

Abstract
When antimicrobials are used empirically, pathogen MICs equal to clinical breakpoints or epidemiological cutoff values must be considered. This is to ensure that the most resistant pathogen subpopulation is appropriately targeted to prevent emergence of resistance. Accordingly, we determined the pharmacokinetic (PK) profile of moxifloxacin at 400 mg/day in 18 patients treated empirically for community-acquired pneumonia. We developed a population pharmacokinetic model to assess the potential efficacy of moxifloxacin and to simulate the maximal MICs for which recommended pharmacokinetic-pharmacodynamic (PK-PD) estimates are obtained. Moxifloxacin plasma concentrations were determined the day after therapy initiation using ultra-high-performance liquid chromatography. Peak drug concentrations (Cmax) and area under the free drug concentration-time curve from 0 to 24 h (fAUC0–24) values predicted for each patient were evaluated against epidemiological cutoff MIC values for Streptococcus pneumoniae, Haemophilus influenzae, and Legionella pneumophila. PK-PD targets adopted were a Cmax/MIC of ≥12.2 for all pathogens, an fAUC0–24/MIC of >34 for S. pneumoniae, and an fAUC0–24/MIC of >75 for H. influenzae and L. pneumophila. Individual predicted estimates for Cmax/MIC and fAUC0–24/MIC as well as simulated maximal MICs resulting in target attainment for oral and intravenous administration of the drug were suitable for S. pneumoniae and H. influenzae but not for L. pneumophila. These results indicate that caution must be taken when moxifloxacin is used as monotherapy to treat community-acquired pneumonia caused by L. pneumophila. In conclusion, this report reveals key information relevant to the empirical treatment of community-acquired pneumonia while highlighting the robust and flexible nature of this population pharmacokinetic model to predict therapeutic success. (Clinical Trials Registration no. NCT01983839.)
INTRODUCTION
With the global trend toward decreased antibiotic susceptibility, it is of great importance to make sure that any antibiotic given targets the most resistant bacterial subpopulation present to prevent further emergence of resistance. A worst-case scenario with pathogen MICs equal to clinical MIC breakpoints must be taken into consideration when antimicrobials are used empirically. Moxifloxacin (Avelox; Bayer), a “fourth-generation” fluoroquinolone, is often used in the empirical treatment of severe community-acquired pneumonia (CAP), which is one of the most common infectious diseases and among the primary causes of death worldwide (1, 2). Even though the majority of CAP cases are managed in the primary care setting, symptoms can be severe and up to 10% of hospitalized patients with CAP need treatment in an intensive care unit (ICU) (3). Streptococcus pneumoniae is the primary pathogen responsible for CAP (4), but many other microorganisms, including Gram-negative and atypical bacteria (e.g., Legionella pneumophila, Mycoplasma pneumoniae, and Chlamydophila pneumoniae), may also be etiological agents.
Moxifloxacin has a broad spectrum of antibacterial activity and is considered effective against the vast majority of CAP pathogens, including Gram-positive, Gram-negative, and atypical bacteria, as well as multidrug-resistant S. pneumoniae (5–9). The drug is well tolerated, and clinical studies have shown that moxifloxacin is superior to, or as effective as, ceftriaxone, amoxicillin-clavulanic acid, and levofloxacin in the treatment of CAP (10–12). The recommended dose of moxifloxacin is 400 mg/day (q.d.). No dosage adjustment is required in elderly patients, obese patients (13), or patients with renal or mild hepatic impairment (14). Furthermore, due to the risk of a prolonged QT interval (a measure of the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle), it is recommended that the daily dose of moxifloxacin should not exceed 400 mg (15–17).
Fluoroquinolones are antimicrobials with concentration-dependent bactericidal activity. For fluoroquinolones, the pharmacokinetic/pharmacodynamic (PK-PD) measures that correlate with clinical and bacteriologic efficacy are the ratio of the peak drug concentration to the MIC of the drug (Cmax/MIC) and the ratio of the 24-h area under the free drug concentration-time curve to the MIC (fAUC0–24/MIC) (18–20). fAUC0–24/MIC has the strongest association with therapeutic efficacy for multiple fluoroquinolones (19–21). Nevertheless, an optimal Cmax/MIC ratio is of importance, both to achieve the optimal bactericidal effect in the treatment with fluoroquinolones and to avoid the emergence of resistance (22–24). A Cmax/MIC of 8 to 20 has been referred to as the optimal value to achieve these goals (22, 25–27). Preston et al. (26) found that a Cmax/MIC of ≥12.2 was predictive of a favorable clinical and microbiological outcome in levofloxacin treatment of pulmonary, urinary tract, and soft-tissue infections. Furthermore, Drusano et al. (22) demonstrated in an animal model that a Cmax/MIC greater than 10 was associated with a positive clinical outcome when treating Pseudomonas aeruginosa sepsis with fluoroquinolones. Fluoroquinolones are often used in the treatment of respiratory tract infections, and a high probability of therapeutic response has been observed with fAUC0–24/MIC ratios of greater than 34 (28). Both in vivo and in vitro studies have shown that a higher fAUC0–24/MIC is required for successful treatment of infections due to Gram-negative bacteria than for infections caused by S. pneumoniae and other Gram-positive bacteria (25, 27, 29, 30). To our knowledge, no in vivo studies regarding the correlation between fAUC0–24/MIC and clinical outcome have been performed for atypical bacteria.
The PK and PD properties of antimicrobials have been the focus of many studies during recent years because these parameters may help predict clinical outcome, lead to improved dosage regimens, and aid in preventing the selection of resistant pathogen mutants. The fact that antibiotic resistance is increasing worldwide emphasizes the importance of choosing the right antimicrobial at the right dose, especially in situations in which antimicrobials are being used empirically. If PK-PD targets cannot be met for the antimicrobial agent chosen, an alternative agent must be considered in the treatment.
This study was designed to determine the pharmacokinetics of moxifloxacin administered at 400 mg q.d. to patients treated empirically for CAP. To accomplish this aim, we established a PK population model. This approach was adopted with the dual purpose of assessing the potential efficacy of the drug and performing Monte-Carlo simulations to characterize the maximal MICs for which PK-PD targets are obtained for pathogens commonly known to cause CAP.
MATERIALS AND METHODS
Study design.
This report details a prospective, observational study conducted at the Department of Infectious Diseases, Aarhus University Hospital, Aarhus, Denmark, between March 2013 and April 2014. All patients were informed about the purpose of the study. Given that this study was undertaken in parallel with standard-of-care treatment of CAP, the Regional Ethical Committee approved the study without requiring signed informed consent (Clinical Trials Registration no. NCT01983839).
Patient population and study drug.
Patients with diagnosed CAP who were empirically prescribed moxifloxacin at 400 mg q.d. by the treating physician, according to national CAP treatment guidelines, had plasma concentrations of moxifloxacin determined. Patients less than 18 years of age were excluded from the study, and the age, gender, and body weight of each enrolled patient were registered. Moxifloxacin was administered either orally (p.o.) or intravenously (i.v.) with infusion over 1 h. On the second day of treatment, each patient had a blood sample drawn immediately before administration of moxifloxacin, 1 h after administration of moxifloxacin, 3 h after administration of moxifloxacin, and 24 h after administration of moxifloxacin, to determine the plasma concentration-time course of the drug. When moxifloxacin was administered i.v., blood samples were drawn 1, 3, and 24 h after the end of infusion. All plasma samples were stored at −20°C until analysis. To assess a possible influence on the moxifloxacin PK parameters, plasma concentrations of alanine aminotransferase (ALAT), albumin, and creatinine were determined on the same day as the plasma concentrations of moxifloxacin.
Microbiological analysis.
In the Department of Clinical Microbiology, sputum and whole-blood samples were cultured according to standard operating procedures (i.e., whole-blood cultures are incubated in Bact/ALERT for up to 6 days). Sputum samples and positive blood cultures were streaked to agar plates, incubated overnight, and investigated for growth of bacterial pathogens. When a bacterial species was isolated, a moxifloxacin MIC was obtained by using E-tests (AB Biodisk, Solna, Sweden) performed on Mueller-Hinton agar plates. In addition, PCR analysis to detect M. pneumoniae, C. pneumoniae, and L. pneumophila in sputum and a legionella urinary antigen test (LUT) and a pneumococci urinary antigen test (PUT) (BinaxNOW) were performed upon request from the treating physicians.
UHPLC analysis.
The free moxifloxacin plasma concentrations were assessed using ultra-high-performance liquid chromatography (UHPLC). Prior to analysis, 300 μl of serum was added to a 96-well ultrafilter plate (Acroprep 30K Omega; Pall Corporation, USA) with a 30-kDa molecular cutoff. After centrifugation for 30 min at 1,000 × g, 15 μl filtrate was mixed with 20 μl 10 mM phosphate buffer (pH 3) (NaH2PO4·H2O adjusted with HCl; Merck, Germany) in a 96-well tray (Kem-En-Tec, Denmark) (0.2 ml Thin-Wall). Standards of moxifloxacin (0.47, 1.88, 7.5, and 15 μg/ml) were prepared by adding moxifloxacin (Sigma-Aldrich, Denmark) to 0.9% NaCl–water and mixed with phosphate buffer. The separation system (Agilent 1290 Infinity; Agilent Technologies, USA) was equipped with a 100-mm-by-2.1-mm C18 column (Kinetex; Phenomenex, USA) (1.7 μm inner diameter), which was preheated to 40°C. For analysis, 5 μl prepared sample or standard was injected into the UHPLC system, and moxifloxacin was separated from other plasma compounds with a gradient of methanol (Sigma-Aldrich, Denmark) in phosphate buffer, changing from 0% to 50% over 4 min. The typical retention time of moxifloxacin was 3.1 min. Detection of moxifloxacin was done with UV detection at 295 nm, and calculation of the concentrations was done with ChemStation Software (Agilent Technologies, USA). Intrarun (total) imprecision values (percent coefficients of variation [%CV]) were 3.0% (6.2%) at 0.9 mg/liter and 4.8% (6.2%) at 3.5 mg/liter. The limit of quantification was defined as the lowest concentration with a CV of <20% and was found to be 0.05 mg/liter.
Population PK modeling, PD evaluation, and simulations.
The plasma concentration-time profiles of moxifloxacin were modeled using NONMEM 7.3 (Icon Development Solutions, Hanover, MD, USA). The first-order conditional estimation (FOCE) method with interaction was used. PK i.v. and p.o. data were modeled simultaneously.
The log-normal distribution of the parameters around the typical value was assumed:
where Pi is the value of the parameter in the individual i, P is the typical value of the parameter in the population, and ηi is the normally distributed interindividual variability (IIV) with mean 0 and variance ω2. Additive, proportional, and combined additive and proportional residual error models were investigated.
One- and two-compartment models with clearance (CL) from the central compartment were evaluated for moxifloxacin disposition. Moxifloxacin absorption was modeled with a first-order absorption rate (ka) with or without a lag time.
Gender was investigated as a categorical covariate, while body weight and creatinine, alanine aminotransferase (ALAT), and albumin plasma concentrations were evaluated as continuous covariates on the moxifloxacin PK parameters using a power function. Additionally, body weight as a covariate was investigated by fixing the exponents according to allometric scaling (0.75 for clearance and 1.0 for volume parameters) (31).
Perl-speaks-NONMEM (PsN) (32) was used for model execution and simulations with a Pirana graphical interphase (33). Model discrimination and selection of best fit to the PK data were based on goodness-of-fit plots and prediction-corrected visual predictive checks (pcVPC) (34) using R (R-project) with the Xpose 4.0 package (35) and the parameters precision and reduction in objective function values (OFV). Inclusion of a parameter was regarded as statistically significant (P < 0.05) for a difference in OFV (dOFV) of −3.84 for nested structures, while P < 0.01 was used as the backwards deletion criterion for final acceptance in the covariate analysis, using a stepwise covariate model approach.
Calculation of the total maximum plasma concentration (Cmax, accounting for 40% plasma protein binding [36]) and the area under the unbound plasma concentration-time curve from 0 to 24 h (fAUC0–24) on day 2 was performed with model-estimated parameters for oral administration according to the following equations:
where tmax is the time for maximum plasma concentration, V is the volume of distribution, k is the elimination rate constant, and F is the absolute oral bioavailability. Calculations based on i.v. infusion, where tmax is 1 h, reduced the Cmax equation to the following:
where Cmax refers to the total and not the free plasma concentration of the drug. This is the conventional approach for presenting Cmax/MIC. To our knowledge, there are no published values for fCmax/MIC (free drug concentration) derived from clinical studies regarding fluoroquinolones. In order to compare our results to this specific PK/PD target, the free plasma concentration measured was transformed to the total plasma concentration. The protein binding rates of moxifloxacin are 30% to 50% (37). We assumed a protein binding rate of 40% on the basis of the measurement made by Stass et al. (36).
The Cmax and fAUC0–24 predicted for each individual were evaluated against the drug MIC for the isolated pathogens. If no pathogen was detected in the sputum or whole-blood cultures, Cmax and fAUC0–24 values were compared to epidemiological cutoff (ECOFF) MIC values for moxifloxacin published by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) for S. pneumoniae (0.5 mg/liter) and Haemophilus influenzae (0.125 mg/liter) (38). We made the same comparison to L. pneumophila. While neither ECOFF MICs nor clinical MIC breakpoints have been defined yet by EUCAST for this pathogen, the Cmax and fAUC0–24 values were compared to ECOFF moxifloxacin MIC values (1.0 mg/liter), based on the wild-type MIC distribution for clinical L. pneumophila serogroup 1 isolates recently published by Bruin et al. (39).
Based on previous studies addressing efficacy breakpoints for fluoroquinolones, the PK-PD targets adopted were a Cmax/MIC of ≥12.2 (26) for all pathogens, an fAUC0–24/MIC of >34 for S. pneumoniae (28), and an fAUC0–24/MIC of >75 for Gram-negative bacteria (21, 27). To our knowledge, there have been no in vivo studies regarding efficacy breakpoints for fluoroquinolones and L. pneumophila. Considering L. pneumophila to be a Gram-negative microorganism, the PK-PD target of fAUC0–24/MIC > 75 was adopted for this pathogen as well.
Typical PK profiles stratified by included covariates were illustrated by plots of population-predicted plasma concentrations versus time at the minimum and maximum values of the covariate. A total of 2,000 trial Monte-Carlo simulations were performed with the final PK model to estimate the maximal MIC value of a pathogen that would result in ≥90% probability of target attainment (PTA) for the PK-PD targets previously mentioned: Cmax/MIC of >12.2, fAUC0–24/MIC of >34, and fAUC0–24/MIC of >75. The 2,000 simulations were performed using a population of 100 patients with a normally distributed body weight around 70 kg (standard deviation [SD], 13 kg; range, 45 to 98 kg [to include only the body weight range validated by the model]). Simulations were performed for moxifloxacin at 400 mg q.d. p.o. and i.v., respectively, providing simulated Cmax and fAUC0–24 values on day 2.
Statistical analysis.
Data were analyzed using Stata version 13 (StataCorp, College Station, TX) and GraphPad Prism 6.0. Continuous variables were defined as median and interquartile range (IQR).
RESULTS
Patient characteristics and microbiology.
Patient characteristics are shown in Table 1. A total of 18 patients (8 men and 10 women) were included in the study. The median age was 73 years (IQR, 66 to 83), and the median body weight was 72.6 kg (IQR, 64.1 to 80.6). A total of 15 patients received moxifloxacin p.o., and 3 patients received moxifloxacin i.v. Four patients only had plasma concentrations of moxifloxacin determined before administration of the drug, 1 h after administration of the drug, and 3 h after administration of the drug. This was due to different clinical examinations being performed at 24 h, which precluded blood sampling at that time.
TABLE 1.
Patient characteristics (n = 18)
| Parameter | Value(s) |
|---|---|
| Age (yrs) | 73 (66–83)a; (18–94)b |
| Gender | |
| Male | 8 (44)c |
| Female | 10 (56)c |
| Body wt (kg) | 72.6 (64.1–80.6)a; (37–100)b |
Median (interquartile range).
Range.
n (%).
None of the patients had pathogens detected in the whole-blood cultures, and only three patients had a pathogen detected in the sputum samples: one sample contained Escherichia coli, one M. pneumonia, and one L. pneumophila. As the M. pneumoniae and L. pneumophila isolates could not be cultured, the moxifloxacin MIC determination was performed only for E. coli (0.023 mg/liter). For one patient, the PUT result was positive but no pathogen was cultured from the sputum or whole blood, and it was not possible to perform a subsequent MIC. Eight of the 14 patients who did not have a pathogen detected in blood, sputum, or urine samples had been prescribed an antibiotic by their primary care physician before admission to the hospital.
PK-PD analysis.
The moxifloxacin plasma concentration-time profiles were described using a one-compartment model with first-order absorption and elimination rates with IIV for CL and F, including a proportional residual error, with adequate (<25%) parameter precision and shrinkage values. Relative to a model with interindividual variability with respect to CL and V, the final model had a drop of 5.7 in the OFV with improved parameter precision. Also, an OMEGA block was tested to assess covariance between CL and F, but no significant improvement was found for this model. The pcVPC (p.o. only), demonstrating adequate prediction of the final model relative to the observed data, and the corresponding pharmacokinetic parameters are shown in Fig. 1 and Table 2, respectively. The model-estimated median values of total Cmax and fAUC0–24 for the current study population were 3.99 mg/liter (IQR, 3.19 to 5.29) and 32.78 mg · h/liter (IQR, 22.75 to 47.31), respectively. Among the available covariates, only allometric scaling of body weight for clearance and volume of distribution provided a substantial explanation of residual and interindividual variability. No other covariate passed the forward inclusion step of a P value of <0.05.
FIG 1.

Prediction-corrected visual predictive check (pcVPC) of final PK model following p.o. administration of 400 mg moxifloxacin. Open circles, observations; solid black line, median interpolated observations; dotted black lines, 10th and 90th percentiles of the observed data.
TABLE 2.
Pharmacokinetic parameters corresponding to the one-compartment population model depicted in Fig. 1a
| Parameter | Estimate (RSE %) | IIV (RSE %) | Description |
|---|---|---|---|
| CL × (wt/70)0.75 (liters/h) | 12 (10) | 21 (26)b | Systemic clearance with allometric wt scaling |
| V × (wt/70) (liters) | 165 (13) | Central vol of distribution with allometric wt scaling | |
| F (%) | 89 (16) | 30 (29)c | Oral bioavailability |
| ka (h−1) | 5.4 (58) | Absorption rate constant | |
| σ1, prop error (%) | 34 (10) | Proportional residual error |
RSE, relative standard error reported on the approximate standard deviation scale; IIV, interindividual variability expressed as coefficient of variation; prop, proportional.
Shrinkage, 20%; Etabar (statistical significance of the mean ETA being zero) P = 0.01.
Shrinkage, 25%; Etabar P = 0.007.
Each of the individual model-predicted Cmax and fAUC0–24 values was divided by the ECOFF MIC for S. pneumoniae (0.5), H. influenzae (0.125), and L. pneumophila (1.0) (Fig. 2). All patients achieved a Cmax/MIC of >12.2 for H. influenzae. This was achieved in only three patients (17%) for S. pneumoniae and in none for L. pneumophila. All patients achieved an fAUC0–24/MIC of >34 for S. pneumoniae and an fAUC0–24/MIC of >75 for H. influenzae. None of the patients achieved an fAUC0–24/MIC of >75 for L. pneumophila. The patient with E. coli in the sputum sample (MIC, 0.023 mg/liter) achieved both PK-PD targets (Cmax/MIC, 287.4; fAUC0–24/MIC, 2,057).
FIG 2.

Model predicted Cmax and fAUC0–24 for each patient compared to the MIC for common respiratory pathogens. Each patient's model-predicted Cmax (A) and fAUC0–24 (B) were divided by the MIC for the indicated pathogen. Values below the PK-PD targets are shaded in gray. PD targets: Cmax/MIC > 12.2 (A); fAUC24/MIC > 34 for S. pneumoniae, fAUC24/MIC > 75 for H. influenzae and L. pneumophila (B). Horizontal lines represent median values.
To gain insights into the influence of body weight on the PK-PD relationship of moxifloxacin, the typical PK profiles for a 37-kg patient versus a 100-kg patient were derived from the final moxifloxacin PK model and are illustrated in Fig. 3. The corresponding Cmax and fAUC0–24 values for a 37-kg patient were 7.3 mg/liter and 50 mg · h/ml, respectively. The corresponding Cmax and fAUC0–24 values for a 100-kg patient were 2.5 mg/liter and 18 mg · h/ml, respectively.
FIG 3.
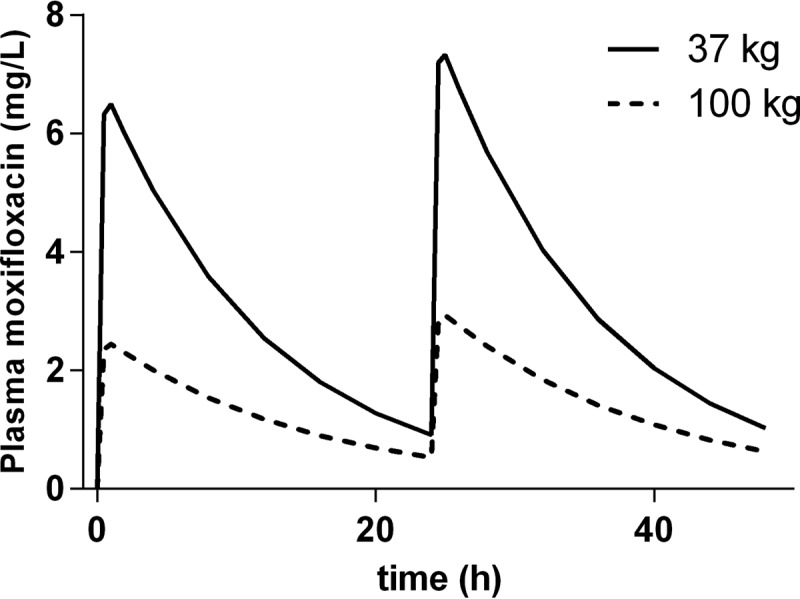
Illustrations of typical PK profiles of moxifloxacin at 400 mg q.d. in a patient weighing 37 and in a patient weighing 100 kg. Each profile is derived from the PK model presented in Fig. 1 and illustrates the differences in plasma concentrations versus time at the minimum and maximum value of body weight in the current population. The free plasma concentration measured was transformed to the total plasma concentration, assuming a protein binding rate of 40%.
Because moxifloxacin can be dosed either orally or via infusion during the empirical treatment of CAP, we simulated PK-PD target attainments as a function of MIC for both dosing strategies. The results are shown in Fig. 4 and Table 3. The maximal MICs resulting in a PTA value of ≥90% for the PK-PD targets adopted were higher in the i.v. simulations than in the p.o. simulations.
FIG 4.

Probabilities of target attainment (PTA) versus MIC derived from simulations of moxifloxacin administration. (A) Simulations of p.o. moxifloxacin at 400 mg q.d. (B) Simulations of i.v. moxifloxacin at 400 mg q.d. fAUC24/MIC > 34 = PK-PD target adopted for Gram-positive pathogens. fAUC24/MIC > 75 = PK-PD target adopted for Gram-negative and atypical pathogens. The dotted lines represent PTA 90%.
TABLE 3.
Estimates from the Monte-Carlo simulations of PTA versus MIC depicted in Fig. 4
| PK-PD target | Maximal MIC for PTA ≥ 90% (mg/liter)a |
|
|---|---|---|
| p.o. simulation | i.v. simulation | |
| Cmax/MIC > 12.2 | 0.21 | 0.29 |
| fAUC24/MIC > 34 | 0.54 | 0.73 |
| fAUC24/MIC > 75 | 0.25 | 0.33 |
Each estimate represents the maximal MIC, corresponding to the PD target specified, for which PTA ≥90% was obtained.
DISCUSSION
In this study, we developed a PK population model to assess the pharmacokinetic profile of moxifloxacin at 400 mg q.d., as used in the empirical treatment of patients with CAP. To our knowledge, this is the first PK population model published for moxifloxacin treatment of patients with CAP. Our results may therefore be of help for clinicians treating CAP patients with this particular drug.
Using ECOFF MICs, our results confirm that moxifloxacin at 400 mg q.d. is still a good choice for treating CAP caused by S. pneumoniae and H. influenzae but that caution should be taken when the drug is used as monotherapy in the treatment of L. pneumophila. As depicted in Fig. 2, none of the patients achieved a Cmax/MIC of >12.2 and an fAUC0–24/MIC of >75 for L. pneumophila. However, Legionella spp. have certain growth requirements which make susceptibility testing for this particular pathogen challenging. BCYE-α [N-(2-acetamido)-2-aminoethanesulfonic acid-buffered charcoal yeast extract alpha] agar plates used to support the growth of Legionella spp. are buffered with charcoal with the primary function of absorbing metabolites which might otherwise inhibit the growth of the pathogen. It is known that the charcoal may inhibit the activity of a number of antibiotics, including fluoroquinolones (40). Performing susceptibility testing for L. pneumophila, using BCYE-α agar plates, might therefore yield elevated MICs, which should be kept in mind when interpreting our results.
The fact that 8 of the 14 patients in this study who did not have a pathogen detected in blood, sputum, or urine samples had been prescribed an antibiotic by their primary care physician before admission to the hospital may have been a factor contributing to the low rate of positive specimen results. In general, the causative pathogen is defined in fewer than half of the patients diagnosed with CAP, due to antibiotic use and variability in disease severity (2).We focused our study on three pathogens associated with CAP while recognizing that many pathogens are capable of causing this disease. The majority of pathogens causing CAP are typically considerably more susceptible to moxifloxacin than the ECOFF MICs used in our calculations and therefore would be successfully treated by the standard dose, namely, 400 mg q.d. For example, we identified a single pathogen in the current study for which it was possible to perform a MIC determination. The MIC for this E. coli isolate was 0.023 mg/liter, resulting in a Cmax/MIC of 287.4 and an fAUC0–24/MIC of 2,057, which are far above the estimates correlated with clinical and microbiological efficacy (21, 26, 27). However, when the susceptibility pattern of local potential pathogens is unknown, adopting the most conservative ECOFF MICs or clinical-breakpoint MICs may be the most appropriate and clinically beneficial approach when choosing the type and dosing of antimicrobial drugs.
Using a population approach, we developed a robust one-compartment model with simultaneous modeling of p.o. and i.v. data which adequately described the PK of moxifloxacin for both administration routes. This was accomplished despite the limitations associated with the relatively small sample size in this study and the restricted number of blood samples from each patient. The model structure and typical PK parameters with body weight as a covariate are comparable to a recently published moxifloxacin PK model developed by Florian et al. on the basis of rich data and a large population (9,235 moxifloxacin concentrations included from 1,045 subjects) (17), strengthening the validity of the current treatment model. In contrast to the work by Florian et al., our model includes not only oral but also i.v. PK data, making the current model useful for both dosing regimens. Provided that the levels of oral bioavailability are comparable between the two studies, oral moxifloxacin PK data from the studied pneumonia patients and healthy volunteers do not appear to be significantly different.
Also, a two-compartment model developed by Kees et al. (41) has been used to describe the disposition of moxifloxacin, providing comparable estimates of systemic clearance (11.3 liters/h) and total volume of distribution (115.2 liters).
The Monte-Carlo simulations of PTA versus MIC provide valuable information about the maximal MICs for which recommended PK-PD estimates are obtained for potential Gram-positive and Gram-negative pathogens, for both p.o. and i.v. administration of moxifloxacin at 400 mg q.d. (Fig. 4 and Table 3). Infusion administration allows higher maximal MICs in order to achieve the PK-PD targets adopted, which suggests that moxifloxacin at 400 mg q.d. should be administered i.v. in cases of severe CAP. However, because of the high bioavailability of the drug, the differences in maximal MICs between p.o. and i.v. administration were relatively small overall. Our results of maximal MICs in the i.v. simulations are similar to those found by Kontou et al. (42), who estimated the maximal MIC that would guarantee an optimal exposure of moxifloxacin at 400 mg q.d. i.v. in patients with severe lower respiratory tract infections. Specifically, they identified maximal MICs of 0.79 and 0.32 mg/liter for Gram-positive and Gram-negative bacteria, respectively. Kees et al. (41) calculated PTA values for pathogen drug MICs ranging from 0.064 to 1 mg/liter in ICU patients treated with moxifloxacin at 400 mg q.d. In their study, an fAUC24/MIC of >34 was obtained for pathogens with a MIC of up to 0.25 mg/liter and an fAUC24/MIC of >75 was obtained for pathogens with a MIC of up to 0.125 mg/liter. The values that they measured were lower than ours, which may be explained by the fact that changes in PK parameters are common in critically ill patients who need treatment in the ICU (43, 44). Our PK model indicates that the typical PK profile is related to the body weight of the patient. Theoretically, this means that patients with a low body weight are more likely to benefit from moxifloxacin at 400 mg q.d. than patients with a high body weight when they are infected with pathogens exhibiting a relatively high drug MIC.
In conclusion, our report demonstrates that a robust PK model for moxifloxacin at 400 mg q.d. can be created using a population approach that incorporates sparse blood sampling data. In this era of emerging multidrug bacterial resistance, choosing the right antimicrobial at the right dose is of utmost importance. This poses a significant challenge during the empirical treatment of infections, as the susceptibility of the causative pathogen to the antimicrobial drug is unknown. In these situations, a PK model such as ours may outline the potential efficacy of a drug and simulate the maximal MICs for which certain PK-PD estimates are obtained while accounting for variables, including drug administration route and patient body weight.
ACKNOWLEDGMENTS
Kristina Öbrink-Hansen is supported by a Ph.D. grant from the Faculty of Health Science, University of Aarhus, Aarhus, Denmark.
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Paul W. Denton for assistance in the drafting of the manuscript.
REFERENCES
- 1.Wiemken TL, Peyrani P, Ramirez JA. 2012. Global changes in the epidemiology of community-acquired pneumonia. Semin Respir Crit Care Med 33:213–219. doi: 10.1055/s-0032-1315633. [DOI] [PubMed] [Google Scholar]
- 2.Polverino E, Torres Marti A. 2011. Community-acquired pneumonia. Minerva Anestesiol 77:196–211. [PubMed] [Google Scholar]
- 3.Niederman MS, Mandell LA, Anzueto A, Bass JB, Broughton WA, Campbell GD, Dean N, File T, Fine MJ, Gross PA, Martinez F, Marrie TJ, Plouffe JF, Ramirez J, Sarosi GA, Torres A, Wilson R, Yu VL; American Thoracic Society. 2001. Guidelines for the management of adults with community-acquired pneumonia. Diagnosis, assessment of severity, antimicrobial therapy, and prevention. Am J Respir Crit Care Med 163:1730–1754. doi: 10.1164/ajrccm.163.7.at1010. [DOI] [PubMed] [Google Scholar]
- 4.Welte T, Torres A, Nathwani D. 2012. Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 67:71–79. doi: 10.1136/thx.2009.129502. [DOI] [PubMed] [Google Scholar]
- 5.Blondeau JM, Vaughan D, Laskowski R, Borsos S; Canadian Antimicrobial Study Group. 2001. Susceptibility of Canadian isolates of Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae to oral antimicrobial agents. Int J Antimicrob Agents 17:457–464. doi: 10.1016/S0924-8579(01)00334-X. [DOI] [PubMed] [Google Scholar]
- 6.Blondeau JM. 1999. A review of the comparative in-vitro activities of 12 antimicrobial agents, with a focus on five new ‘respiratory quinolones’. J Antimicrob Chemother 43(Suppl B):1–11. doi: 10.1093/jac/43.suppl_2.1. [DOI] [PubMed] [Google Scholar]
- 7.Saravolatz L, Manzor O, Check C, Pawlak J, Belian B. 2001. Antimicrobial activity of moxifloxacin, gatifloxacin and six fluoroquinolones against Streptococcus pneumoniae. J Antimicrob Chemother 47:875–877. doi: 10.1093/jac/47.6.875. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Zhao X, Drlica K. 2002. Selection of Streptococcus pneumoniae mutants having reduced susceptibility to moxifloxacin and levofloxacin. Antimicrob Agents Chemother 46:522–524. doi: 10.1128/AAC.46.2.522-524.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lister PD, Sanders CC. 2001. Pharmacodynamics of moxifloxacin, levofloxacin and sparfloxacin against Streptococcus pneumoniae. J Antimicrob Chemother 47:811–818. doi: 10.1093/jac/47.6.811. [DOI] [PubMed] [Google Scholar]
- 10.Welte T, Petermann W, Schurmann D, Bauer TT, Reimnitz P; MOXIRAPID Study Group. 2005. Treatment with sequential intravenous or oral moxifloxacin was associated with faster clinical improvement than was standard therapy for hospitalized patients with community-acquired pneumonia who received initial parenteral therapy. Clin Infect Dis 41:1697–1705. doi: 10.1086/498149. [DOI] [PubMed] [Google Scholar]
- 11.Finch R, Schurmann D, Collins O, Kubin R, McGivern J, Bobbaers H, Izquierdo JL, Nikolaides P, Ogundare F, Raz R, Zuck P, Hoeffken G. 2002. Randomized controlled trial of sequential intravenous (i.v.) and oral moxifloxacin compared with sequential i.v. and oral co-amoxiclav with or without clarithromycin in patients with community-acquired pneumonia requiring initial parenteral treatment. Antimicrob Agents Chemother 46:1746–1754. doi: 10.1128/AAC.46.6.1746-1754.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anzueto A, Niederman MS, Pearle J, Restrepo MI, Heyder A, Choudhri SH. 2006. Community-acquired pneumonia recovery in the elderly (CAPRIE): efficacy and safety of moxifloxacin therapy versus that of levofloxacin therapy. Clin Infect Dis 42:73–81. doi: 10.1086/498520. [DOI] [PubMed] [Google Scholar]
- 13.Kees MG, Weber S, Kees F, Horbach T. 2011. Pharmacokinetics of moxifloxacin in plasma and tissue of morbidly obese patients. J Antimicrob Chemother 66:2330–2335. doi: 10.1093/jac/dkr282. [DOI] [PubMed] [Google Scholar]
- 14.Balfour JA, Lamb HM. 2000. Moxifloxacin: a review of its clinical potential in the management of community-acquired respiratory tract infections. Drugs 59:115–139. doi: 10.2165/00003495-200059010-00010. [DOI] [PubMed] [Google Scholar]
- 15.Lapi F, Wilchesky M, Kezouh A, Benisty JI, Ernst P, Suissa S. 2012. Fluoroquinolones and the risk of serious arrhythmia: a population-based study. Clin Infect Dis 55:1457–1465. doi: 10.1093/cid/cis664. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Cass JD, Bradley JA, Dahm CM, Sun Z, Kadyszewski E, Engwall MJ, Zhou J. 2005. QT prolongation and proarrhythmia by moxifloxacin: concordance of preclinical models in relation to clinical outcome. Br J Pharmacol 146:792–799. doi: 10.1038/sj.bjp.0706389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Florian JA, Tornoe CW, Brundage R, Parekh A, Garnett CE. 2011. Population pharmacokinetic and concentration-QTc models for moxifloxacin: pooled analysis of 20 thorough QT studies. J Clin Pharmacol 51:1152–1162. doi: 10.1177/0091270010381498. [DOI] [PubMed] [Google Scholar]
- 18.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10; quiz, 11–12. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 19.Lode H, Borner K, Koeppe P. 1998. Pharmacodynamics of fluoroquinolones. Clin Infect Dis 27:33–39. doi: 10.1086/514623. [DOI] [PubMed] [Google Scholar]
- 20.Craig WA. 2001. Does the dose matter? Clin Infect Dis 33(Suppl 3):S233–S237. doi: 10.1086/321854. [DOI] [PubMed] [Google Scholar]
- 21.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis 44:79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 22.Drusano GL, Johnson DE, Rosen M, Standiford HC. 1993. Pharmacodynamics of a fluoroquinolone antimicrobial agent in a neutropenic rat model of Pseudomonas sepsis. Antimicrob Agents Chemother 37:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peloquin CA, Cumbo TJ, Nix DE, Sands MF, Schentag JJ. 1989. Evaluation of intravenous ciprofloxacin in patients with nosocomial lower respiratory tract infections. Impact of plasma concentrations, organism, minimum inhibitory concentration, and clinical condition on bacterial eradication. Arch Intern Med 149:2269–2273. [PubMed] [Google Scholar]
- 24.Blaser J, Stone BB, Groner MC, Zinner SH. 1987. Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob Agents Chemother 31:1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Odenholt I, Cars O. 2006. Pharmacodynamics of moxifloxacin and levofloxacin against Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae and Escherichia coli: simulation of human plasma concentrations after intravenous dosage in an in vitro kinetic model. J Antimicrob Chemother 58:960–965. doi: 10.1093/jac/dkl356. [DOI] [PubMed] [Google Scholar]
- 26.Preston SL, Drusano GL, Berman AL, Fowler CL, Chow AT, Dornseif B, Reichl V, Natarajan J, Corrado M. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125–129. doi: 10.1001/jama.279.2.125. [DOI] [PubMed] [Google Scholar]
- 27.Forrest A, Nix DE, Ballow CH, Goss TF, Birmingham MC, Schentag JJ. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother 37:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambrose PG, Bhavnani SM, Owens RC Jr. 2003. Clinical pharmacodynamics of quinolones. Infect Dis Clin North Am 17:529–543. doi: 10.1016/S0891-5520(03)00061-8. [DOI] [PubMed] [Google Scholar]
- 29.Ambrose PG, Grasela DM, Grasela TH, Passarell J, Mayer HB, Pierce PF. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob Agents Chemother 45:2793–2797. doi: 10.1128/AAC.45.10.2793-2797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacy MK, Lu W, Xu X, Tessier PR, Nicolau DP, Quintiliani R, Nightingale CH. 1999. Pharmacodynamic comparisons of levofloxacin, ciprofloxacin, and ampicillin against Streptococcus pneumoniae in an in vitro model of infection. Antimicrob Agents Chemother 43:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holford NH. 1996. A size standard for pharmacokinetics. Clin Pharmacokinet 30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 32.Lindbom L, Ribbing J, Jonsson EN. 2004. Perl-speaks-NONMEM (PsN)–a Perl module for NONMEM related programming. Comput Methods Programs Biomed 75:85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Keizer RJ, Karlsson MO, Hooker A. 2013. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol 2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. 2011. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonsson EN, Karlsson MO. 1999. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64. [DOI] [PubMed] [Google Scholar]
- 36.Stass H, Kubitza D. 1999. Pharmacokinetics and elimination of moxifloxacin after oral and intravenous administration in man. J Antimicrob Chemother 43(Suppl B):83–90. [DOI] [PubMed] [Google Scholar]
- 37.Ulldemolins M, Roberts JA, Rello J, Paterson DL, Lipman J. 2011. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin Pharmacokinet 50:99–110. doi: 10.2165/11539220-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 38.European Committee on Antimicrobial Susceptibility Testing (EUCAST). 2014. MIC distributions. http://mic.eucast.org/Eucast2/ Accessed 18 September 2014.
- 39.Bruin JP, Ijzerman EP, den Boer JW, Mouton JW, Diederen BM. 2012. Wild-type MIC distribution and epidemiological cut-off values in clinical Legionella pneumophila serogroup 1 isolates. Diagn Microbiol Infect Dis 72:103–108. doi: 10.1016/j.diagmicrobio.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Marques T, Piedade J. 1997. Susceptibility testing by E-test and agar dilution of 30 strains of Legionella spp. isolated in Portugal. Clin Microbiol Infect 3:365–368. doi: 10.1111/j.1469-0691.1997.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 41.Kees MG, Schaeftlein A, Haeberle HA, Kees F, Kloft C, Heininger A. 2013. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J Antimicrob Chemother 68:1331–1337. doi: 10.1093/jac/dkt040. [DOI] [PubMed] [Google Scholar]
- 42.Kontou P, Manika K, Chatzika K, Papaioannou M, Sionidou M, Pitsiou G, Kioumis I. 4 July 2013, posting date Pharmacokinetics of moxifloxacin and high-dose levofloxacin in severe lower respiratory tract infections. Int J Antimicrob Agents doi: 10.1016/j.ijantimicag.2013.04.028. [DOI] [PubMed] [Google Scholar]
- 43.Varghese JM, Roberts JA, Lipman J. 2010. Pharmacokinetics and pharmacodynamics in critically ill patients. Curr Opin Anaesthesiol 23:472–478. doi: 10.1097/ACO.0b013e328339ef0a. [DOI] [PubMed] [Google Scholar]
- 44.Udy AA, Roberts JA, De Waele JJ, Paterson DL, Lipman J. 2012. What's behind the failure of emerging antibiotics in the critically ill? Understanding the impact of altered pharmacokinetics and augmented renal clearance. Int J Antimicrob Agents 39:455–457. doi: 10.1016/j.ijantimicag.2012.02.010. [DOI] [PubMed] [Google Scholar]