

Abstract
In Salmonella enterica, PmrD is a connector protein that links the two-component systems PhoP-PhoQ and PmrA-PmrB. While Escherichia coli encodes a PmrD homolog, it is thought to be incapable of connecting PhoPQ and PmrAB in this organism due to functional divergence from the S. enterica protein. However, our laboratory previously observed that low concentrations of Mg2+, a PhoPQ-activating signal, leads to the induction of PmrAB-dependent lipid A modifications in wild-type E. coli (C. M. Herrera, J. V. Hankins, and M. S. Trent, Mol Microbiol 76:1444–1460, 2010, http://dx.doi.org/10.1111/j.1365-2958.2010.07150.x). These modifications include phosphoethanolamine (pEtN) and 4-amino-4-deoxy-l-arabinose (l-Ara4N), which promote bacterial resistance to cationic antimicrobial peptides (CAMPs) when affixed to lipid A. Here, we demonstrate that pmrD is required for modification of the lipid A domain of E. coli lipopolysaccharide (LPS) under low-Mg2+ growth conditions. Further, RNA sequencing shows that E. coli pmrD influences the expression of pmrA and its downstream targets, including genes coding for the modification enzymes that transfer pEtN and l-Ara4N to the lipid A molecule. In line with these findings, a pmrD mutant is dramatically impaired in survival compared with the wild-type strain when exposed to the CAMP polymyxin B. Notably, we also reveal the presence of an unknown factor or system capable of activating pmrD to promote lipid A modification in the absence of the PhoPQ system. These results illuminate a more complex network of protein interactions surrounding activation of PhoPQ and PmrAB in E. coli than previously understood.
INTRODUCTION
Bacteria often encounter adverse conditions that threaten survival in an unpredictable environment. The first line of defense for most Gram-negative bacteria is the outer membrane, which contains lipopolysaccharide (LPS) in the outer leaflet that interfaces with the surroundings (1). LPS is a multicomponent macromolecule anchored to the bacterial membrane via its lipid A domain, a potent activator of the host innate immune response (2, 3). In the presence of environmental stressors, numerous Gram negatives have evolved machinery to modify the lipid A moiety with chemical groups that promote bacterial survival by creating a fortified, more resistant outer membrane (4).
Lipid A modifications often are regulated by complex two-component system (TCS) protein networks that coordinate detection of various signals with targeted transcriptional regulation. A typical TCS consists of a sensor histidine kinase that detects specific environmental signals and a cognate response regulator, which carries out changes in expression of a subset of genes known as its regulon. Upon recognition of a given signal, the sensor first autophosphorylates and then phosphorylates the response regulator, causing it to activate or repress gene expression within the regulon. When the signal is no longer present or detectable, the sensor deactivates the response regulator by dephosphorylation, thereby terminating transcriptional control of the affected genes (2, 5, 6). The research here involves two such systems, PhoP-PhoQ (PhoPQ) and PmrA-PmrB (PmrAB), where the former protein in each pair is the response regulator and the latter is the sensor histidine kinase. PhoQ responds to cues, including depletion of Mg2+ and the presence of cationic antimicrobial peptides (CAMPs) (7–9). Although its in vivo relevance is still to be fully elucidated, micromolar Mg2+ is a strong activating signal for PhoQ that is commonly used in the laboratory. PmrB senses CAMPs, mildly acidic pH, and high Fe3+ concentrations (10–12). These two major TCSs are widely distributed across Gram-negative bacteria, particularly in enteric genera, including Salmonella, Escherichia, Klebsiella, Shigella, and Citrobacter. Activation of PhoPQ and/or PmrAB affects numerous cellular processes, including, in certain organisms, the induction of various lipid A modifications (Fig. 1A).
FIG 1.

E. coli and S. enterica modify lipid A in response to environmental signals, altering the integrity of the outer membrane. (A, left) Typically, E. coli produces a lipid A structure comprised of a β-(1′,6)-linked disaccharide of glucosamine that is bis-phosphorylated and hexa-acylated (3). Select glucosamine carbons are numbered in red. (Right) Lipid A can be modified by numerous enzymes and under various conditions. Modifications include addition of l-Ara4N by ArnT (green) and pEtN by EptA (red), addition of a phosphate group at the 1-phosphate by LpxT (brown), removal of the 3′-linked acyl chains by LpxR (purple), hydroxylation of the 3′-secondary acyl chain by LpxO (gray), removal of the 3-linked acyl chain by PagL (pink), and addition of a secondary palmitate chain at the 2-linked primary acyl chain by PagP (blue). Asterisks designate enzymes found in S. enterica but not E. coli K-12. pEtN and l-Ara4N positions may be reversed and/or double addition of residues may occur (32, 45). (B) Model for PmrD-mediated cross talk between PhoPQ and PmrAB. Inner membrane sensor PhoQ is activated by low Mg2+ or CAMPs in the periplasm (7). It autophosphorylates and then activates the cytosolic response regulator, PhoP, which increases transcription of the pmrD gene. The PmrD protein binds to phospho-PmrA, a response regulator associated with sensor PmrB. PmrD mechanically inhibits dephosphorylation of PmrA by PmrB, allowing continued transcription of PmrA-dependent genes, including eptA and arnT. Ultimately, PmrD promotes addition of pEtN and l-Ara4N to the lipid A under PhoPQ-activating conditions, leading to polymyxin resistance. Our results implicate a second system (question mark) that is able to activate PmrD in an E. coli phoPQ double mutant.
For instance, the PmrAB regulon includes two genes involved in lipid A modification: eptA, coding for a phosphoethanolamine (pEtN) transferase, and arnT, coding for a 4-amino-4-deoxy-l-arabinose (l-Ara4N) transferase (13–16). Enzymes EptA and ArnT function at the inner membrane, transferring their respective amine-containing residues to the lipid A phosphate groups before transport across the periplasm (Fig. 1B) (14, 16). Decoration of lipid A with pEtN or l-Ara4N groups masks the charge of one or both of its anionic phosphates. This reduces the net negative charge of the molecule and the outer membrane as a whole, which helps protect the bacterium from positively charged CAMPs (Fig. 1B) (14, 15).
In Salmonella enterica, PmrAB-dependent genes also can be activated indirectly in a process that requires cross talk through PhoPQ (17). The vehicle for interaction between these two-component systems is PmrD, a small PhoP-activated protein that binds to and mechanically blocks dephosphorylation of activated phospho-PmrA by its cognate sensor, PmrB (18–20) (Fig. 1B). In effect, PmrD helps maintain PmrA in its active state, allowing it to continue influencing the transcription of genes in its regulon. Consequently, eptA and arnT also are transcribed when environmental conditions exclusively activate PhoPQ, such as in low concentrations of Mg2+. In this way, disparate environmental signals that activate PhoPQ or PmrAB ultimately lead to the same phenotypic outcome in S. enterica: lipid A modifications that protect against CAMPs (17).
Conversely, cross talk between PhoPQ and PmrAB is thought not to occur in E. coli. Winfield and Groisman previously demonstrated that when wild-type K-12 strain MG1655 is grown in low Mg2+, PmrA-dependent genes are not activated and bacterial survival is extremely impaired upon exposure to CAMP polymyxin B (21). These cellular responses instead require direct activation of PmrAB, suggesting that in E. coli, this system is not connected to PhoPQ. This phenotypic difference first was attributed to functional divergence between the Salmonella and E. coli PmrD proteins, suggesting that PmrD is an inactive connector of PhoPQ and PmrAB in the latter organism (21). However, it was later found that E. coli PmrD promotes the connection of PhoPQ and PmrAB when expressed heterologously in S. enterica (22). Ultimately, E. coli PmrB was demonstrated to possess vigorous phosphatase activity that exceeds that of its S. enterica homolog. Together, these findings suggested that in E. coli, PhoPQ and PmrAB are disconnected not because PmrD is inactive but because PmrB dephosphorylates PmrA faster than PmrD can protect it (22).
Former studies have analyzed gene expression and CAMP survival data to define a role for PmrD in E. coli but have not involved phenotypic analysis of lipid A. The lipid A profile can reveal the presence or absence of common structural modifications, including pEtN and l-Ara4N; therefore, it is a direct read-out for activation of modification machinery and CAMP resistance. The structure of this molecule determines the biochemical status of the outer membrane and should agree with gene expression and survival data. Here, we isolate lipid A from wild-type and pmrD mutant E. coli to discover unexpectedly that (i) pEtN/l-Ara4N modifications are present on lipid A isolated from wild-type E. coli grown in micromolar concentrations of Mg2+, and (ii) under the same low-Mg2+ conditions, pmrD is required for and actively promotes these PmrA-dependent modifications. In line with this, we proceed to confirm that pmrD is important for bacterial survival upon exposure to polymyxin B and influences the expression of PmrA-dependent genes, as shown by RNA sequencing (RNA-seq) analysis. Intriguingly, our findings also uncover the existence of a second PhoPQ-independent system that activates PmrD and, as a result, the addition of PmrA-dependent pEtN and l-Ara4N groups to the lipid A under low-Mg2+ growth conditions. In all, this research establishes that communication does occur between PhoPQ and PmrAB in E. coli through PmrD but in a more complex manner than expected.
MATERIALS AND METHODS
Chemicals and other materials.
32Pi was obtained from PerkinElmer. Precoated glass-backed thin-layer chromatography (TLC) plates were purchased from EMD Chemicals. Luria-Bertani (LB) agar and LB broth were from Becton-Dickinson. Polymyxin B sulfate was from Sigma. All other chemicals were reagent grade and purchased from Fisher.
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are summarized in Table S1 in the supplemental material. Primary plate cultures of E. coli were grown from glycerol stock on LB agar, broth medium, or N minimal medium [0.1 M Bis-Tris, pH 7.5 or 5.8, 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 1 mM KH2PO4, 0.10% Casamino Acids, 0.20% glucose, 0.0002% thiamine, 15 μM FeSO4, 10 μM or 10 mM MgSO4] at 37°C with the appropriate antibiotic: ampicillin (100 μg/ml), kanamycin (30 μg/ml), or chloramphenicol (30 μg/ml).
Recombinant DNA techniques.
Primers used in this study are listed in Table S2 in the supplemental material. PCR was performed using reagents from Agilent, and PCR cleanup was done with the QIAquick PCR purification kit (Qiagen). DNA fragments were extracted from agarose gels using the QIAquick gel extraction kit (Qiagen). Restriction endonucleases, T4 DNA ligase, and shrimp alkaline phosphatase were purchased from New England BioLabs. All enzymes were used according to the manufacturers' instructions.
Total RNA isolation and quantitative reverse transcription-PCR (RT-PCR).
E. coli strains were grown overnight in LB liquid culture. The following day, bacteria were spun down, washed with N minimal media without Mg2+ or iron, and inoculated at an optical density at 600 nm (OD600) of 0.1 into N minimal media, pH 7.5, containing 10 μM MgSO4. Cultures were grown to an OD600 of approximately 0.6, and then cells were harvested and total RNA extracted with the Qiagen RNeasy minikit, followed by treatment with RNase-free DNase (Qiagen). cDNA synthesis was performed with the high-capacity cDNA reverse transcription kit (AB Applied Biosystems). The quantification of target genes by quantitative PCR (qPCR) was performed using 2× SYBR green PCR master mix (AB Applied Biosystems) and specific primers for each transcript (see Table S2 in the supplemental material). Data analysis was performed using an ABI 7900HT Fast real-time PCR system and the Software Sequence Detection Systems (SDS), version 2.4 (AB Applied Biosystems). The relative expression ratio of the target transcript was calculated in comparison to the gyrB transcript as the reference gene by following the Pfaffl method (23).
Construction of Illumina libraries, RNA sequencing, and data analysis.
Total RNA was depleted of rRNA using the Ribo-Zero rRNA removal kit for Gram-negative bacteria (Epicentre). Illumina libraries were built from rRNA-depleted total RNA using the NEBNext ultra directional RNA library prep kit for Illumina. RNA sequencing data were mapped to the E. coli W3110 reference genome in CLC Bio Genomics Workbench software, and expression values were determined using RPKM. Baggerly's test on the proportion of counts between samples determined a P value and weighted-proportions fold changes per gene (24). A weighted-proportion absolute change of 4-fold and an false discovery rate (FDR)-corrected P value of ≤0.01 were used as cutoffs as previously described (25). Reads mapping to the last 21 bp of the pmrD gene in the W3110 pmrD mutant were discarded based on the use of the Keio pmrD mutant as the source of the W3110 mutant strain.
Polymyxin B survival assays.
E. coli strains were grown overnight in LB liquid culture. The following day, bacteria were spun down, washed with N minimal medium without Mg2+ or iron, and inoculated at an OD of 0.1 into N minimal medium under one of the following conditions: (i) pH 7.5, 10 μM MgSO4 or (ii) pH 5.8, 10 μM MgSO4. Cultures were grown to an OD600 of 0.6 and split in half; half of the culture was treated with either 2.5 or 5 μg/ml polymyxin B, and the other half was treated with an equivalent volume of phosphate-buffered saline (PBS). Cultures were incubated at 37°C for 1 h and then serially diluted and plated on LB agar. Survival values were calculated by dividing the number of bacteria after treatment with polymyxin B relative to those incubated in the presence of PBS and then multiplied by 100.
Generation of E. coli mutants.
All mutant strains, with the exception of the W3110 phoPQ mutant, were generated by P1 vir phage transduction from individual Keio collection mutants for pmrD, pmrA, or pmrB as previously described (26, 27). These strains include W3110 pmrD, pmrA, phoPQ pmrD, phoPQ pmrA, and phoPQ pmrB, and MG1655 pmrD mutants. Candidate colonies were evaluated using a primer within the kanamycin cassette of each mutation (primer 6) and an outside primer specific to an up- or downstream neighboring gene (primer 3 for pmrD, primer 4 for pmrA, and primer 5 for pmrB). The W3110 phoPQ mutant was generated in the DY330 strain based on the λ Red recombination system (28) using primers 18 and 19 as previously described (12).
Cloning of pmrD coding sequence and RBS from pET21a into low-copy-number plasmid pWSK29.
W3110 pmrD (primers 1 and 2) was cloned into vector pET21a (Novagen) behind the T7lac promoter. The PCR product from pmrD amplification was digested with NdeI and BamHI and ligated overnight into pET21a at 16°C using T4 DNA ligase (New England BioLabs) to generate pET21apmrD. The plasmid next was cut with XbaI and XhoI, excising the pmrD coding region along with a ribosomal binding site (RBS). The resultant fragment was ligated into pWSK29 to give pWSK29pmrD. The vector was transformed into XL-1 Blue (Stratagene) for propagation and various K-12 strains for expression of pmrD. Based on consistency in sequence between strains, the same two primers were used to clone pmrD from strains W3110, MG1655, enterohemorrhagic E. coli (EHEC), and enterotoxigenic E. coli (ETEC). All cloned vectors were sequenced prior to use.
Isolation and analysis of lipid A species from 32P-labeled cells.
Various strains were grown either in LB or N minimal medium at 37°C with 2.5 μCi/ml 32Pi. Bacteria were harvested at an OD of ∼0.8 and washed with 5 ml phosphate-buffered saline. 32P-labeled lipid A was isolated as described previously and spotted onto a silica gel TLC plate (∼10,000 cpm per lane) (29). Lipids were separated using a chloroform, pyridine, 88% formic acid, and water solvent system (50:50:16:5, vol/vol/vol/vol). TLC plates were exposed to a PhosphorImager screen and analyzed using a Bio-Rad Molecular Imager in conjunction with Quantity One software.
Mass spectrometry of lipid A species.
Mass spectra of purified lipids were acquired in the negative-ion linear mode using a matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometer as previously described using a MALDI-TOF/TOF mass spectrometer (ABI 4700 Proteomics Analyzer) (30).
RESULTS
pEtN and l-Ara4N lipid A modifications are induced in wild-type E. coli grown in low Mg2+ and are pmrD dependent.
Our laboratory has routinely demonstrated that E. coli grown in either rich media (such as Luria broth) or minimal media containing high concentrations of Mg2+ (1 to 10 mM) produces 1-diphosphate lipid A. The 1-diphosphate structural variant arises when the inner membrane kinase LpxT (Fig. 1A) transfers a phosphate group from undecaprenyl pyrophosphate to the 1-phosphate group of lipid A (12, 31). Conversely, when E. coli is cultured in low concentrations of Mg2+ (10 to 100 μM), we see lipid A containing PmrA-dependent lipid A modifications (pEtN and l-Ara4N) present in the radiolabeled profile (12). Here, we sought to repeat this result and build on it to investigate a potential role for pmrD in the modification of E. coli lipid A. To this end, 32P-radiolabeled lipid A was isolated from wild-type, pmrD mutant, and complemented pmrD mutant E. coli strains in the W3110 K-12 background grown in both high (10 mM) and low (10 μM) concentrations of Mg2+ N minimal medium (Fig. 2A). As previously observed, wild-type E. coli grown in low Mg2+ integrated both pEtN and l-Ara4N modifications in single and various double combinations (Fig. 2A, lane 4), similarly to WD101, a strain constitutively expressing pmrA (14). Lipid A species modified at both phosphates may have either double pEtN, double l-Ara4N, or one of each group (14, 16, 32). In notable opposition to the wild-type lipid A profile, these modifications were completely absent from the pmrD mutant grown under the same conditions (Fig. 2A, lane 5), while complementation of the pmrD mutant restored the presence of pEtN/l-Ara4N modifications (Fig. 2A, lane 6). Conversely, lipid A profiles of both wild-type and pmrD mutant strains grown in high Mg2+ revealed a 1-diphosphate phenotype (Fig. 2A, lanes 1 and 2). pEtN/l-Ara4N modifications are absent from this growth condition, presumably because high Mg2+ suppresses PhoPQ and pmrD transcription (7). However, when pmrD is expressed in trans in a pmrD mutant strain grown in high Mg2+, pEtN/l-Ara4N-modified lipid A is observed instead of the 1-diphosphate variant (Fig. 2A, lane 3). This demonstrates that exogenous expression of pmrD from a low-copy-number plasmid is sufficient to induce PmrA-dependent lipid A modifications under a growth condition where they are not observed otherwise.
FIG 2.

pmrD is required for addition of pEtN and l-Ara4N to lipid A under low-Mg2+ conditions. Lipid A species observed in the profiles are depicted as cartoons; l-Ara4N, green hexagon; pEtN, red circle. Positive-control strain WD101 produces lipid A constitutively modified with pEtN and l-Ara4N. (A) E. coli W3110 strains were grown in N minimal medium with either 10 mM Mg2+ (high; lanes 1 to 3) or 10 μM Mg2+ (low; lanes 4 to 6). W3110 pmrD was expressed from low-copy-number plasmid pWSK29 in the W3110 pmrD mutant. (B) E. coli MG1655 strains were grown in N minimal medium with 10 μM Mg2+. MG1655 pmrD was expressed from low-copy-number plasmid pWSK29 in the MG1655 pmrD mutant. (C) W3110 pmrD from EHEC or ETEC was expressed from low-copy-number plasmid pWSK29 in the W3110 pmrD mutant in N minimal medium with 10 μM Mg2+.
We also investigated the lipid A profile of the closely related K-12 E. coli strain MG1655 to determine if our observations were strain specific. MG1655 was chosen based on its use in previous studies that questioned the functionality of PmrD and the link between PhoPQ and PmrAB in E. coli (21). Our results establish that pEtN and l-Ara4N groups also are added to lipid A when this K-12 strain is grown in low Mg2+ and that pmrD is required for their presence (Fig. 2B). Further, expression of pmrD from both EHEC and ETEC in the W3110 pmrD mutant complemented lipid A modification to the same extent as the endogenous W3110 gene (Fig. 2C, lanes 3 and 4). This suggests that pathogenic E. coli strains also encode functional PmrD proteins that connect with the PmrAB system.
All results shown in Fig. 2A were confirmed by MALDI-TOF mass spectrometry (Fig. 3). Strains grown in high Mg2+ produced major molecular ions at approximately m/z 1797, indicating the presence of hexa-acylated, bis-phosphorylated lipid A (Fig. 3A, B, and C), the predominant species observed in the radiolabeled profiles (Fig. 2A, lanes 1 and 2). Wild-type E. coli grown in low Mg2+ produced several major peaks corresponding to three main lipid A species: hexa-acylated, bis-phosphorylated (m/z 1797.0), pEtN-lipid A (m/z 1919.9), and l-Ara4N-lipid A (m/z 1927.1) (Fig. 3D). Modified species were absent from lipid A of the pmrD mutant strain grown in low Mg2+, which generated a major molecular ion only at m/z 1796.1, corresponding to hexa-acylated lipid A (Fig. 3E). This spectrum was identical to that of a pmrA mutant in low Mg2+ (see Fig. S1 in the supplemental material). The complementation of the pmrD mutant in low Mg2+ led to the restoration of the singly modified lipid A species observed in the wild-type strain as well as the doubly modified species (pEtN2-lipid A at m/z 2042.2 and pEtN/l-Ara4N-lipidA at m/z 2051.2) (Fig. 3F). This further demonstrates the role of pmrD in inducing PmrAB-dependent lipid A modification with pEtN and l-Ara4N when E. coli is grown in low Mg2+. Taken together, these data show that pmrD promotes pEtN and l-Ara4N lipid A modification under low-Mg2+ growth conditions across different E. coli strains.
FIG 3.

MALDI-TOF mass spectrometry of lipid A from E. coli W3110 wild-type, pmrD mutant, and complemented pmrD mutant strains in high and low Mg2+. Structures and corresponding exact masses are provided for reference. (A to C) Lipid A from W3110 wild-type, pmrD mutant, and complemented pmrD mutant strains grown in 10 mM Mg2+ (high) generate major molecular ion peaks at m/z 1,796.4, 1,796.1, and 1,797.3, respectively. These peaks correspond to hexa-acylated, bis-phosphorylated lipid A. The complemented pmrD mutant also produces minor peaks corresponding to the single addition of pEtN (m/z 1920.3) and l-Ara4N (m/z 1,927.3). (D) Lipid A from W3110 wild type grown in 10 μM Mg2+ (low) produces a molecular ion at m/z 1797.0, representing a hexa-acylated, bis-phosphorylated species. It also generates major peaks corresponding to lipid A singly modified with pEtN (m/z 1919.9) and l-Ara4N (m/z 1927.1), as well as double modification with one of each residue at each phosphate (m/z 2050.0). The peak at m/z 1848.1 corresponds to 1-dephosphorylated hexa-acylated lipid A with one l-Ara4N molecule. (E) pmrD mutant lipid A generates a major molecular ion at m/z 1796.1 when this strain is grown in low Mg2+, indicating the presence of hexa-acylated, bis-phosphorylated lipid A. (F) When grown in low Mg2+, the complemented pmrD mutant produces major ions corresponding to single (m/z 1920.2) and double (m/z 2042.2) addition of pEtN, single (m/z 1927.3) addition of l-Ara4N, and addition of one pEtN and one l-Ara4N (m/z 2051.2). Minor peaks at m/z 1717 and 1769 in panels B, D, E, and F correspond to lipid A species lacking a phosphate group or bearing a shorter acyl chain, respectively. Red labels indicate species with a pEtN group, and green labels indicate species with an l-Ara4N residue.
pEtN/l-Ara4N lipid A modifications are PmrA dependent but only partially PhoPQ dependent.
Previous research has defined the epistatic relationships among PmrD, PhoPQ, and PmrAB in S. enterica (18, 19). We initially analyzed the 32P-labeled lipid A profiles of phoPQ and pmrA mutant strains to explore whether similar relationships exist between these proteins in E. coli (Fig. 4). It has been shown that in E. coli, pmrD expression and protein production are induced in low Mg2+ in a phoP-dependent manner (21). Given this, we expected that deletion of phoPQ from the genome would result in the loss of pEtN and l-Ara4N lipid A modifications, since pmrD is required for addition of these groups, and pmrD expression is dependent on this two-component system.
FIG 4.

pEtN/l-Ara4N lipid A modifications are PmrA dependent but only partially PhoPQ dependent. Lipid A species observed in the profiles are depicted as cartoons; l-Ara4N, green hexagon; pEtN, red circle. Positive-control strain WD101 produces lipid A constitutively modified with pEtN and l-Ara4N. E. coli W3110 strains were grown in 10 μM Mg2+ (low) N minimal media, and radiolabeled lipid A was isolated and separated by TLC.
Surprisingly, when we isolated lipid A from a phoPQ double mutant grown in low-Mg2+ medium, it displayed a partially modified profile containing mostly single and minimal double pEtN and l-Ara4N additions (Fig. 4, lane 2). Densitometry analysis of the chromatographically separated, radiolabeled lipid A in Fig. 4 showed that approximately 40% of phoPQ mutant lipid A was modified versus 89% in the wild-type strain (see Table S3 in the supplemental material). To confirm mutation of phoP and phoQ in the mutant, we performed semiquantitative RT-PCR to demonstrate that neither gene was expressed when the strain was grown in low Mg2+ N minimal medium (data not shown). As a follow-up, we generated a phoPQ pmrD triple mutant to determine if the modifications we observed in the lipid A profile of the phoPQ mutant were pmrD dependent. Indeed, we did not observe modifications in the phoPQ pmrD triple mutant but could restore them upon in trans expression of pmrD in this triple mutant (Fig. 4, lanes 3 and 4). These results imply that an unknown factor or system also can activate pmrD expression, leading to completion of the pmrA-dependent lipid A modification pathway in the absence of phoPQ. As expected, a pmrA mutant was unable to modify its lipid A with pEtN and l-Ara4N, and expression of pmrD in trans could not bypass this phenotype (Fig. 4, lanes 5 and 6). These results confirm that PmrD activity occurs upstream of PmrA but challenge previous findings by showing that pmrD expression is not strictly PhoPQ dependent.
pmrD is transcriptionally active in a phoPQ mutant, and its expression is not influenced by PmrAB.
Given the unexpected presence of pEtN/l-Ara4N modifications in the phoPQ mutant lipid A profile, we reasoned that a PhoPQ-independent factor or system must be activating pmrD transcription in this double mutant. We suspected that PmrA or PmrB could be candidates for transcriptional control of pmrD as a means to ensure expression of eptA and arnT in the absence of phoPQ. Additionally, S. enterica PmrA negatively regulates pmrD transcription, so such an interaction would not be unprecedented (33).
To address this possibility, we isolated RNA from the wild-type, phoPQ mutant, and phoPQ pmrA and phoPQpmrB triple mutant strains grown in low Mg2+. We then determined the expression levels of several key genes by quantitative PCR analysis (Fig. 5). rRNA rrsG showed comparable expression levels across all strains relative to the wild type. Expression of PhoPQ-dependent response regulator rstA was drastically reduced (20-fold) in the three mutant strains. On the contrary, pmrD expression was reduced by approximately 1.7-fold relative to the wild type but showed 12-fold higher expression than rstA in the phoPQ mutant. Since rstA expression is entirely dependent on PhoPQ, these data demonstrate that pmrD depends only partially on PhoPQ for transcriptional activation in agreement with the radiolabeled lipid A profiles shown in Fig. 4. pmrD expression was not significantly different among phoPQ, phoPQpmrA, and phoPQpmrB mutants. We next measured pmrA mRNA levels in the phoPQ mutant strain, which approximately matched the level observed in the wild type. Finally, PmrA-activated arnT showed roughly the same expression level as pmrD in the phoPQ mutant relative to the wild type and was, as expected, almost undetectable in the triple mutant strains. These results confirm that pmrD is expressed in a phoPQ mutant but also suggest that PmrA and PmrB do not exert transcriptional control over pmrD in the absence of phoPQ.
FIG 5.
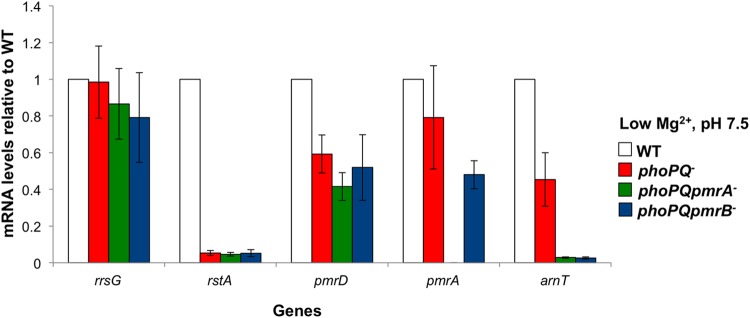
pmrD expression in a phoPQ mutant is not influenced by PmrAB. E. coli W3110 wild-type, phoPQ mutant, phoPQ pmrA mutant, and phoPQ pmrB mutant strains were grown in N minimal medium with 10 μM Mg2+ (low), and RNA was isolated. Relative expression levels of each gene were determined by quantitative PCR. Results are representative of three biological replicates.
PmrD plays a role in expression of pmrA, thereby indirectly affecting expression of downstream pmrA-dependent genes.
To confirm that the presence of pmrD affects PmrA-dependent gene expression as it does lipid A profiles, we performed RNA-seq analysis on W3110 wild-type and pmrD mutant strains grown in low Mg2+ and in high Mg2+. Transcript levels for eptA, arnT, and pmrA all showed statistically significant upregulation in low versus high Mg2+ in the wild-type strain, compared with static, statistically insignificant changes in expression between the two Mg2+ conditions for the pmrD mutant (Table 1). This pattern also held for PmrA-dependent genes involved in synthesis of l-Ara4N (ugd and arnABCD). Conversely, in both strains, a PmrA-independent but PhoP-dependent gene, rstA (34), was expressed at similar levels in both low and high Mg2+. This analysis was generated from the sequenced library of one biological replicate of each strain; a second biological replicate yielded similar results. RNA-seq data for both biological replicates can be found in Tables S4 and S5 in the supplemental material.
TABLE 1.
Select RNA-seq data comparing gene expression in wild-type and pmrD mutant E. coli strains grown in low- versus high-magnesium minimal medium
| Gene | Product | Notes | Gene expression in: |
|||
|---|---|---|---|---|---|---|
| WT |
pmrD mutant |
|||||
| Fold change | P value | Fold change | P value | |||
| arnT | 4-Amino-4-deoxy-l-arabinose transferase | pmrA and pmrD dependent | 6.69 | <10−9 | 1.13 | 0.88 |
| eptA | Phosphoethanolamine transferase | pmrA and pmrD dependent | 32.05 | <10−9 | 1.54 | 0.6 |
| pmrA | DNA-binding response regulator in two-component regulatory system with pmrB | pmrA and pmrD dependent | 7.58 | <10−9 | −1.25 | 0.45 |
| ugd | UDP glucose 6-dehydrogenase | pmrA and pmrD dependent | 20.22 | <10−9 | 1.47 | 0.24 |
| arnA (yfbG) | Fused UDP-L-Ara4N formyltransferase/UDP-glucuronic acid C-4′-decarboxylase | pmrA and pmrD dependent | 10.2 | <10−9 | −1.32 | 0.31 |
| arnB (yfbE) | UDP-4-amino-4-deoxy-l-arabinose oxoglutarate aminotransferase | pmrA and pmrD dependent | 69.60 | <10−9 | −1.89 | 0.11 |
| arnC (yfbF) | Undecaprenyl phosphate 4-deoxy-4-formamido-l-arabinose transferase | pmrA and pmrD dependent | 110 | <10−9 | −2.24 | 0.02 |
| arnD (yfbH) | 4-Deoxy-4-formamido-l-arabinose-phosphoundecaprenol deformylase | pmrA and pmrD dependent | 18.4 | <10−9 | −1.41 | 0.49 |
| rstA | DNA-binding response regulator in two-component regulatory system with RstB | phoP dependent; pmrA and pmrD independent | 52 | <10−9 | 64.19 | <10−9 |
| gyrA | Gyrase subunit A | Housekeeping gene; pmrA and pmrD independent | −1.8 | <10−9 | −1.53 | <10−9 |
| rpsD | Ribosomal protein | Housekeeping gene; pmrA and pmrD independent | −1.3 | <10−9 | −1.62 | <10−9 |
RNA-seq data were confirmed by quantitative PCR (see Fig. S2 in the supplemental material). For example, relative expression levels of pmrA and its downstream targets, arnT and eptA, decreased significantly in the pmrD mutant strain versus that of the wild type; there was a 10-fold decline between strains for pmrA, 23-fold for arnT, and 72-fold for eptA. In contrast, rstA expression showed no significant change in expression between strains. These results demonstrate that PmrA-dependent gene expression is robust in low Mg2+ and PmrD plays a role in expression of the PmrA regulon.
Polymyxin B resistance in E. coli is pmrD dependent in low Mg2+ but not in mildly acidic pH.
pEtN and l-Ara4N lipid A modifications are especially important for bacterial survival in an environment where host-produced CAMPs are present. These chemical groups mask the negative charge of the lipid A molecule imparted by the phosphates that flank the glucosamine disaccharide. Thus, the overall charge of the bacterial cell surface becomes more neutral and better repels positively charged antimicrobial peptides than an unmodified outer membrane (10, 35) (Fig. 1B). We reasoned that an E. coli pmrD mutant would survive poorly with respect to the wild type when exposed to the CAMP polymyxin B due to the inability of the pmrD mutant to incorporate protective lipid A modifications (Fig. 2A).
Accordingly, we determined the ability of wild-type, pmrD mutant, and complemented pmrD mutant strains to survive in various concentrations of polymyxin B. To start, we grew these strains in low Mg2+ N minimal medium (Fig. 6A), the same condition known to induce pEtN/l-Ara4N modifications in the wild type. Under these parameters, we noticed a statistically significant 41.4% decline in survival between the wild type and the pmrD mutant after a 1-h exposure to 2.5 μg/ml polymyxin B. This difference was no longer observed upon the expression of pmrD in the mutant strain. Repetition of the same assay conditions with 5 μg/ml polymyxin B increased the disparity between the wild type and the pmrD mutant even further, with a statically significant 68% decline in survival between the two strains that also could be reversed by overexpression of pmrD (Fig. 6A).
FIG 6.

pmrD plays a role in polymyxin B resistance. (A) E. coli W3110 wild type, pmrD mutant, and the complemented pmrD mutant were grown in N minimal medium with 10 μM Mg2+ (low) at pH 7.5. Strains were challenged with 0, 2.5, or 5 μg/ml polymyxin B or phosphate-buffered saline for 1 h, serially diluted, and plated for survival. The wild-type strain survived at 98.7% and 78.75% in 2.5 and 5 μg/ml polymyxin B, respectively. The pmrD mutant survived at 57.3% and 10.68% when exposed to 2.5 and 5 μg/ml polymyxin B, respectively. (B) The experiment described in panel A was performed at pH 5.8. The wild type survived at 95.8% and 105.6% in 2.5 and 5 μg/ml polymyxin B, respectively. The pmrD mutant survived at levels that were not statistically significantly different from those of the wild type: 106% and 111.8% in 2.5 and 5 μg/ml polymyxin B, respectively. Results are representative of 3 biological replicates. *, P < 0.05.
We next determined survival at mildly acidic pH 5.8, a signal that induces pEtN and l-Ara4N lipid A modifications regardless of Mg2+ concentration and independently of the PhoPQ system (11, 12). While both PmrB and PhoQ can be activated at pH 5.8, the PmrA-dependent lipid A modifications seen under this condition likely are due predominantly to the activation of PmrB, since they are observed even when PhoQ is repressed (7, 11, 18, 36). Therefore, since this condition autonomously activates PmrAB and downstream lipid A modifications, it follows that the presence of pmrD should be irrelevant to bacterial survival, since its function is bypassed. Wild-type and pmrD mutant strains did not show a statistically significant difference in survival when grown at pH 5.8 in low Mg2+ at either 2.5 or 5 μg/ml polymyxin B (Fig. 6B). The survival data presented here clearly illustrate the necessity of pmrD for polymyxin B resistance when Mg2+ is limiting and underscore that its activity occurs upstream of PmrAB.
DISCUSSION
A rapid response is crucial for bacterial survival when environmental conditions change. For Gram-negative organisms, biochemical modification of outer membrane components can result in altered membrane characteristics tailored to defend against specific external insults. The lipid A portion of LPS is a common target for modification, as modulation of its charge and subtleties of its structure can drastically impact membrane integrity (2–4, 37).
When abundant in the environment, divalent cations such as Mg2+ and Ca2+ bind to LPS, creating ionic bridges between neighboring negative lipid A molecules; this contributes considerable stability to the outer membrane (37). Conversely, when divalent cations are limited, the absence of this crucial reinforcement results in a vulnerable membrane and initiation of a bacterial response to address its weakened defenses (7). In Salmonella, this signal is sensed by PhoQ and subsequently propagated to the PmrAB system via PmrD; this cascade ultimately leads to the activation of enzymes EptA and ArnT that add polar residues to the lipid A (14, 16). While limiting Mg2+ is an important signal that negatively affects the Gram-negative bacterial cell, it is unclear if Salmonella and E. coli encounter environments specifically depleted of Mg2+, particularly in the course of infection (8, 38–40). Although decoration of lipid A with pEtN and l-Ara4N results in a stronger membrane barrier that can be advantageous in low Mg2+, the primary benefit of these positively charged modifications more likely is linked with protection against CAMPs (15, 41). It is thought that low Mg2+ promotes activation of PhoQ in a manner that mimics activation of this sensor kinase by CAMPs. CAMPs compete with and displace divalent cations from PhoQ, allowing it to assume an active conformation; low Mg2+ concentrations may similarly promote this active conformation by allowing PhoQ to remain free from the membrane (8). In any case, the results presented here clearly demonstrate that this lipid A modification machinery is robustly activated by low Mg2+ growth conditions, which suggests there exists an evolutionary pressure to modify the membrane under this condition.
Based on our research, this circuitry in E. coli is more complicated than previously thought. There has been a long-held belief in the field that cross talk does not occur between PhoPQ and PmrAB in this organism, based on one group's finding that PmrA-dependent genes are not transcribed when the organism is grown in PhoPQ-activating low Mg2+ (21). Previous findings from our laboratory, however, detected the presence of pEtN and l-Ara4N additions on lipid A isolated from wild-type E. coli grown in 10 μM magnesium (12). Further, Hagiwara et al. suggested that PhoPQ and PmrD are necessary for full transcriptional induction of PmrA-dependent genes (42). These data are confirmed in the current work, which also highlights the necessity of E. coli pmrD for these PmrAB-dependent lipid A modifications, expression of PmrA-dependent genes, and polymyxin B resistance in low Mg2+ conditions. Therefore, while previous research has labeled E. coli PmrD largely as an inactive two-component system connector protein, our findings demonstrate that it is active and necessary for lipid A modification.
Importantly, we uncovered a second, as-yet unidentified system or factor that activates pmrD under low Mg2+ conditions in the absence of phoPQ. pEtN and l-Ara4N were clearly integrated into lipid A of the phoPQ double mutant (Fig. 4), which was surprising given that pmrD expression had previously been shown to be strictly PhoP dependent in E. coli (21). This phenotype could result either from activation of pmrD in a PhoPQ-independent manner or a second connector protein having redundant activity with PmrD. Given that subsequent deletion of pmrD from the phoPQ mutant removes these modified species from the profile and evidence that pmrD is transcriptionally active in the phoPQ mutant (Fig. 5), we conclude that the former possibility is more likely. It is difficult to predict whether this second system contributes to lipid A modification in the wild-type strain or if it functions predominantly when PhoPQ is absent. Progress is ongoing in our laboratory to define the molecular machinery behind these findings. In all, these results suggest that E. coli has wired a second route to activate pmrD in the absence of its primary transcriptional activator, PhoP. This underlines the importance of pEtN/l-Ara4N modifications in low Mg2+ and maintains the necessity of PmrD in their addition to lipid A.
In the last decade, a number of small proteins have been reported to mediate cross talk between various TCSs in E. coli. For instance, EvgAS activates a 65-amino-acid inner membrane protein, SafA (formerly known as B1500), which then complexes with and activates PhoQ (43). This connection is thought to diversify survival strategies in an acidic environment, as EvgAS likely is involved in the acid resistance response. More recently, PmrB of the PmrAB TCS has been demonstrated as a phosphodonor for quorum-sensing response regulator QseB in the absence of its cognate sensor, QseC (44). PmrB can phosphorylate QseB but is inefficient at dephosphorylating it. Therefore, in the absence of QseC, QseB is overactive, leading to aberrant transcription cascades and loss of virulence. These studies underscore that communication between TCSs often involves multilayered networks. Our findings also reflect this, introducing a new layer of complexity into the PhoPQ-PmrD-PmrAB story.
This cascade has been characterized extensively in S. enterica, but the current work focuses on the dynamics of this system in E. coli. Our results strongly support that (i) PmrD maintains the PhoPQ-PmrAB connection in this organism and (ii) a second system can respond to low Mg2+ in the absence of phoPQ to ensure activation of PmrD and fulfillment of the lipid A modification pathway. In all, these findings expand the repertoire of signals integrated and machinery employed by E. coli to respond to and defend against CAMPs.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) (AI064184 and AI076322 to M.S.T.) and the Army Research Office (grant W911NF-12-1-0390 to M.S.T.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.05052-14.
REFERENCES
- 1.Funahara Y, Nikaido H. 1980. Asymmetric localization of lipopolysaccharides on the outer membrane of Salmonella typhimurium. J Bacteriol 141:1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Needham BD, Trent MS. 2013. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11:467–481. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitfield C, Trent MS. 2014. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 4.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 6.Laub MT. 2011. The role of two-component signal transduction systems in bacterial stress responses, p 45–58. In Storz G, Hengge R (ed), Bacterial stress responses, 2nd ed Wiley, New York, NY. [Google Scholar]
- 7.García Véscovi E, Soncini FC, Groisman EA. 1996. Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell 84:165–174. doi: 10.1016/S0092-8674(00)81003-X. [DOI] [PubMed] [Google Scholar]
- 8.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. 2005. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122:461–472. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 9.Gunn JS, Richards SM. 2007. Recognition and integration of multiple environmental signals by the bacterial sensor kinase PhoQ. Cell Host Microbe 1:163–165. doi: 10.1016/j.chom.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Wösten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. 2000. A signal transduction system that responds to extracellular iron. Cell 103:113–125. doi: 10.1016/S0092-8674(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 11.Perez JC, Groisman EA. 2007. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol Microbiol 63:283–293. doi: 10.1111/j.1365-2958.2006.05512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrera CM, Hankins JV, Trent MS. 2010. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol Microbiol 76:1444–1460. doi: 10.1111/j.1365-2958.2010.07150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. 1998. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 14.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CR. 2001. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem 276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CR. 2001. Lipid A modifications in polymyxin-resistant Salmonella typhimurium: PmrA-dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J Biol Chem 276:43111–43121. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 16.Lee H, Hsu F-F, Turk J, Groisman EA. 2004. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J Bacteriol 186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunn JS, Miller SI. 1996. PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J Bacteriol 178:6857–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kox LF, Wösten MM, Groisman EA. 2000. A small protein that mediates the activation of a two-component system by another two-component system. EMBO J 19:1861–1872. doi: 10.1093/emboj/19.8.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato A, Groisman EA. 2004. Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev 18:2302–2313. doi: 10.1101/gad.1230804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo S-C, Lou Y-C, Rajasekaran M, Chang Y-W, Hsiao C-D, Chen C. 2013. Structural basis of a physical blockage mechanism for the interaction of response regulator PmrA with connector protein PmrD from Klebsiella pneumoniae. J Biol Chem 288:25551–25561. doi: 10.1074/jbc.M113.481978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winfield MD, Groisman EA. 2004. Phenotypic differences between Salmonella and Escherichia coli resulting from the disparate regulation of homologous genes. Proc Natl Acad Sci U S A 101:17162–17167. doi: 10.1073/pnas.0406038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen HD, Jewett MW, Groisman EA. 2011. Ancestral genes can control the ability of horizontally acquired loci to confer new traits. PLoS Genet 7:e1002184. doi: 10.1371/journal.pgen.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baggerly KA, Deng L, Morris JS, Aldaz CM. 2003. Differential expression in SAGE: accounting for normal between-library variation. Bioinformatics 19:1477–1483. doi: 10.1093/bioinformatics/btg173. [DOI] [PubMed] [Google Scholar]
- 25.Davies BW, Bogard RW, Young TS, Mekalanos JJ. 2012. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 27.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran AX, Karbarz MJ, Wang X, Raetz CRH, McGrath SC, Cotter RJ, Trent MS. 2004. Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A. J Biol Chem 279:55780–55791. doi: 10.1074/jbc.M406480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stead CM, Beasley A, Cotter RJ, Trent MS. 2008. Deciphering the unusual acylation pattern of Helicobacter pylori lipid A. J Bacteriol 190:7012–7021. doi: 10.1128/JB.00667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Touzé T, Tran AX, Hankins JV, Mengin-Lecreulx D, Trent MS. 2008. Periplasmic phosphorylation of lipid A is linked to the synthesis of undecaprenyl phosphate: periplasmic dephosphorylation of undecaprenyl-PP. Mol Microbiol 67:264–277. doi: 10.1111/j.1365-2958.2007.06044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibbons HS, Kalb SR, Cotter RJ, Raetz CRH. 2005. Role of Mg2+ and pH in the modification of Salmonella lipid A after endocytosis by macrophage tumour cells. Mol Microbiol 55:425–440. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 33.Kato A, Latifi T, Groisman EA. 2003. Closing the loop: the PmrA/PmrB two-component system negatively controls expression of its posttranscriptional activator PmrD. Proc Natl Acad Sci U S A 100:4706–4711. doi: 10.1073/pnas.0836837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minagawa S, Ogasawara H, Kato A, Yamamoto K, Eguchi Y, Oshima T, Mori H, Ishihama A, Utsumi R. 2003. Identification and molecular characterization of the Mg2+ stimulon of Escherichia coli. J Bacteriol 185:3696–3702. doi: 10.1128/JB.185.13.3696-3702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. 2000. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun 68:6139–6146. doi: 10.1128/IAI.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prost LR, Daley ME, Le Sage V, Bader MW, Le Moual H, Klevit RE, Miller SI. 2007. Activation of the bacterial sensor kinase PhoQ by acidic pH. Mol Cell 26:165–174. doi: 10.1016/j.molcel.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-del Portillo F, Foster JW, Maguire ME, Finlay BB. 1992. Characterization of the micro-environment of Salmonella typhimurium-containing vacuoles within MDCK epithelial cells. Mol Microbiol 6:3289–3297. doi: 10.1111/j.1365-2958.1992.tb02197.x. [DOI] [PubMed] [Google Scholar]
- 39.Miller SI, Kukral AM, Mekalanos JJ. 1989. A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci U S A 86:5054–5058. doi: 10.1073/pnas.86.13.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alpuche Aranda CM, Swanson JA, Loomis WP, Miller SI. 1992. Salmonella typhimurium activates virulence gene transcription within acidified macrophage phagosomes. Proc Natl Acad Sci U S A 89:10079–10083. doi: 10.1073/pnas.89.21.10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murata T, Tseng W, Guina T, Miller SI, Nikaido H. 2007. PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar typhimurium. J Bacteriol 189:7213–7222. doi: 10.1128/JB.00973-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagiwara D, Yamashino T, Mizuno T. 2004. A genome-wide view of the Escherichia coli BasS-BasR two-component system implicated in iron-responses. Biosci Biotechnol Biochem 68:1758–1767. doi: 10.1271/bbb.68.1758. [DOI] [PubMed] [Google Scholar]
- 43.Eguchi Y, Itou J, Yamane M, Demizu R, Yamato F, Okada A, Mori H, Kato A, Utsumi R. 2007. B1500, a small membrane protein, connects the two-component systems EvgS/EvgA and PhoQ/PhoP in Escherichia coli. Proc Natl Acad Sci U S A 104:18712–18717. doi: 10.1073/pnas.0705768104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guckes KR, Kostakioti M, Breland EJ, Gu AP, Shaffer CL, Martinez CR III, Hultgren SJ, Hadjifrangiskou M. 2013. Strong cross-system interactions drive the activation of the QseB response regulator in the absence of its cognate sensor. Proc Natl Acad Sci U S A 110:16592–16597. doi: 10.1073/pnas.1315320110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Z, Ribeiro AA, Raetz CRH. 2000. High-resolution NMR spectroscopy of lipid A molecules containing 4-amino-4-deoxy-l-arabinose and phosphoethanolamine substituents different attachment sites on lipid A molecules from NH4VO3-treated Escherichia coli versus kdsA mutants of Salmonella typhimurium. J Biol Chem 275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.