of the Leishmania cellular membrane, forming aqueous pores leading to increased membrane permeability and subsequent ion imbalance, which kill the parasite; however, the high cost and therapeutic complications of AmB limit its use (10). AmB resistance is rare under clinical conditions (11), but Purkait et al. (12) reported one AmB-resistant (AmBr) clinical isolate of L. donovani, in which they encountered the overexpression of ATP-binding cassette (ABC) transporters, the upregulation of the thiol metabolic pathway, and altered membrane permeability. In recent years, miltefosine (MIL) has been introduced as the first oral agent against VL, but factors, such as its teratogenic potential and decreasing efficacy, might limit its use (13). The antileishmanial activity of MIL involves the intracellular accumulation of the drug, which is regulated by a phospholipid translocase, the MIL transporter complex (14, 15). The exact mode of antileishmanial action is still unclear, although it has been found to cause apoptosis-like processes in L. donovani (16), affect phospholipid composition (17), and inhibit cytochrome c oxidase (18). MIL resistance in clinical isolates is still rare so far, but it can easily be induced experimentally. Under these conditions, it is associated with inactivating point mutations in the MIL transporter complex (14, 15, 19) and the altered expression of genes related to DNA repair and replication, lipid metabolism, protein synthesis, transport activity, and antioxidant defense (20). Another antileishmanial drug is paromomycin (PMM), an aminoglycoside antibiotic that inhibits protein synthesis in Leishmania (21) and has been used for VL treatment in regions that are endemic for the disease (22). PMM resistance in Leishmania has been reported only experimentally and is associated with altered membrane fluidity, decreased drug uptake, and increased expression of ABC transporters (23, 24).
Environmental changes, drug resistance, and immunosuppression contribute to the emergence and spread of VL. Chemotherapy, together with vector control, remains the mainstay of VL control (25). In this context, the WHO recommends to use drug combinations of existing antileishmanial agents in order to reduce the duration, cost, and toxicity of treatment, prolong the therapeutic life span of existing drugs, and delay the emergence of resistance. Combinations have recently been tested systematically in clinical trials (26, 27), but additional studies are needed to monitor the long-term efficacy of combination therapy and determine the potential risk of the emergence of resistance. The recent findings of experimental resistance in L. donovani to several combinations of different antileishmanial drugs, after 10 weeks of drug pressure, are of great concern (28). In order to prevent and monitor the emergence of resistance against combination therapy, it is essential to identify the molecular adaptations developed by the parasites that are resistant to drug combinations.
Several “omic” technologies offer unprecedented opportunities for global characterization of pathogens. Metabolomics is particularly relevant for studies on drug resistance, as the metabolome is regarded as the closest representation of the resistance phenotype. Furthermore, this profiling technology is being increasingly used for experimental research on trypanosomatids, since the upstream “omics” (genomics and transcriptomics) are complicated by (i) limitations in the functional annotation of identified sequences and (ii) the fact that their gene expression is regulated at the posttranscriptional level. Hence, these studies might have limitations when studying the rapid effect of drugs or the mechanism behind rapidly acquired drug resistance (29). In the present study, we implemented an untargeted metabolomic approach to identify the metabolic changes in isogenic lines experimentally resistant to several drug combinations (CTR lines) and their respective single-resistant lines (single-R lines). We addressed both quantitative and qualitative differences in the metabolomes of the CTR lines and experimentally validated the main emerging hypotheses.
MATERIALS AND METHODS
Chemicals.
Trivalent antimony (SbIII), amphotericin B (AmB), paromomycin (PMM), propidium iodide (PI), DPH (1,6-diphenylhexa-1,3,5-triene), cholic acid, bovine serum albumin (BSA), thiazolyl blue tetrazolium (MTT), H2O2, menadione sodium bisulfite, and l-proline were obtained from Sigma-Aldrich (St. Louis, MO). Miltefosine (MIL) was purchased from Zentaris GmbH (Frankfurt am Main, Germany). Hexadecylphospho[1,2-ethylene-14C]choline ([14C]MLF) (35.9 μCi/mmol) was synthesized by Amersham Pharmacia Biotech (Buckinghamshire, United Kingdom). [3H]Paromomycin (156.25 μCi/mmol) was custom synthesized by Moravek Biochemicals, Inc. For the serial dilution test (described in the supplemental material), an MIL batch from Haupt Pharma was included. All drug stock solutions were prepared in RPMI 1640-modified medium (Invitrogen) supplemented with 20% (vol/vol) heat-inactivated fetal calf serum (hiFCS) (Invitrogen). Phosphate-buffered saline (PBS) (pH 7.4), Sytox green, dichlorofluorescein diacetate (H2DCFDA), MitoSOX red, and RNase A were purchased from Invitrogen.
Leishmania culture conditions.
All lines were grown at 28°C in RPMI 1640 plus 20% hiFCS at pH 7.2 and 28°C, and the resistant lines were maintained in the continuous presence of drugs, as described in reference 28. The following abbreviations will be used for the single-R and CTR lines (the drug[s] to which they are resistant is mentioned in parentheses): S line (SbIII), A line (AmB), M line (MIL), P line (PMM), AM line (AmB/MIL), AP line (AmB/PMM), AS line (AmB/SbIII), MP line (MIL/PMM), and SP line (SbIII/PMM).
Metabolite extraction.
Parasite cultures were initiated by inoculating 1 × 106 parasites/ml from a log-phase culture in 5 ml of RPMI 1640 medium plus 20% hiFCS. Four independently growing cultures of each line were further treated as biological replicates (BR). The different lines were grown synchronously, while the growth was monitored daily by microscopic counting; the different lines were all harvested in the stationary-growth stage for metabolite extraction. The metabolite extraction protocol was done as previously described (30).
Liquid chromatography and mass spectrometry analysis.
Elution of the guard column (20 mm by 2.1 mm inside diameter [i.d.], 5 μm; Merck/SeQuant) and Zic-HILIC high-performance liquid chromatography (HPLC) column (150 mm by 2.1 mm i.d., 3.5 μm, 100 Å; Merck/SeQuant) was carried out with a gradient of (A) 0.1% formic acid in acetonitrile and (B) 0.1% formic acid in water. The flow rate was 100 μl/min, with an injection volume of 10 μl. Gradient elution was performed as described in t'Kindt et al. (30). High-resolution mass measurements were obtained with an Exactive Orbitrap mass spectrometer (Thermo Fisher) at the Scottish Metabolomics Facility (Glasgow Polyomics, University of Glasgow, Glasgow, Scotland). Fast polarity switching allows for simultaneous measurements in positive and negative ion electrospray mode. The electrospray ionization (ESI) source voltage was optimized to 4.5 kV and 3.5 kV (for the positive and negative modes, respectively), and capillary voltage was set to 40 V and 30 V (for the positive and negative modes, respectively). The source temperature was set to 275°C, and the sheath and auxiliary gas flow rates were set to 40 and 5 machine-specific units, respectively. Full-scan spectra were acquired over an m/z-range of 70 to 1,400 Da, with the mass resolution set to 50,000 full width at half maximum (FWHM). All spectra were collected in continuous single mass spectrometry (MS) mode. A sample list setup was performed as described previously (7, 31), and data processing and metabolite identification were performed in mzMatch, as described extensively in Berg et al. (7). Briefly, the selected mass chromatograms were putatively identified by matching the masses progressively to those from metabolite-specific databases (mass accuracy, <2 ppm, after correction for loss or gain of a proton in negative-mode or positive-mode ESI, respectively). In a first round of identification, an in-house Leishmania database was used (based on LeishCyc and further completed with identifications from Lipid Metabolites and Pathways Strategy [MAPS] [30, 32]). The remaining unidentified peaks were subjected to a second round of matching against the complete Lipid MAPS (32) and then against KEGG (33). The putative identifications for the lipids were manually annotated with the total number of carbons, the number of double bonds in side chains, and the number of side chains. The rejected metabolites from previous rounds were then matched against a contaminant database (34), allowing the removal of typical impurities and buffer components often detected in metabolomics experiments, a peptide database, and the Human Metabolome Database (35). This iterative process was used in order to restrict the number of potential matches to the most likely matches (36).
The few samples that displayed unacceptable analytical variation (signal intensity drift) were removed from further analysis (M_BR1, S_BR2, and AM_BR3). Data processing yielded a total of 257 putative metabolites. Relative quantification was based on raw peak heights and expressed relative to the (average) peak height of the untreated wild-type line maintained in culture (WTM). This relative expression was called the fold change.
Measurement of reactive oxygen species production.
The generation of intracellular reactive oxygen species (ROS) with or without drug pressure was measured using the cell-permeable nonfluorescent probe dichlorofluorescein diacetate (H2DCFDA), as described in Manzano et al. (37). Briefly, parasites (1 × 107/ml) of the different L. donovani lines (control [WTM], A, M, S, AS, and SP) were left untreated or exposed to 0.1 μM AmB, 10 μM MIL, or 100 μM SbIII for 48 h at 28°C in culture medium. Next, the promastigotes were washed with HBS buffer (21 mM HEPES, 0.7 mM Na2HPO4, 137 mM NaCl, 5 mM KCl, and 6 mM d-glucose [pH 7.1]) and incubated in the same buffer supplemented with 40 nM H2DCFDA for 30 min at 28°C. The fluorescent dichlorofluorescein (DCF) product obtained after esterase cleavage and oxidation was measured by flow cytometry using a FACScan flow cytometer (Becton-Dickinson, San Jose, CA; excitation, 488 nm; emission, 535 nm). On the other hand, we used the cell-permeable fluorogenic probe MitoSOX red, which selectively targets mitochondria, to detect mitochondrial ROS production, as described in Manzano et al. (37). Thus, parasites (1 × 107/ml) of the different L. donovani lines were loaded with 5 μM MitoSOX for 2 h at 28°C and then washed and resuspended in culture medium with or without 0.1 μM AmB, 10 μM MIL, or 100 μM SbIII for 48 h at 28°C. After washing, the fluorescence of oxidized MitoSOX red was measured by flow cytometry using a FACScan flow cytometer (excitation, 488 nm; emission, 580 nm).
Susceptibility of Leishmania lines to oxidative stress induced by menadione and H2O2.
The 50% effective concentration (EC50) and resistance index (EC50 ratio for resistant and WTM parasites) to menadione and H2O2 were determined for the WTM, A, M, P, S, AS, and SP lines using an MTT colorimetric assay after incubating the promastigotes for 72 h at 28°C in the presence of increasing concentrations of menadione or H2O2, as described previously (38, 39).
Susceptibility of Leishmania lines to AmB, MIL, or SbIII in the presence of proline.
WTM line promastigotes (4 × 106 parasites/ml) were incubated in RPMI 1640 medium with or without 200 mM proline for 120 h. During the last 72 h of this incubation, AmB, MIL, or SbIII was added to determine the EC50 to these drugs using an MTT colorimetric assay, as described in Kennedy et al. (38).
DNA content analysis.
DNA content was analyzed by flow cytometry, as described in reference 40. Briefly, parasites (4 × 106 promastigotes/ml) of the different L. donovani lines (WTM, A, M, S, AS, and SP) were incubated with and without their corresponding drugs at 0.1 μM AmB, 10 μM MIL, or 100 μM SbIII for 48 h at 28°C in culture medium. Next, 1 × 107 promastigotes were washed twice with PBS buffer, fixed in ice-cold methanol for 3 min on ice, resuspended in 500 μl of 1 μg/ml PI and 100 μg/ml RNase A in PBS, and incubated for 1 h in the dark at room temperature. Dye fluorescence was measured by flow cytometry using a FACScan flow cytometer (Becton Dickinson, San Jose, CA), and fluorescence emission was quantified using the CellQuest software.
Preparation of parasite surface membrane-enriched fraction.
Leishmania promastigotes of different lines (109 cells/ml) were disrupted in lysis buffer (10 mM Tris-HCl, 2 mM EDTA, 25 μg/ml leupeptin [pH 8.0]) for 45 min in a prechilled high-pressure cavitator. The parasite lysates were centrifuged at 1,000 × g to eliminate cell debris, and the obtained supernatant was centrifuged at 8,000 × g at 4°C for 30 min to obtain parasite surface membrane-enriched fractions. The supernatant was removed and the pellets washed twice with 10 mM HEPES and 145 mM NaCl (pH 7.4) buffer before being resuspended in the same buffer. The alkaline phosphatase tartrate-resistant activity was determined as a control of the membrane-enriched fractions, as described in reference 41.
Membrane fluidity assessment.
The membrane fluidity of the different L. donovani lines was measured as described in Manzano et al. (42). Briefly, Leishmania plasma membrane samples (0.06 to 0.11 mg/ml final protein concentration) in 10 mM HEPES and 145 mM NaCl (pH 7.4) buffer were incubated with DPH (1,6-diphenylhexa-1,3,5-triene) probe in N,N′-dimethylformamide (DMF) in the dark for 30 min at a 1:2,500 probe-to-protein weight ratio. The final DMF concentration in the membrane suspension was always <0.05%. Steady-state fluorescence anisotropy (r), as defined by Lakowicz (43), was determined by measuring the vertical and horizontal components of the fluorescence emission with excitation polarized vertically in a Cary Eclipse spectrofluorimeter (Agilent Technologies). The grating factor (GF) is specific for the instrument and is defined by the ratio of the fluorescence intensities with polarizers that are horizontal and vertical (excitation and emission, respectively) and horizontal and horizontal, respectively. The slit widths for both excitation and emission were 5 nm. The excitation wavelength was 360 nm, and emission was monitored at 430 nm. The temperature was fixed to 28°C. The data for each experiment were calculated as the average of 10-s anisotropy measurements.
Plasma membrane integrity.
Sytox green dye was used to assess plasma membrane integrity, as described previously, with some modifications (40). Briefly, parasites (1 × 107 promastigotes) of the different L. donovani lines (WTM, A, M, S, AS, and SP) were left untreated or treated with increasing concentrations of their corresponding drugs (AmB, MIL, or SbIII) for 48 h at 28°C in culture medium, washed twice with HBS buffer, and then incubated with 2 μM Sytox green for 15 min at 28°C. After washing, the retained fluorescence was measured by flow cytometry using a FACScan flow cytometer (excitation, 488 nm; emission, 525 nm).
Accumulation of AmB, MIL, PMM, or SbIII in Leishmania lines.
The AmB accumulation assays were performed as described in Purkait et al. (12), with the modification that AmB-resistant (A and AS) and -susceptible (WTM) promastigotes (1 × 108 parasites/ml) were incubated in RPMI 1640 medium with 0.1 μM AmB for 1 h before the washing steps. The supernatants were analyzed by ultraperformance liquid chromatography-mass spectrometry (UPLC-MS), as previously described, with some modifications (44). Chromatography analysis was carried out on a Waters UPLC Acquity H-Class system. The reverse-phase chromatography separation was performed on a Cortecs UPLC C18 1.6-μm column (2.1 by 50 mm) (Waters Corp., Milford, MA, USA), with the column temperature maintained at 40°C. The gradient elution for UPLC was performed with two solvents: solvent A, H2O plus 0.1% formic acid, and solvent B, acetonitrile containing 0.1% formic acid. The gradient began with 90% eluent A and decreased to 5% A over 5.0 min before returning to 90% A at 7.10 min. The UPLC was connected to a triple quadrupole tandem-mass detector Xevo TQS (Waters Corp.) with an electrospray ionization (ESI) source for mass spectrometry detection, monitoring the ion for AmB at 906.73 m/z. The mass spectrometry parameters are listed in Table S1 in the supplemental material. A stock standard solution of AmB was prepared in methanol-dimethyl sulfoxide (DMSO) (9:1 [vol/vol]) at a concentration of 0.2 mg/ml and stored in 4.0-ml plastic vials at −20°C covered with aluminum foil to protect the AmB from light. Different working standard solutions of AmB were prepared by a dilution of the above-mentioned stock solutions in pure methanol and were kept at −20°C. For measuring the internalization of [14C]MIL, the following adjustments were made from the procedure of Sánchez-Cañete et al. (45): 2 × 107 promastigotes per ml of WTM or M lines were incubated in culture medium with 2.5 μM [14C]MIL (0.09 μCi/ml) for 1 h at 28°C before the washing steps. To determine the accumulation of [3H]PMM, 2 × 107 promastigotes per ml of the WTM, P, or SP line were incubated in HBS supplemented with 1% d-glucose plus 10 μM [3H]PMM (1.56 μCi/ml) for 1 h at 28°C. Next, the samples were washed with PBS and incubated with 500 μM PMM on ice for 15 min to eliminate the radioactivity bound to the cell surface. After a second PBS wash, the protein concentration and counts per min were determined. Antimony accumulation was measured by inductively coupled plasma MS (ICP-MS), as described in Manzano et al. (37), with a specific adaptation of the protocol: promastigotes (1 × 108 per ml) of the WTM, S, AS, and SP lines were incubated with 100 μM SbIII in RPMI 1640 culture medium for 1 h at 28°C prior to the washing steps and ICP-MS analysis.
Statistical analysis.
For the metabolomics data analysis, a two-sample t test assuming unequal variance was combined with a multiple testing correction by the Benjamini-Hochberg method to calculate whether or not the fold changes were statistically significant and to keep the false-discovery rate (q) at <5% (P < 0.05 and p < q). For all other experiments, the statistical comparisons between the groups were performed using Student's t test. Metabolite changes were considered to be biologically significant when the ratio of signal intensity between the resistant line and the WTM line (fold change) was >2 (significant increase) or <0.5 (significant decrease) and statistically significant (P < 0.05 and P < q); other ratios were considered to be nonsignificant changes.
RESULTS
Metabolomics. (i) Quantitative analysis.
The same L. donovani promastigote strain, MHOM/ET/67/HU3, was previously made resistant to (i) single drugs (single-R lines) SbIII (S line), AmB (A line), MIL (M line), and PMM (P line) and (ii) drug combinations (combination therapy-resistant [CTR] lines) AmB/MIL (AM line), AmB/PMM (AP line), AmB/SbIII (AS line), MIL/PMM (MP line), and SbIII/PMM (SP line) by in vitro selection through a stepwise adaptation process, as described in García-Hernández et al. (28). The wild-type original (WTO) parental strain was included in the metabolomics assay; to ensure that no large metabolic changes occurred during in vitro parasite maintenance, a wild-type line maintained in culture (WTM) was compared with the WTO line. In total, 257 metabolites were putatively identified in all lines, of which 133 had a mass accuracy of <1 ppm and 124 had a mass accuracy between 1 and 2 ppm. The 257 metabolites represented the following classes (in order of quantitative representation; see Fig. S1 in the supplemental material): glycerophospholipids (GPLs), amino acids and derivatives, fatty acyls, purines and pyrimidines, carbohydrates, sphingolipids and sphingoid bases, steroids and derivatives, glycerolipids, vitamins, and cofactors. No significant changes were detected comparing the metabolome of the WTO to the WTM stock, showing that keeping the WT line in culture for several passages does not alter metabolism considerably, and that the detection of differential metabolites (Δmb) in this study is only due to drug resistance and/or drug pressure. Most significant changes (compared to WTM) were found in the lines resistant to SbIII and its combinations (number of changes): S (29), AS (31), SP (49), and also A (32) (Fig. 1). Fewer significant changes were detected in the M (20), P (14), MP (11), AP (5), and AM (3) resistant lines (Fig. 1). Whereas in the A and SP resistant lines, both significantly increased and decreased metabolites were present, Δmb were most often increased in all other resistant lines (Fig. 1). The amount of Δmb was not necessarily related to the level of resistance of the corresponding lines (see, for instance, lines P and MP, which showed the highest resistance index values but lowest numbers of Δmb; Fig. 1).
FIG 1.

Number of differential metabolites in single-R and CTR L. donovani lines compared to those in the WTM line. The resistance index (RI) against AmB, MIL, SbIII, and PMM, respectively, is mentioned for each single-R line (A, M, S, and P) and CTR line (RI/RI) (AS, SP, AM, MP, and AP) (adapted from García-Hernández et al. [28]). Differential metabolites (Δmb) are either increased or decreased (fold change, <0.5 or >2 and P < 0.05 and P < q).
A principal component analysis (PCA) based on all 257 identified metabolites enabled the visualization of the major differences between the samples (Fig. 2). The first principal component (PC1, 22.49%) was not able to separate the samples, but the second and third principal components (PC2, 17.38%; PC3, 11.06%) gathered the WTM, WTO, P, AP, and AM lines together in one cluster, which can be explained by the low number of Δmb in these lines (Fig. 1). The other lines are more clearly separated (A, S, AS, MP, and SP), whereas the M line is close to the central cluster but contains more Δmb.
FIG 2.
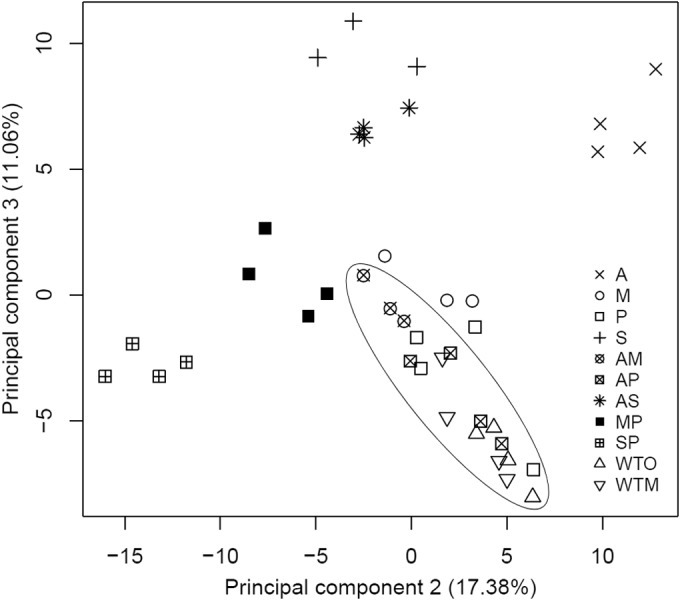
Principal component analysis distinguishes single-R and CTR L. donovani promastigote lines. This analysis was based on the quantitative measurements of all 257 putatively identified compounds. In this study, three biological replicates were removed due to signal intensity drift: M_BR1, S_BR2, and AM_BR3. Separate PCA plots for each CTR and their respective single-R lines can be found in Fig. S2 in the supplemental material.
The metabolic profiles were also analyzed individually for each of the five CTR lines and compared with the single-R lines (Table 1; see also Fig. S2 in the supplemental material). Particular attention was given to three parameters: (i) the proportion of new metabolic changes in the CTR lines (number of new changes observed in a CTR line, i.e., changes not found in the single-R lines/total number of changes observed in the CTR line), (ii) the proportion of metabolites shared with single-R lines (i.e., the number of Δmb shared between CTR lines and each of the single-R lines/total number of Δmb encountered in the two single-R lines), and (iii) the dominant character of the single-R-associated changes in the CTR line (determined by the single-R line sharing the highest number of Δmb with the CTR line). In terms of new metabolic changes, CTR lines can be separated into three main classes: (i) no new changes at all (AP), (ii) new Δmb in the CTR line but fewer than the number of shared Δmb with the single-R lines (AM and AS), and (iii) new Δmb in the CTR line and more than the shared Δmb with the single-R lines (MP and SP). Interestingly, this classification fits with resistance index values being the highest in the MP and SP lines. The analysis of the proportion of Δmb shared with single-R lines further discriminates classes ii and iii mentioned above: class ii AM shares very little with A and M, while class ii AS shares a lot with A and S; similarly, class iii MP shares very little with M and P, while class iii SP shares a lot with S and P. Two CTR lines (AS and SP) show a strong dominance of one of the single-R adaptations, in both cases being related to the S line. Of note, this is not associated with high resistance index to SbIII (around 2 in both AS and SP). Reciprocally, the two CTR lines involving the P line and showing highest resistance index values to PMM (MP, 14; SP, 18) do not show a dominance of the P line among shared Δmb.
TABLE 1.
Metabolic profiles of the five L. donovani CTR lines
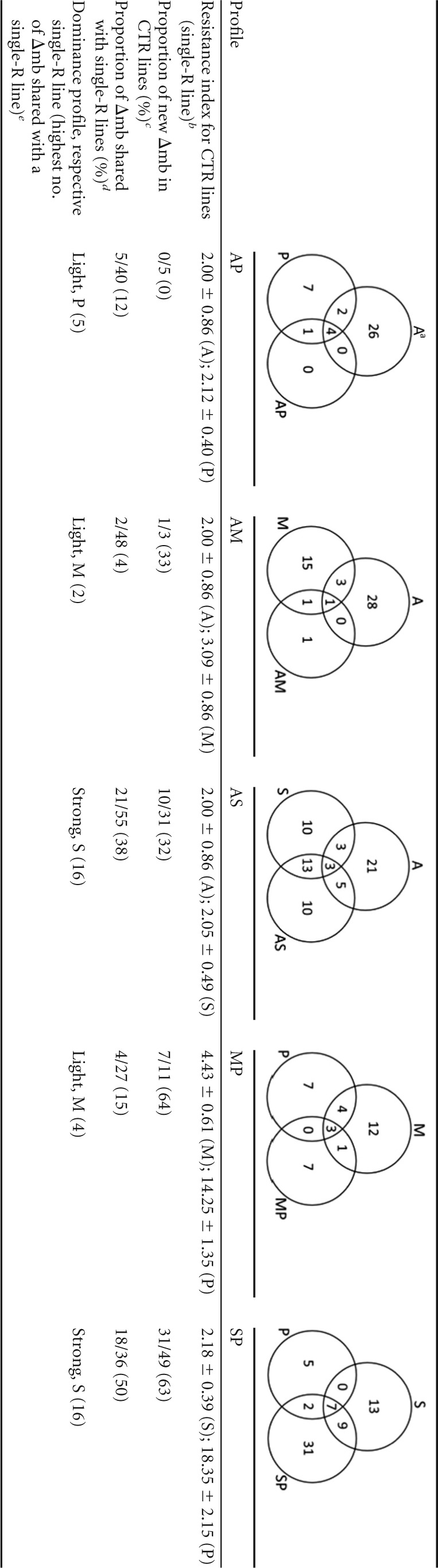
The Venn diagrams show the shared and new differential metabolites (Δmb) for each CTR strain in relation to the respective single-R strains.
The resistance indexes of the CTR line were calculated by dividing the EC50 for each resistant line by that for the WTM line and are the means ± the standard deviations (SD) of the resistance index values from three independent experiments (28).
The proportion of new Δmb in CTR lines was calculated by dividing the number of new changes observed in a CTR line by the total number of changes observed in the CTR line.
The proportion of metabolites shared with single-R lines was calculated by dividing the number of Δmb shared between CTR lines and each of the single-R lines by the total number of Δmb encountered in the two single-R lines.
The dominance profile was determined by the single-R line sharing the highest number of Δmb with the CTR line (≤5 shared Δmb, light; >10 shared Δmb, strong).
(ii) Qualitative analysis.
For a qualitative interpretation, we further focused on the CTR lines AS and SP since (i) most metabolic changes were detected in these lines (Table 1), and (ii) they show the most distinct profiles compared to that of WTM (Fig. 1). In the AS and SP lines, we detected metabolic changes that were shared between both CTR lines, here grouped as CTR collective metabolic changes (Fig. 3 and 4; see Table S2 in the supplemental material). The AS and SP lines shared 13 significantly increased metabolites and 3 significantly decreased metabolites (see Table S2). These collective metabolic changes were detected in the proline biosynthesis pathway (Fig. 3), the transsulfuration pathway (Fig. 3), and lipid metabolism (Fig. 4). One aromatic amino acid and a set of 5 scattered metabolites were also detected (Fig. 3, grouped as others). Both heatmaps (Fig. 3 and 4) indicate that these collective metabolic changes were in general more pronounced in the SP line. Several of these CTR collective metabolites were also retrieved as differential in the other CTR lines (see Table S3 in the supplemental material). For example, significant changes in proline metabolism were also found in the MP line, and a similar (although not significant) trend was shared with the other CTR lines AM and AP.
FIG 3.

Metabolic profiles of differential metabolites belonging to the proline biosynthesis pathway, transsulfuration pathway (TSU), tryptophan degradation (Trp degr.), acylglycines, and others in heatmap format. The samples are presented along the bottom. Averages were taken for the intensities of the biological replicates per line, and the intensities were rescaled between 0 (blue) and 100 (yellow). On the right, a first classification is made for the metabolites based on the metabolic pathway to which they belong. A second classification is made based on whether they are shared between the AS and SP line (CTR-collective) or are specific for one CTR line (AS or SP). The asterisk indicates that another isomer was detected for this metabolite (see Table S4 in the supplemental material for more details). AdoMet, S-adenosylmethionine; N-Ac-phenylalanine, N-acetylphenylalanine; 2-Me-erythritol-4-P, 2-methyl-erythritol-4-phosphate; sedoheptulose-7-P, sedopheptulose-7-phosphate. AdoMet was found to be increased significantly in both AS and SP lines but not by 2-fold in AS (fold change, 1.78). Tryptophan (Trp) was found to be increased by 2.58-fold in the AS line, with a P value of 0.0146 but with P > q.
FIG 4.

Metabolic profiles of differential metabolites belonging to the lipid metabolism in heatmap format. The samples are presented along the bottom. Averages were taken for the intensities of the biological replicates per line, and the intensities were rescaled between 0 (blue) and 100 (yellow). On the right, a classification is made based on whether they are shared between the AS and SP line (CTR collective) or specific for one CTR line (AS or SP). An asterisk indicates that another isomer was detected for this metabolite (see Table S4 in the supplemental material for more details). The abbreviations for glycerophosphocholines (GPCs) and glycerophosphoethanolamines (GPEs) should be interpreted as follows: GPL(x:y/z), where x represents the number of carbons in the fatty acid side chain(s), y represents the number of double bonds, and z represents the number of side chains. Glycerol-3-P, glycerol-3-phosphate; sterol A, a secosteroid with a monoisotopic mass of 398.318484 Da; sterol B, a secosteroid with a monoisotopic mass of 412.333833 Da (an ergostatetraene-diol and its isomers). Mevalonate was found to be decreased significantly in both AS and SP lines but not by 0.5-fold in AS (fold change, 0.53; P < 0.05).
Increased levels of aromatic amino acids, proline, indole acrylate, and the acylglycine caproylglycine were shared by the AS line and the respective single-R lines A and S (not always significantly; see Table 2). However, a clear additive resistance mechanism was not retrieved in the AS line, as reflected in Table 1 (Δmb in AS is not the sum of Δmb in A and S), and this is further supported by the fact that (i) 10 new metabolic changes (i.e., 32%) were found in the AS line (not present in the respective single-R lines), (ii) the important changes in the GPL metabolism observed in the A line (12 ΔGPLs) were completely absent in the AS line, and (iii) the AS line seems to be more adapted to antimonial drug pressure (Table 1, 13 shared Δmb between AS and S versus 5 shared Δmb between AS and A). For example, the increase of metabolites of the transsulfuration pathway (AdoMet and methionine) in the AS line is shared with the S line only. In both the A and AS lines, a sterol with a monoisotopic mass of 398.318484 Da (corresponding to a cholestatetraene-diol or its isomer hydroxy-cholestatriene-one, so far not present in the Leishmania databases at KEGG or MetaCyc) was found to be increased 2-fold. Thirteen metabolic changes were found in the AS line, which remained unaltered in the other CTR lines (specific metabolic changes in ovothiol, indole acrylate, methionine, pyrroline-carboxylate, caproylglycine) and were located in the same metabolic pathways as the CTR collective metabolic changes (Fig. 3 and 4; see also Table S2 in the supplemental material), proline biosynthesis, transsulfuration pathway, and lipid metabolism.
TABLE 2.
Key differential metabolites involved in the two hypotheses generated from the untargeted metabolomics study in L. donovani linesa
| Metaboliteb | Metabolic pathwayc | Fold change in resistant line vs WT lined |
|||||
|---|---|---|---|---|---|---|---|
| A/WTM | M/WTM | S/WTM | P/WTM | AS/WTM | SP/WTM | ||
| Tryptophan1 | Trp degradation pathway, aromatic amino acid | 3.22 | 3.44 | 2.12 | 1.29 | 2.58 | 0.89 |
| Indole acrylate1 | Trp degradation pathway | 3.33 | 3.43 | 2.12 | 1.36 | 2.49 | 0.90 |
| Caproylglycine1 | N-acylglycines | 2.67 | 1.26 | 3.08 | 1.66 | 2.37 | 1.00 |
| Valerylglycine1 | N-acylglycines | 1.97 | 0.80 | 1.32 | 1.11 | 1.17 | 0.38 |
| Proline1 | Proline biosynthesis pathway | 3.31 | 4.98 | 7.01 | 3.01 | 8.84 | 14.74 |
| Methionine1 | Precursor of AdoMet and homocysteine | 1.81 | 1.34 | 2.07 | 1.41 | 2.08 | 1.15 |
| AdoMet1 | Transsulfuration pathway | 0.72 | 1.82 | 2.81 | 1.44 | 1.78 | 2.41 |
| Pyridoxine1 | Vit B6 metabolism, transsulfuration pathway | 2.30 | 2.96 | 2.41 | 3.49 | 2.46 | 4.64 |
| Ovothiol1 | Unclassified | 1.30 | 0.57 | 2.34 | 1.18 | 2.79 | 1.26 |
| Tyrosine1 | Aromatic amino acid | 3.45 | 1.32 | 1.45 | 1.01 | 1.33 | 0.77 |
| N-acetyl-Phe1 | Acetylated aromatic acid | 3.70 | 1.29 | 8.46 | 1.40 | 6.40 | 2.66 |
| Mevalonate2 | Precursor of sterol biosynthesis | 0.11 | 0.522 | 0.31 | 0.42 | 0.53 | 0.16 |
| Glycerol-3-P2 | Precursor of glycerolipids | 0.78 | 1.67 | 1.94 | 1.70 | 2.24 | 2.89 |
| GPC(30:0/2) 2 | GPL | 0.81 | 1.21 | 2.39 | 1.69 | 1.84 | 2.18 |
| GPC(30:2/2) 2 | GPL | 0.55 | 0.80 | 1.53 | 1.39 | 1.48 | 2.06 |
| 3-Dehydrosphinganine2 | Sphingolipid precursor (sphingoid base) | 0.37 | 1.50 | 0.31 | 0.45 | 0.34 | 0.24 |
For each metabolite, the metabolic pathway is listed, and the fold changes in each single resistant line (A, M, S, P) and two CTR lines (AS, SP) compared to the WTM line are shown.
The superscripts 1 or 2 refer to involvement in hypothesis 1, protection against oxidative stress, or hypothesis 2, changes in membrane fluidity. More significant changes in the GPL metabolism can be found in Table S4 in the supplemental material.
Trp, tryptophan; GPL, glycerophospholipid.
Fold changes (>2, increase; <0.5, decrease) in bold are significant changes (P < 0.05 and P < q).
The SP line is the only CTR line in which there are more metabolic changes present than in the respective single-R lines (S [29] + P [14] was less than that in SP [46]; Fig. 1). We detected both significantly increased (14) and decreased (11) metabolites that were specific for this CTR line (except for one GPL and valerylglycine shared with the MP line) and again belonged to the aforementioned pathways of proline biosynthesis, lipid metabolism, and an(other) acylglycines (see Table S2 in the supplemental material). As seen in Fig. 4, it is clear that the proportion of new SP-specific metabolic changes (i.e., those not shared with the respective single-R lines) in the SP line (63%) is higher than that in the AS line (32%). Details on the metabolic changes in the single-R lines and other CTR lines, including correspondence with the respective single-R lines, can be found in Tables S3 and S4 in the supplemental material.
Experimental validation of main hypotheses.
The metabolic changes described above led us to develop two major hypotheses: (i) there was increased protection against oxidative stress (predicted among others by changes in the metabolites of both the proline biosynthesis and tryptophan degradation pathway, the transsulfuration pathway, and two aromatic amino acids) and (ii) there were alterations in the membranes (predicted by changes in metabolites of the lipid metabolism and a precursor of the steroid biosynthesis). Both hypotheses were validated with a series of functional experiments.
Protection against oxidative stress.
First, we studied the accumulation of ROS upon drug exposure in the control (WTM) and resistant Leishmania lines. For this purpose, we used the H2DCFDA dye, the fluorescence intensity of which is indicative of the levels of overall intracellular ROS. WTM parasites incubated with or without 0.1 μM AmB, 10 μM MIL, or 100 μM SbIII exhibited an increase in H2DCFDA fluorescence signals (Fig. 5A), confirming increased ROS production upon exposure to these antileishmanial drugs. In contrast, levels of ROS generation in the resistant lines were similar in both the absence and presence of antileishmanial drugs (Fig. 5A). Since the major source of oxidants in eukaryotes is the mitochondria, we used MitoSOX red dye (mitochondrial superoxide indicator) to measure the specific mitochondrial accumulation of superoxide, which is indicative of the ROS levels in the mitochondria. As expected, AmB, MIL, and SbIII induced an increase in MitoSOX red fluorescence levels in the WTM line but not in the resistant lines (Fig. 5B). The results obtained on ROS production showed an increased capacity for protection against oxidative stress in drug-resistant Leishmania lines; this might be due to decreased ROS production but also to a more efficient way of dealing with ROS. Therefore, in a second phase, the WTM and resistant Leishmania lines were directly tested for susceptibility to external oxidative stress by checking their susceptibilities to H2O2 and another stress-inducing agent, menadione. The results indicated that the A, M, P, and AS resistant lines showed a similar tolerance to menadione as that of the WTM line (37.58 μM) (Fig. 6A). In contrast, the S and SP resistant lines exhibited a higher tolerance to menadione than did the WTM line (1.54- and 1.41-fold, respectively) (Fig. 6A). Regarding H2O2, all resistant lines except the SP line showed a higher tolerance to H2O2 than did the WTM line (Fig. 6B); specifically, the highest EC50s were found for the S and AS lines (1.74- and 1.86-fold higher than the WTM line, respectively). Together, these experiments show that the CTR lines possess a more efficient mechanism to deal with ROS. In a next step, we demonstrated the effect of proline on drug susceptibility. Therefore, we incubated the WTM line in culture medium supplemented or not with 200 mM proline before analyzing the tolerances to different oxidative stress-inducing drugs. The results showed no differences in the tolerances to AmB and MIL in the presence of proline (Table 3). In contrast, the tolerance to SbIII increased 2-fold in the presence of proline (Table 3).
FIG 5.
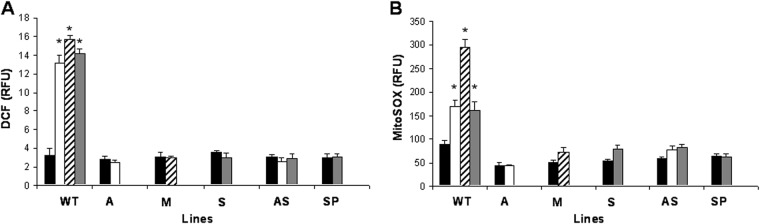
Drug-induced ROS generation in L. donovani lines. The L. donovani lines control (WTM), A, M, S, AS, and SP were left untreated (black columns) or exposed to 0.1 μM AmB (white columns), 10 μM MIL (diagonal line columns), or 100 μM SbIII (gray columns) for 48 h. The parasites were incubated with 40 nM H2DCFDA for 30 min at 28°C (A) or 5 μM MitoSOX for 2 h at 28°C (B). The fluorescence intensity was determined by flow cytometry analysis and expressed as relative fluorescence units (RFU). The data are the means ± standard deviation (SD) values from three independent experiments. Significant differences versus the control were determined by Student's t test (*, P < 0.001).
FIG 6.
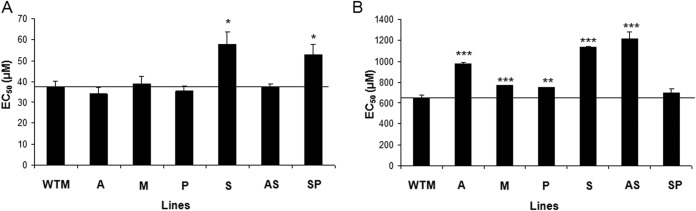
Drug susceptibility profile to menadione and H2O2 in L. donovani lines. Promastigote forms of L. donovani lines (WTM and resistant lines A, M, P, S, AS, and SP) were grown as described in Materials and Methods for 72 h at 28°C in the presence of increasing concentrations of menadione (A) or H2O2 (B). Cell viability was determined using an MTT-based assay. The data are the mean EC50 ± SD from three independent experiments. Significant differences versus the WTM were determined using Student's t test (*, P < 0.01; **, P < 0.005; ***, P < 0.001). The black line indicates the threshold at which the levels of EC50 have been modified versus WTM.
TABLE 3.
Effect of proline on drug susceptibility in L. donovania
| Line | EC50 μM (RI)b |
||
|---|---|---|---|
| AmB | MIL | SbIII | |
| WTM | 0.063 ± 0.002 | 3.45 ± 0.19 | 96.94 ± 8.91 |
| WTM + proline | 0.060 ± 0.002 (0.95 ± 0.07) | 3.24 ± 0.06 (0.94 ± 0.07) | 191.96 ± 8.62 (1.98 ± 0.14)c |
Promastigote forms of WTM were grown with or without 200 mM proline and incubated for 120 h at 28°C in the presence of increasing concentrations of oxidative stress-inducing drugs, as described in Materials and Methods. Cell viability was determined using an MTT-based assay.
The resistance index (RI) was calculated by dividing the EC50 for WTM grown with proline by the EC50 for WTM grown without proline, which is shown in parentheses. The data are the means of EC50 or RI ± SD from three independent experiments.
Significant differences were determined using Student's t test. The result of 191.96 ± 8.62 was significant at a P value of <0.001.
It was previously shown that SbIII, MIL, and AmB (but not PMM) trigger apoptosis-like cell death (ALCD) associated with oxidative stress in sensitive Leishmania parasites, and resistant lines were tolerant to both (46). We thus verified if the above-mentioned protection against oxidative stress was related to a decreased ALCD. Therefore, we determined the effect of drug pressure on genomic DNA degradation in the different Leishmania lines using PI fluorescence and flow cytometry analysis. If no DNA degradation was detected in the resistant lines, this would further support the increased protection against oxidative stress and resistance against the corresponding drug-induced ALCD. The fluorescence values for the cells with DNA degradation were lower than those for G1 cells (i.e., there was a subG1 peak in the DNA histograms) (47). After 48 h of incubation with 0.1 μM AmB, 10 μM MIL, or 100 μM SbIII, 41.28, 21.24, and 11.06% of the WTM parasites, respectively, had DNA accumulation in the subG1 region compared with only 3.14% of the untreated control parasites in the WTM line (see Fig. S3 in the supplemental material). In contrast, the A and AS resistant lines treated with 0.1 μM AmB showed 2.65 and 2.25%, respectively, of the parasites with DNA in the subG1 region, compared with 3.09 and 2.07%, respectively, of the untreated parasites of these lines (see Fig. S3). Similarly, the M resistant line incubated with 10 μM MIL exhibited 1.63% of the parasites in subG1 and 1.68% in the case of untreated parasites. Finally, the S, AS, and SP resistant lines treated with 100 μM SbIII had 1.95, 3.05, and 3.46%, respectively, of the parasite DNA in the subG1 region compared with 1.78, 2.07, and 3.31%, respectively, of the untreated parasites (see Fig. S3). Together, our results indicate that there was no genomic DNA fragmentation in Leishmania resistant lines under drug pressure.
Membrane alterations.
The fluidity of the plasma membrane was evaluated by fluorescence anisotropy using DPH as a fluorescent probe. Significant differences between the DPH anisotropy values for the different parasite plasma membranes were found at 28°C (Table 4). Since the anisotropy values for DPH give an estimation of its free rotation in the lipid bilayer, higher fluorescence anisotropy should correspond to a decrease in membrane fluidity. Accordingly, the membranes from all resistant lines were significantly less fluid than were those of the WTM line, except for the M line cells, which showed similar anisotropy values as those of the WTM line. Among the resistant lines, A showed the lowest fluidity, followed by the SP, P, AS, and S lines (Table 4). To check if membrane fluidity and drug accumulation are correlated, the intracellular accumulations of AmB, MIL, PMM, and SbIII in the L. donovani WTM and resistant lines were measured. In the case of AmB and MIL, we did not observe significant changes in the intracellular accumulation of AmB (in the A and AS lines) or MIL (in the M line) (Table 5). The accumulation of PMM was slightly lower (but not significant) in the P and SP lines (Table 5). Finally, the S line accumulated similar levels of SbIII as did the control line, whereas SbIII accumulation was significantly increased in the AS and SP lines (Table 5).
TABLE 4.
Membrane fluidity of L. donovani WTM and resistant linesa
| Line | Anisotropy valuesb |
|---|---|
| WTM | 0.220 ± 0.010 |
| A | 0.290 ± 0.001** |
| M | 0.230 ± 0.028 |
| P | 0.251 ± 0.001* |
| S | 0.244 ± 0.003* |
| AS | 0.246 ± 0.003* |
| SP | 0.266 ± 0.001* |
Steady-state fluorescence anisotropy of DPH incorporated in plasma membranes of different Leishmania lines.
The data are the means ± SD from 2 to 4 independent experiments. Significant differences versus the WTM line were determined by Student's t test (*, P < 0.05; **, P < 0.01).
TABLE 5.
Membrane fluidity and drug accumulation in L. donovani WTM and resistant linesa
| Line | Anisotropy values | % drug accumulationb |
|||
|---|---|---|---|---|---|
| AmB | MIL | PMM | SbIII | ||
| WTM | 0.220 ± 0.010 | 100 ± 7.23 | 100 ± 8.43 | 100 ± 7.43 | 100 ± 1.06 |
| A | 0.290 ± 0.001** | 100.28 ± 0.69 | |||
| M | 0.230 ± 0.028 | 101.01 ± 4.42 | |||
| P | 0.251 ± 0.001* | 86.05 ± 7.80 | |||
| S | 0.244 ± 0.003* | 109.48 ± 11.52 | |||
| AS | 0.246 ± 0.003* | 100.25 ± 0.54 | 233.23 ± 11.79*** | ||
| SP | 0.266 ± 0.001* | 91.47 ± 9.48 | 145.42 ± 8.22*** | ||
Steady-state fluorescence anisotropy of DPH was incorporated in plasma membranes of different Leishmania lines. The drug accumulation assay was performed after incubating Leishmania lines with 0.1 μM AmB, 2.5 μM MIL, 10 μM PMM, or 100 μM SbIII, as described in Materials and Methods. Significant differences versus WTM line were determined by Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
The data are the means ± SD from three independent experiments.
Additionally, as AmB, MIL, and SbIII are known to affect, as a secondary effect, the plasma membrane integrity in Leishmania parasites (4, 12, 19), we have determined the entrance of the impermeable dye Sytox green into the cytoplasm of WTM and drug-resistant lines. At the highest concentrations assayed (0.4 μM AmB, 20 μM MIL, and 400 μM SbIII), we detected an increased Sytox green fluorescence in the control (WTM) parasites but not in the resistant lines (see Fig. S4 in the supplemental material), an observation demonstrating the integrity of the plasma membrane of single-R and CTR lines and supporting their resistance.
DISCUSSION
The scarcity of new leads in the pipeline for chemotherapy of leishmaniasis highlights the importance of preserving the current drugs (25). Drug combination therapy is regarded as one of the last useful options for the treatment of visceral leishmaniasis (VL), in order to circumvent drug resistance. The search for the best combination of drugs requires, among other things, the definition of the parasitic changes associated with resistance (48). We present here the first metabolomics study that investigated the metabolic features of experimentally induced CTR lines and their respective single-R lines (28). This study attempted to elucidate the mechanisms underlying the ability of Leishmania parasites to develop resistance to combinations of current antileishmanial drugs.
Since the CTR lines were selected from the WT line using both drugs simultaneously, like in a clinical context (and not by first inducing resistance to one drug and then to a second drug), different metabolic adaptation mechanisms were expected to appear in the CTR lines compared to those in the single-R lines. This is illustrated by the fact that (i) a straightforward additive effect (e.g., if AS would be the sum of A and S) is absent and (ii) new metabolic changes appear in some CTR lines that are absent in the respective single lines. We proposed a scheme for classifying the degree of metabolic adaptations among the CTR lines (Table 1), taking into account the new changes specific for the CTR lines, those shared with single-R lines, and possible dominance by the single-R lines. This reveals the overall character of each CTR line and highlights a range of adaptations among the five lines studied here, with AP and AM at one end (low number of changes, no or few new CTR-specific changes at all, and light dominance of P and M, respectively) and SP at the other end (high number of changes, both [i] shared with S or P and [ii] SP specific and showing strong S dominance). This range of metabolic changes and, more particularly, the rate of CTR specific changes reflected well the degree of resistance (as measured by the resistance index) of the CTR lines, with AP, AM, and AS showing the less pronounced resistance character (RI, 2 to 3) and MP and SP being the most resistant. Interestingly, the two combinations with S led to a strong dominance of S-related changes in the respective CTR lines, but this was not related to strong resistance against SbIII, as in both AS and SP, the resistance index against SbIII was 2.
Of note, all CTR lines shared metabolic adaptations (CTR collective changes) in proline metabolism, as high (3- to 14-fold) and/or significant increases were detected for proline and the dipeptide (iso)leucyl-proline. In addition, in the AS and SP lines, specific metabolic changes belonging to this pathway were also found (in AS for the precursor pyrroline-5-carboxylate, and in SP for proline betaine), indicating an even more extensive adaptation in these CTR lines. Proline has been shown to enhance cell survival during environmental stress (e.g., drug pressure). In Trypanosoma cruzi, it was shown to be vital during metacyclogenesis, a process that has been linked to nutrient stress (49), and to modulate resistance toward ROS and drugs (e.g., nifurtimox and benznidazole) (50). This finding was confirmed by a recent study showing that the overexpression of T. cruzi proline dehydrogenase led to resistance to hydrogen peroxide (51). In Trypanosoma brucei, proline was found to be involved in the cytosolic malic enzyme pathway responsible for oxidative stress management through cytosolic NADPH homeostasis (52). In L. donovani, it was shown that (i) proline is required for volume recovery during osmotic stress responses (53) and (ii) intracellular proline levels were increased in purine-starved cells (54). In that second study, a proteome analysis showed a decrease in proline dehydrogenase and an increase in pyrroline-5-carboxylate synthetase-like protein and pyrroline-5-carboxylate reductase, which both led to higher levels of proline. Interestingly, the corresponding metabolite pyrroline-5-carboxylate was found to be increased in our study. Martin et al. (54) also showed that purine starvation augmented the interconversion of hypoxanthine and ampicillin (AMP) to imipenem (IMP), both key nucleotides of the purine salvage pathway of L. donovani (55). High levels of hypoxanthine were also found in the resistant lines of our study (IMP and AMP were not detected). Although the precise role of proline remains unclear, our findings and the above-mentioned reports all point toward proline as a general stress-response metabolite, and it has been demonstrated that proline can act as a free radical scavenger in vitro (56). Hence, in drug-resistant L. donovani lines, increased levels of proline might protect against oxidative stress caused by drug pressure. We have indeed shown that proline plays a protective role against drug-induced oxidative stress in the case of SbIII (a 2-fold increase in susceptibility was observed) but not in the case of MIL and AmB.
Furthermore, several other more specific metabolic adaptations were observed in the resistant lines, pointing toward increased protection against oxidative stress. First, the metabolites of the transsulfuration pathway (AdoMet and pyridoxine) were increased in the AS and SP lines. Also, methionine was found to be increased in the AS line. It is noteworthy to mention that a proteomics study on AmB-resistant (AmBr) L. infantum also found increased expression of AdoMet synthetase and S-adenosylhomocysteine hydrolase, leading to the production of AdoMet and homocysteine, respectively (57). Via AdoMet decarboxylase, AdoMet is converted to S-adenosylmethionamine, which donates an aminopropyl group to putrescine, resulting in the production of spermidine, one of the precursors of trypanothione. The cofactor vitamin B6 (pyridoxine) is needed for the conversion of homocysteine into cysteine. Further downstream, no differential metabolites were detected, which might be explained by the easily oxidizable nature of thiol-containing metabolites. Second, in the S, A, M, and AS lines, increased levels of tryptophan and its degradation product indole acrylate were observed. In Escherichia coli, this has been linked to resistance against antibiotic treatment by triggering protective responses against oxidative stress, predominantly during the transition to stationary stage (58). In addition, in the A, S, AS, and SP lines, increased levels of aromatic amino acids were detected, of which, for example, tyrosine is needed for the production of methionine via a transamination reaction (59). Ovothiol, which was increased in the AS line only, as well as two increased metabolites of the pentose phosphate pathway in the S line, might provide redox equivalents to protect against oxidative stress (60).
Thus, collective metabolic changes (found in all CTR lines) and specific ones (found in certain CTR lines) all point in the same direction, i.e., toward the protection of the parasites against oxidative stress. This hypothesis was experimentally verified by first measuring ROS production (i) in the complete parasitic cell with a dichlorofluorescein diacetate-based assay and (ii) in the mitochondria with MitoSOX red in both WTM and resistant lines under drug pressure. All resistant lines showed the absence of a significant increase in ROS production under drug pressure, confirming the suggested increased capacity to protect against drug-induced oxidative stress. This can be due to decreased ROS production but also to a more efficient way of handling ROS (e.g., by proline). By challenging WTM and resistant lines with external ROS sources, such as H2O2, we showed that all resistant lines (except the SP line) possess more efficient ROS detoxification and are more resistant to oxidative stress. Less efficient protection was observed against menadione (only the S and SP lines were more tolerant than the WTM line), which might reflect that different mechanisms of resistance to different ROS species exist among the different lines (which is supported by the metabolomics findings), since menadione generates superoxide and H2O2 generates peroxide (61). A different response toward these two ROS-inducing compounds was also described in Saccharomyces cerevisiae (62).
Among the drugs used here, three of them (AmB, Sb, and MIL) are reported to trigger ALCD associated with ROS in susceptible lines (46); furthermore, parasites showing single resistance to these drugs were shown to be tolerant to ALCD, a known trigger of oxidative stress (46). We thus analyzed ALCD here by DNA fragmentation and showed that the CTR lines AS and SP and the corresponding single-R lines did not show signs of ALCD under pressure of the respective drugs. These findings complement previous ones by García-Hernández et al. (28), and all point to the resistant lines being protected against the oxidative stress that originated during drug-induced apoptosis.
A second class of major adaptations encountered here concerns lipid metabolism (Table 2). In all resistant lines, a general trend was observed concerning changes in the saturation levels of the GPLs and increases or decreases in fatty acyls but without any significant overlap with all CTR lines. More particularly, the CTR lines AS and SP shared increases in a building block of the glycerophospholipid synthesis (glycerol-3-P) (also shared with MP), two fatty acyls, and decreases in two precursor metabolites of the sterol biosynthesis (copaene and mevalonate; mevalonate is produced from acetyl-coenzyme A [CoA] in the mitochondrion) and a sphingolipid precursor (3-dehydrosphinganine). Although ergosterol (the target molecule of AmB) was not found to be significantly altered in the resistant lines, a sterol with monoisotopic mass of 398.318484 Da was found to be increased in the AS and A lines (including a trend in the AP and AM lines) but was decreased in the SP line (including a similar trend in the MP line). Changes in the lipidome have been reported in the case of parasites that are resistant toward AmB (12), SbIII (30, 63), and MIL (64) and might have a significant effect on membranes, consequently interfering with the mode of action of drugs (in the case of AmB and MIL) or the uptake of drugs and salvage metabolites. This hypothesis on the changes in the plasma membrane was assessed experimentally through studies of membrane fluidity. Membrane fluidity was tested with a fluorescence anisotropy assay and showed that all membranes from the resistant lines (except M) present lower fluidity. Similar results (lower fluidity) have been obtained and suggested in the resistance of Leishmania parasites to tafenoquine (42) and MIL (65) but are different from the results obtained with resistance to PMM (24), antimonials (7, 66), and AmB (12), for which membrane fluidity was found to be higher. Our results are supported by the fact that an increase in six low unsaturated GPLs and a decrease in six high unsaturated GPLs were detected in the A line, which was expected to result in a decrease in membrane fluidity. More generally, a more rigid plasma membrane will hamper drugs from entering, solubilizing, and diffusing into the membrane and might alter the functioning of transporters; hence, this indirectly influences the accumulation of the drugs (67). In our experiments, membrane fluidity changes were not accompanied by significant changes in drug uptake for AmB, MIL, and PMM. However, the lines that were resistant to drug combinations with SbIII (AS and SP lines) were surprisingly accompanied by a significant increase in SbIII uptake, possibly due to increased drug sequestration. Together, these results highlight the importance and central role of protection to oxidative stress in the resistance of these lines.
In conclusion, this study highlighted a range of metabolic adaptations among the four single-R and five CTR lines. In the single-R lines, metabolic changes were clearly linked to the mode of action of the respective drug. For example, MIL and AmB both disrupt the plasma membrane as a primary mode of action; hence, clear changes were observed in the GPL compositions of these single-R lines. For the M line, however, this was not translated into significant changes in membrane fluidity or drug accumulation in the M line, which possibly reflects that this line is a more tolerant but not (yet) fully resistant line (resistance index, 1.81). The observed phenotypic adaptation of increased protection against oxidative stress might be interpreted as a first (and perhaps still reversible) adaptation toward the drug, in expectation of more structural and irreversible adaptations later, such as membrane changes. In agreement with the hypothesis of a multifactorial mode of action of sodium stibogluconate (SSG) (4), the S line showed adaptations in different metabolic pathways (fatty acyls, amino acids, and the pentose phosphate pathway). The mode of action of PMM is more targeted at the protein level, perhaps explaining why not many differential metabolites were detected (14) in the P line, although the highest resistance index was obtained for this line (11-fold). In theory, combination therapy should delay the development of drug resistance if the two combined drugs have a different mode of action by targeting different metabolic pathways (68). After showing in vitro that L. donovani can rapidly develop resistance against combined drugs (28), the current report demonstrates that CTR lines develop the same type of phenotypic adaptations, despite showing different metabolic changes. This might constitute an alarming finding, as it might interfere with the mode of action of all drugs that are currently used for the treatment of VL. The present metabolomics study profiled the stationary life stage of promastigotes, characterized by the presence of metacyclic parasites that are preparing for intracellular survival as amastigotes; however, further work should be dedicated to the analysis of this stage. Similarly, resistance induction in the amastigote and promastigote stages should be compared, as the mechanisms might differ (69). Ultimately, the obtained results should be further complemented by characterizing clinical isolates from patients failing combination therapy.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Research Foundation Flanders (FWO) (grants G.0B81.12 and 11O1614N to B.C.), the Inbev-Baillet Latour Fund (grant 620001 to M.B.), the Belgian Science Policy Office (TRIT, contract P7/41), and by the Spanish Grants Proyecto de Excelencia, Junta de Andalucia reference no. CTS-7282 (to F.G.), SAF2012-34267 (to F.G.), and SAF2011-28102 (to S.C.). The funders had no role in the decision to publish or the preparation of the manuscript.
We thank Andris Jankevics (Faculty of Life Sciences, Manchester Institute of Biotechnology, University of Manchester, United Kingdom) for his assistance with the data analysis and the Scottish Metabolomics Facility (ScotMet) for the measurements of the samples on their LC-MS platform.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04231-14.
REFERENCES
- 1.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M, WHO Leishmaniasis Control Team . 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sundar S. 2001. Drug resistance in Indian visceral leishmaniasis. Trop Med Int Health 6:849–854. doi: 10.1046/j.1365-3156.2001.00778.x. [DOI] [PubMed] [Google Scholar]
- 3.Sundar S, More DK, Singh MK, Singh VP, Sharma S, Makharia A, Kumar PC, Murray HW. 2000. Failure of pentavalent antimony in visceral leishmaniasis in India: report from the center of the Indian epidemic. Clin Infect Dis 31:1104–1107. doi: 10.1086/318121. [DOI] [PubMed] [Google Scholar]
- 4.Ashutosh, Sundar S, Goyal N. 2007. Molecular mechanisms of antimony resistance in Leishmania. J Med Microbiol 56:143–153. doi: 10.1099/jmm.0.46841-0. [DOI] [PubMed] [Google Scholar]
- 5.Mookerjee Basu J, Mookerjee A, Sen P, Bhaumik S, Sen P, Banerjee S, Naskar K, Choudhuri SK, Saha B, Raha S, Roy S. 2006. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother 50:1788–1797. doi: 10.1128/AAC.50.5.1788-1797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schonian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M. 2011. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res 21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg M, Vanaerschot M, Jankevics A, Cuypers B, Maes I, Mukherjee S, Khanal B, Rijal S, Roy S, Opperdoes F, Breitling R, Dujardin JC. 2013. Metabolic adaptations of Leishmania donovani in relation to differentiation, drug resistance, and drug pressure. Mol Microbiol 90:428–442. doi: 10.1111/mmi.12374. [DOI] [PubMed] [Google Scholar]
- 8.Gradoni L, Soteriadou K, Louzir H, Dakkak A, Toz SO, Jaffe C, Dedet JP, Campino L, Canavate C, Dujardin JC. 2008. Drug regimens for visceral leishmaniasis in Mediterranean countries. Trop Med Int Health 13:1272–1276. doi: 10.1111/j.1365-3156.2008.02144.x. [DOI] [PubMed] [Google Scholar]
- 9.Thakur CP, Pandey AK, Sinha GP, Roy S, Behbehani K, Olliaro P. 1996. Comparison of three treatment regimens with liposomal amphotericin B (AmBisome) for visceral leishmaniasis in India: a randomized dose-finding study. Trans R Soc Trop Med Hyg 90:319–322. doi: 10.1016/S0035-9203(96)90271-0. [DOI] [PubMed] [Google Scholar]
- 10.Singh N, Kumar M, Singh RK. 2012. Leishmaniasis: current status of available drugs and new potential drug targets. Asian Pac J Trop Med 5:485–497. doi: 10.1016/S1995-7645(12)60084-4. [DOI] [PubMed] [Google Scholar]
- 11.Burza S, Sinha PK, Mahajan R, Sanz MG, Lima MA, Mitra G, Verma N, Das P. 2014. Post Kala-Azar dermal leishmaniasis following treatment with 20 mg/kg liposomal amphotericin B (Ambisome) for primary visceral leishmaniasis in Bihar, India. PLoS Negl Trop Dis 8:e2611. doi: 10.1371/journal.pntd.0002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, Pandey K, Ravidas V, Kumar M, De T, Singh D, Das P. 2012. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother 56:1031–1041. doi: 10.1128/AAC.00030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, Beijnen JH, Vanaerschot M, Decuypere S, Dhakal SS, Das ML, Karki P, Singh R, Boelaert M, Dujardin JC. 2013. Increasing failure of miltefosine in the treatment of Kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis 56:1530–1538. doi: 10.1093/cid/cit102. [DOI] [PubMed] [Google Scholar]
- 14.Pérez-Victoria FJ, Gamarro F, Ouellette M, Castanys S. 2003. Functional cloning of the miltefosine transporter. A novel P-type phospholipid translocase from Leishmania involved in drug resistance. J Biol Chem 278:49965–49971. doi: 10.1074/jbc.M308352200. [DOI] [PubMed] [Google Scholar]
- 15.Pérez-Victoria FJ, Sánchez-Cañete MP, Castanys S, Gamarro F. 2006. Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J Biol Chem 281:23766–23775. doi: 10.1074/jbc.M605214200. [DOI] [PubMed] [Google Scholar]
- 16.Paris C, Bertoglio J, Breard J. 2007. Lysosomal and mitochondrial pathways in miltefosine-induced apoptosis in U937 cells. Apoptosis 12:1257–1267. doi: 10.1007/s10495-007-0052-1. [DOI] [PubMed] [Google Scholar]
- 17.Imbert L, Ramos RG, Libong D, Abreu S, Loiseau PM, Chaminade P. 2012. Identification of phospholipid species affected by miltefosine action in Leishmania donovani cultures using LC-ELSD, LC-ESI/MS, and multivariate data analysis. Anal Bioanal Chem 402:1169–1182. doi: 10.1007/s00216-011-5520-3. [DOI] [PubMed] [Google Scholar]
- 18.Luque-Ortega JR, Rivas L. 2007. Miltefosine (hexadecylphosphocholine) inhibits cytochrome c oxidase in Leishmania donovani promastigotes. Antimicrob Agents Chemother 51:1327–1332. doi: 10.1128/AAC.01415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vincent IM, Weidt S, Rivas L, Burgess K, Smith TK, Ouellette M. 2014. Untargeted metabolomic analysis of miltefosine action in Leishmania infantum reveals changes to the internal lipid metabolism. Int J Parasitol Drugs Drug Resist 4:20–27. doi: 10.1016/j.ijpddr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulshrestha A, Sharma V, Singh R, Salotra P. 2014. Comparative transcript expression analysis of miltefosine-sensitive and miltefosine-resistant Leishmania donovani. Parasitol Res 113:1171–1184. doi: 10.1007/s00436-014-3755-6. [DOI] [PubMed] [Google Scholar]
- 21.Fernández MM, Malchiodi EL, Algranati ID. 2011. Differential effects of paromomycin on ribosomes of Leishmania mexicana and mammalian cells. Antimicrob Agents Chemother 55:86–93. doi: 10.1128/AAC.00506-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundar S, Agrawal N, Arora R, Agarwal D, Rai M, Chakravarty J. 2009. Short-course paromomycin treatment of visceral leishmaniasis in India: 14-day vs 21-day treatment. Clin Infect Dis 49:914–918. doi: 10.1086/605438. [DOI] [PubMed] [Google Scholar]
- 23.Jhingran A, Chawla B, Saxena S, Barrett MP, Madhubala R. 2009. Paromomycin: uptake and resistance in Leishmania donovani. Mol Biochem Parasitol 164:111–117. doi: 10.1016/j.molbiopara.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhandari V, Sundar S, Dujardin JC, Salotra P. 2014. Elucidation of cellular mechanisms involved in experimental paromomycin resistance in Leishmania donovani. Antimicrob Agents Chemother 58:2580–2585. doi: 10.1128/AAC.01574-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dujardin JC, Gonzalez-Pacanowska D, Croft SL, Olesen OF, Spath GF. 2010. Collaborative actions in anti-trypanosomatid chemotherapy with partners from disease endemic areas. Trends Parasitol 26:395–403. doi: 10.1016/j.pt.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Olliaro PL. 2010. Drug combinations for visceral leishmaniasis. Curr Opin Infect Dis 23:595–602. doi: 10.1097/QCO.0b013e32833fca9d. [DOI] [PubMed] [Google Scholar]
- 27.Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, Chakravarty J, Vaillant M, Verma N, Pandey K, Kumari P, Lal CS, Arora R, Sharma B, Ellis S, Strub-Wourgaft N, Balasegaram M, Olliaro P, Das P, Modabber F. 2011. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet 377:477–486. doi: 10.1016/S0140-6736(10)62050-8. [DOI] [PubMed] [Google Scholar]
- 28.García-Hernández R, Manzano JI, Castanys S, Gamarro F. 2012. Leishmania donovani develops resistance to drug combinations. PLoS Negl Trop Dis 6:e1974. doi: 10.1371/journal.pntd.0001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg M, Mannaert A, Vanaerschot M, Van der Auwera G, Dujardin JC. 2013. (Post-) genomic approaches to tackle drug resistance in Leishmania. Parasitology 12:1–14. doi: 10.1017/S0031182013000140. [DOI] [PubMed] [Google Scholar]
- 30.t'Kindt R, Scheltema RA, Jankevics A, Brunker K, Rijal S, Dujardin JC, Breitling R, Watson DG, Coombs GH, Decuypere S. 2010. Metabolomics to unveil and understand phenotypic diversity between pathogen populations. PLoS Negl Trop Dis 4:e904. doi: 10.1371/journal.pntd.0000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg M, Vanaerschot M, Jankevics A, Cuypers B, Breitling R, Dujardin JC. 2012. LC-MS metabolomics from study design to data-analysis—using a versatile pathogen as a test case. Comput Struct Biotechnol J 4:e201301002. doi: 10.5936/csbj.201301002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fahy E, Sud M, Cotter D, Subramaniam S. 2007. LIPID MAPS online tools for lipid research. Nucleic Acids Res 35:W606–W612. doi: 10.1093/nar/gkm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller BO, Sui J, Young AB, Whittal RM. 2008. Interferences and contaminants encountered in modern mass spectrometry. Anal Chim Acta 627:71–81. doi: 10.1016/j.aca.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 35.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. 2007. HMDB: the Human Metabolome Database. Nucleic Acids Res 35:D521–D526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scalbert A, Brennan L, Fiehn O, Hankemeier T, Kristal BS, van Ommen B, Pujos-Guillot E, Verheij E, Wishart D, Wopereis S. 2009. Mass-spectrometry-based metabolomics: limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 5:435–458. doi: 10.1007/s11306-009-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manzano JI, Garcia-Hernandez R, Castanys S, Gamarro F. 2013. A new ABC half-transporter in Leishmania major is involved in resistance to antimony. Antimicrob Agents Chemother 57:3719–3730. doi: 10.1128/AAC.00211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kennedy ML, Cortes-Selva F, Perez-Victoria JM, Jimenez IA, Gonzalez AG, Munoz OM, Gamarro F, Castanys S, Ravelo AG. 2001. Chemosensitization of a multidrug-resistant Leishmania tropica line by new sesquiterpenes from Maytenus magellanica and Maytenus chubutensis. J Med Chem 44:4668–4676. doi: 10.1021/jm010970c. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh S, Goswami S, Adhya S. 2003. Role of superoxide dismutase in survival of Leishmania within the macrophage. Biochem J 369:447–452. doi: 10.1042/BJ20021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carvalho L, Luque-Ortega JR, Lopez-Martin C, Castanys S, Rivas L, Gamarro F. 2011. The 8-aminoquinoline analogue sitamaquine causes oxidative stress in Leishmania donovani promastigotes by targeting succinate dehydrogenase. Antimicrob Agents Chemother 55:4204–4210. doi: 10.1128/AAC.00520-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basselin M, Robert-Gero M. 1998. Alterations in membrane fluidity, lipid metabolism, mitochondrial activity, and lipophosphoglycan expression in pentamidine-resistant Leishmania. Parasitol Res 84:78–83. [DOI] [PubMed] [Google Scholar]
- 42.Manzano JI, Carvalho L, Garcia-Hernandez R, Poveda JA, Ferragut JA, Castanys S, Gamarro F. 2011. Uptake of the antileishmania drug tafenoquine follows a sterol-dependent diffusion process in Leishmania. J Antimicrob Chemother 66:2562–2565. doi: 10.1093/jac/dkr345. [DOI] [PubMed] [Google Scholar]
- 43.Lakowicz JR. 1983. Principles of fluorescence spectroscopy. Plenum Press, New York, NY. [Google Scholar]
- 44.Al-Quadeib BT, Radwan MA, Siller L, Mutch E, Horrocks B, Wright M, Alshaer A. 2014. Therapeutic monitoring of amphotericin B in Saudi ICU patients using UPLC MS/MS assay. Biomed Chromatogr 28:1652–1659. doi: 10.1002/bmc.3198. [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Cañete MP, Carvalho L, Perez-Victoria FJ, Gamarro F, Castanys S. 2009. Low plasma membrane expression of the miltefosine transport complex renders Leishmania braziliensis refractory to the drug. Antimicrob Agents Chemother 53:1305–1313. doi: 10.1128/AAC.01694-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreira W, Leprohon P, Ouellette M. 2011. Tolerance to drug-induced cell death favours the acquisition of multidrug resistance in Leishmania. Cell Death Dis 2:e201. doi: 10.1038/cddis.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139:271–279. doi: 10.1016/0022-1759(91)90198-O. [DOI] [PubMed] [Google Scholar]
- 48.Canuto GA, Castilho-Martins EA, Tavares MF, Rivas L, Barbas C, Lopez-Gonzalvez A. 2014. Multi-analytical platform metabolomic approach to study miltefosine mechanism of action and resistance in Leishmania. Anal Bioanal Chem 406:3459–3476. doi: 10.1007/s00216-014-7772-1. [DOI] [PubMed] [Google Scholar]
- 49.Magdaleno A, Ahn IY, Paes LS, Silber AM. 2009. Actions of a proline analogue, l-thiazolidine-4-carboxylic acid (T4C), on Trypanosoma cruzi. PLoS One 4:e4534. doi: 10.1371/journal.pone.0004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sayé M, Miranda MR, di Girolamo F, de los Milagros Camara M, Pereira CA. 2014. Proline modulates the Trypanosoma cruzi resistance to reactive oxygen species and drugs through a novel d,l-proline transporter. PLoS One 9:e92028. doi: 10.1371/journal.pone.0092028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paes LS, Suarez Mantilla B, Zimbres FM, Pral EM, Diogo de Melo P, Tahara EB, Kowaltowski AJ, Elias MC, Silber AM. 2013. Proline dehydrogenase regulates redox state and respiratory metabolism in Trypanosoma cruzi. PLoS One 8:e69419. doi: 10.1371/journal.pone.0069419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allmann S, Morand P, Ebikeme C, Gales L, Biran M, Hubert J, Brennand A, Mazet M, Franconi JM, Michels PA, Portais JC, Boshart M, Bringaud F. 2013. Cytosolic NADPH homeostasis in glucose-starved procyclic Trypanosoma brucei relies on malic enzyme and the pentose phosphate pathway fed by gluconeogenic flux. J Biol Chem 288:18494–18505. doi: 10.1074/jbc.M113.462978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inbar E, Schlisselberg D, Suter GM, Rentsch D, Zilberstein D. 2013. A versatile proline/alanine transporter in the unicellular pathogen Leishmania donovani regulates amino acid homoeostasis and osmotic stress responses. Biochem J 449:555–566. doi: 10.1042/BJ20121262. [DOI] [PubMed] [Google Scholar]
- 54.Martin JL, Yates PA, Soysa R, Alfaro JF, Yang F, Burnum-Johnson KE, Petyuk VA, Weitz KK, Camp DG Jr, Smith RD, Wilmarth PA, David LL, Ramasamy G, Myler PJ, Carter NS. 2014. Metabolic reprogramming during purine stress in the protozoan pathogen Leishmania donovani. PLoS Pathog 10:e1003938. doi: 10.1371/journal.ppat.1003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berg M, Van der Veken P, Goeminne A, Haemers A, Augustyns K. 2010. Inhibitors of the purine salvage pathway: a valuable approach for antiprotozoal chemotherapy? Curr Med Chem 17:2456–2481. doi: 10.2174/092986710791556023. [DOI] [PubMed] [Google Scholar]
- 56.Kaul S, Sharma SS, Mehta IK. 2008. Free radical scavenging potential of l-proline: evidence from in vitro assays. Amino Acids 34:315–320. doi: 10.1007/s00726-006-0407-x. [DOI] [PubMed] [Google Scholar]
- 57.Brotherton MC, Bourassa S, Legare D, Poirier GG, Droit A, Ouellette M. 2014. Quantitative proteomic analysis of amphotericin B resistance in Leishmania infantum. Int J Parasitol Drugs Drug Resist 4:126–132. doi: 10.1016/j.ijpddr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vega NM, Allison KR, Khalil AS, Collins JJ. 2012. Signaling-mediated bacterial persister formation. Nat Chem Biol 8:431–433. doi: 10.1038/nchembio.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Achouri Y, Noel G, Van Schaftingen E. 2007. 2-Keto-4-methylthiobutyrate, an intermediate in the methionine salvage pathway, is a good substrate for CtBP1. Biochem Biophys Res Commun 352:903–906. doi: 10.1016/j.bbrc.2006.11.111. [DOI] [PubMed] [Google Scholar]
- 60.Krauth-Siegel RL, Leroux AE. 2012. Low-molecular-mass antioxidants in parasites. Antioxid Redox Signal 17:583–607. doi: 10.1089/ars.2011.4392. [DOI] [PubMed] [Google Scholar]
- 61.Pratap SK, Zaidi A, Anwar S, Bimal S, Das P, Ali V. 2014. Reactive oxygen species regulates expression of iron-sulfur cluster assembly protein IscS of Leishmania donovani. Free Radic Biol Med 75:195–209. doi: 10.1016/j.freeradbiomed.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 62.Flattery-O'Brien JA, Dawes IW. 1998. Hydrogen peroxide causes RAD9-dependent cell cycle arrest in G2 in Saccharomyces cerevisiae whereas menadione causes G1 arrest independent of RAD9 function. J Biol Chem 273:8564–8571. doi: 10.1074/jbc.273.15.8564. [DOI] [PubMed] [Google Scholar]
- 63.de Azevedo AF, Dutra JL, Santos ML, Santos DA, Alves PB, de Moura TR, de Almeida RP, Fernandes MF, Scher R, Fernandes RP. 2014. Fatty acid profiles in Leishmania spp. isolates with natural resistance to nitric oxide and trivalent antimony. Parasitol Res 113:19–27. doi: 10.1007/s00436-013-3621-y. [DOI] [PubMed] [Google Scholar]
- 64.Saint-Pierre-Chazalet M, Ben Brahim M, Le Mojec L, Bories C, Rakotomanga M, Loiseau PM. 2009. Membrane sterol depletion impairs miltefosine action in wild-type and miltefosine-resistant Leishmania donovani promastigotes. J Antimicrob Chemother 64:993–1001. doi: 10.1093/jac/dkp321. [DOI] [PubMed] [Google Scholar]
- 65.Rakotomanga M, Saint-Pierre-Chazalet M, Loiseau PM. 2005. Alteration of fatty acid and sterol metabolism in miltefosine-resistant Leishmania donovani promastigotes and consequences for drug-membrane interactions. Antimicrob Agents Chemother 49:2677–2686. doi: 10.1128/AAC.49.7.2677-2686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mukhopadhyay R, Mukherjee S, Mukherjee B, Naskar K, Mondal D, Decuypere S, Ostyn B, Prajapati VK, Sundar S, Dujardin JC, Roy S. 2011. Characterisation of antimony-resistant Leishmania donovani isolates: biochemical and biophysical studies and interaction with host cells. Int J Parasitol 41:1311–1321. doi: 10.1016/j.ijpara.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 67.Eckford PD, Sharom FJ. 2008. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: effects on ATPase activity, drug binding and transport. Biochemistry 47:13686–13698. doi: 10.1021/bi801409r. [DOI] [PubMed] [Google Scholar]
- 68.van Griensven J, Balasegaram M, Meheus F, Alvar J, Lynen L, Boelaert M. 2010. Combination therapy for visceral leishmaniasis. Lancet Infect Dis 10:184–194. doi: 10.1016/S1473-3099(10)70011-6. [DOI] [PubMed] [Google Scholar]
- 69.Hendrickx S, Inocêncio da Luz RA, Bhandari V, Kuypers K, Shaw CD, Lonchamp J, Salotra P, Carter K, Sundar S, Rijal S, Dujardin JC, Cos P, Maes L. 2012. Experimental induction of paromomycin resistance in antimony-resistant strains of L. donovani: outcome dependent on in vitro selection protocol. PLoS Negl Trop Dis 6:e1664. doi: 10.1371/journal.pntd.0001664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.