

Abstract
The cytochrome bc1 complex (cyt bc1) is the third component of the mitochondrial electron transport chain and is the target of several potent antimalarial compounds, including the naphthoquinone atovaquone (ATV) and the 4(1H)-quinolone ELQ-300. Mechanistically, cyt bc1 facilitates the transfer of electrons from ubiquinol to cytochrome c and contains both oxidative (Qo) and reductive (Qi) catalytic sites that are amenable to small-molecule inhibition. Although many antimalarial compounds, including ATV, effectively target the Qo site, it has been challenging to design selective Qi site inhibitors with the ability to circumvent clinical ATV resistance, and little is known about how chemical structure contributes to site selectivity within cyt bc1. Here, we used the proposed Qi site inhibitor ELQ-300 to generate a drug-resistant Plasmodium falciparum clone containing an I22L mutation at the Qi region of cyt b. Using this D1 clone and the Y268S Qo mutant strain, P. falciparum Tm90-C2B, we created a structure-activity map of Qi versus Qo site selectivity for a series of endochin-like 4(1H)-quinolones (ELQs). We found that Qi site inhibition was associated with compounds containing 6-position halogens or aryl 3-position side chains, while Qo site inhibition was favored by 5,7-dihalogen groups or 7-position substituents. In addition to identifying ELQ-300 as a preferential Qi site inhibitor, our data suggest that the 4(1H)-quinolone scaffold is compatible with binding to either site of cyt bc1 and that minor chemical changes can influence Qo or Qi site inhibition by the ELQs.
INTRODUCTION
Malaria is a devastating parasitic disease that affects >200 million people every year and is a leading cause of mortality in the developing world (1). Malaria is caused by Plasmodium parasites, which are transmitted by the bites of infected Anopheles mosquitoes and progress through a series of biologically distinct stages within the hepatocytes and red blood cells of the human host. The most lethal malarial parasite, Plasmodium falciparum, has developed resistance to many frontline antimalarials, including the quinolines quinine, chloroquine, and mefloquine, as well as the antifolates pyrimethamine and sulfadoxine. In many regions of the world, the treatment of multidrug-resistant malaria relies on the use of artemisinin-based combination therapies (ACTs), such as artesunate-amodiaquine and dihydroartemisinin-piperaquine. Unfortunately, artemisinin resistance has emerged in regions of Southeast Asia (2), which threatens to derail the ACT treatment strategy and further restrict therapeutic options for patients in malarious regions. As a result, there is a pressing need for new antimalarial compounds, particularly those that effectively inhibit drug-resistant parasites and function throughout multiple stages of the parasite life cycle to provide combined treatment, prophylaxis, and transmission-blocking activity against malaria (3).
Complex III of the mitochondrial electron transport chain, also known as the cytochrome bc1 complex (cyt bc1), is a validated target for multistage antimalarial therapy. Structurally, there are two known binding sites for antimalarial compounds within cyt bc1: an oxidative (Qo) site and a reductive (Qi) site. Inhibition at either site is sufficient to block the catalytic cycle of cyt bc1 (Q cycle), which ultimately leads to pyrimidine starvation and cell death in P. falciparum (4, 5). To date, pyridones (6, 7), naphthoquinones (8, 9), acridones (10), quinolones (11–13), and benzene sulfonamides (14) have been identified as potent inhibitors of Plasmodium cyt bc1 (15). This includes atovaquone (ATV), which targets the Qo site of the P. falciparum cytochrome bc1 complex (16).
Although atovaquone is a potent and well-tolerated antimalarial drug, its clinical utility is limited by the rapid emergence of resistant parasites when used as monotherapy (17). For this reason, atovaquone is coformulated with proguanil (Malarone) to counter the emergence of resistance and improve clinical efficacy. In the P. falciparum clinical isolate Tm90-C2B, a point mutation at the Qo site of cytochrome b (i.e., Y268S) results in a 3,000-fold loss of ATV sensitivity. As a result, this parasite line has been used as a screening tool for the development of new cyt bc1 inhibitors able to circumvent ATV resistance mutations. Our group has developed an extensive library of compounds based on the general structure of the cyt bc1 inhibitor endochin (11, 12, 18, 19). As was previously reported, several of these endochin-like quinolones (ELQs), including the preclinical candidate ELQ-300 (12), demonstrate remarkable potency against Tm90-C2B parasites, which may suggest preferential Qi site inhibition.
To date, very few Qi site inhibitors of cyt bc1 have been identified, and it has been especially difficult to isolate Qi-selective compounds with activity against P. falciparum. Sequencing studies have shown that the P. falciparum Qi site is structurally distinct from that of other species (20). As a result, even antimycin A, the prototype picomolar inhibitor of the Qi site in bacteria, yeast, and mammalian cells (21), demonstrates decreased activity against P. falciparum, with an in vitro 50% inhibitory concentration (IC50) in the nanomolar range (22). The uniqueness of the P. falciparum Qi site may confer several therapeutic advantages. In addition to retaining potency against ATV-resistant Qo site mutant parasites, Qi site inhibitors may be uniquely selective for parasite cyt bc1 and would thus constitute a novel class of antimalarial compounds.
A major obstacle to the development of Qi site inhibitors for Plasmodium spp. is the lack of effective screening tools to identify Qi-selective compounds. Although studies in yeast have suggested that the quinolone compounds ELQ-271 (23) and HDQ [1-hydroxyl-2-dodecyl-4(1H)-quinolone] (24) function as Qi site inhibitors, no P. falciparum Qi site mutants have been available for verification. Furthermore, with such a small group of effective Qi-targeting antimalarials, it has not yet been possible to make any consistent associations between chemical structure and Qi site preference. In this paper, we introduce a new P. falciparum clone containing a mutation at the cyt bc1 Qi site, and we used this mutant in combination with Tm90-C2B to conduct a detailed assessment of Qi versus Qo targeting within the ELQ library. We identify several structural features that are associated with preferential Qi site activity and provide insight into the ongoing development of new multistage antimalarial inhibitors of cyt bc1.
MATERIALS AND METHODS
Chemicals and chemistry methods.
Unless indicated otherwise, all chemicals and reagents used in this study were from Sigma-Aldrich (St. Louis, MO). The ELQs described in this paper were described previously (12, 18, 19) or were synthesized by methods developed in our laboratory and published elsewhere (11, 25).
In vitro selection of ELQ-300-resistant P. falciparum clones.
A clonal population of P. falciparum Dd2 parasites was maintained at 5% final hematocrit in an atmosphere of 90% N2, 5% CO2, and 5% O2 at 37°C in complete culturing medium (10.4 g liter−1 RPMI 1640 with 2.1 mM glutamine, 5.94 g liter−1 HEPES, 5 g liter−1 AlbuMAX II, 50 mg liter−1 hypoxanthine, 2.1 g liter−1 sodium bicarbonate, and 43 mg liter−1 gentamicin). On day 0 of selection, an initial inoculum of 109 parasites was cultured in the presence of drug at 25 nM. On days 4, 5, and 7, the drug concentration was increased to 32, 40, and 70 nM, respectively, until the cultures were cleared of parasites. The medium was changed daily until the parasites were microscopically undetectable (as assessed by an examination of Giemsa-stained slides) and subsequently every 2 days for the remainder of the experiment. Upon recrudescence, the population of parasites was cloned by limiting dilution (0.8 infected red blood cell [RBC]/well) at 1.8% hematocrit in a 96-well flat-bottom tissue culture plate in the presence of 70 nM ELQ-300. On day 21 of cloning, 5 μl of parasite culture from each well was mixed with a solution containing 0.1 μl/ml SYBR green I and 0.1 μM MitoTracker Deep Red (Life Technologies) and incubated for 20 min prior to an analysis of parasitemia on an Accuri C6 flow cytometer (26).
Sequencing of cytochrome b.
DNA was isolated from the parasites at the mid- to late trophozoite stage. The parasites were saponin lysed with 0.02% saponin in 1× phosphate-buffered saline (PBS). The pellets were resuspended to a volume of 200 μl with PBS-20 mM EDTA, and DNA was isolated using the QIAamp DNA blood minikit (Qiagen), according to the blood protocol. PCR was performed using the Herculase II fusion enzyme (Agilent Technologies). Primer 1 was 5′-CCAGACGCTTTAAATGGATG-3′, and primer 2 was 5′-GTTTGCTTGGGAGCTGTAATC-3′. The PCR products were purified using SV gel and the PCR cleanup kit (Promega). DNA sequencing was performed by Genewiz.
P. falciparum culture.
Laboratory strains of P. falciparum were cultured in human erythrocytes by standard methods under a low-oxygen atmosphere (5% O2, 5% CO2, and 90% N2) in an environmental chamber. The parasites were maintained in fresh human erythrocytes suspended at 2% hematocrit in complete medium at 37°C. The stock cultures were subpassaged every 3 to 4 days by transferring infected RBCs to a flask containing complete medium and uninfected RBCs.
SYBR green I assay.
In vitro antimalarial activity was assessed using a published SYBR green I fluorescence-based method (27). The drugs were added to 96-well plates using 2-fold serial dilutions in HEPES-modified RPMI (described above). Asynchronous P. falciparum parasites were diluted in uninfected RBCs and added to the wells to give a final volume of 200 μl at 2% hematocrit and 0.2% parasitemia. The plates were incubated for 72 h at 37°C. The parasites were then lysed using SYBR green I lysis buffer containing 0.2 μl/ml SYBR green I in 20 mM Tris (pH 7.5), 5 mM EDTA, 0.008% (wt/vol) saponin, and 0.08% (vol/vol) Triton X-100. The plates were incubated in the dark for 30 to 60 min, and the SYBR green I signal was then quantified using a SpectraMax Gemini EM plate reader with excitation and emission bands centered at 497 and 520 nm, respectively. The 50% inhibitory concentrations (IC50) were determined via nonlinear analysis using the GraphPad Prism software. All final IC50 values represent the averages from at least three independent experiments, with each compound run in triplicate.
Molecular modeling.
The 1KYO (Qo) and 1EZV (Qi) crystal structures of Saccharomyces cerevisiae cyt bc1 were obtained from the Protein Data Bank and modified using the Protein Data Bank Viewer, as follows: Qi site, F225L and K228L, and Qo site, C133V, C134L, V135P, Y136W, H141Y, and L275F. The adjusted Qo residues were selected because of their importance in sensitizing S. cerevisiae to known antimalarial inhibitors, including ATV and the quinolone compound RCQ06 (28). The Qi site modifications were made to account for the reduced sensitivity to the Qi site inhibitor antimycin A that is observed in P. falciparum (20). The ELQ models were created using ChemDraw 3D, and docking was performed using the CLC Drug Discovery Workbench software. The Qo and Qi binding pockets were centered on the crystal-bound substrates stigmatellin (Qo) and coenzyme Q6 (Qi). One thousand docking iterations were run per compound at each site.
RESULTS
Development and characterization of the ELQ-300-resistant P. falciparum D1 clone.
Drug-resistant P. falciparum strains provide a wealth of information about the functions of antimalarial compounds and the adaptive mechanisms that allow parasites to avoid their effects. In vitro, drug resistance is generally developed through one of two standard methods, (i) high-concentration drug pressure or (ii) incremental drug pressure, which begins at low concentrations and increases in response to parasite growth over time. Biologically, the first method is used as an indication of resistance propensity, while the second is primarily a tool to generate resistant parasites for further study.
To generate the D1 clone, we cultured P. falciparum Dd2 parasites in the presence of a low concentration of ELQ-300 (25 nM, approximately 4× the IC50). We monitored parasitemia daily and gradually increased the ELQ-300 concentration until parasite growth was fully inhibited and parasitemia dropped below the limit of detection. After 2 weeks at this final concentration of 70 nM, parasites were detected, and three clones were isolated by a limiting dilution (29). DNA sequencing of the cytochrome b gene revealed an I22L mutation in all clones, which mapped to the P. falciparum Qi site. The D1 clone was used for subsequent studies and was found to be 25-fold less sensitive to ELQ-300 than was the parental Dd2 strain. As would be expected for a Qi site mutant, no loss of potency was observed for the Qo site inhibitors ATV and myxothiazol (22) or for the non-cyt bc1-targeting compound chloroquine (CQ) (Table 1).
TABLE 1.
Sensitivities of the P. falciparum Dd2, Tm90-C2B, and D1 strains to ATV, ELQ-300, CQ, and the canonical Qi and Qo site inhibitors antimycin A and myxothiazola
| Compound | IC50 value (nM) or ratio forb: |
||||
|---|---|---|---|---|---|
| Dd2 | Tm90-C2B | C2B/Dd2 | D1 | D1/Dd2 | |
| ATV | 0.4 | >2,500 | >7,800 | 0.7 | 1.7 |
| ELQ-300 | 6.6 | 4.6 | 0.7 | 160 | 24 |
| CQ | 71 | 91 | 1.3 | 110 | 1.5 |
| Antimycin A | 72 | 39 | 0.5 | 35 | 0.5 |
| Myxothiazol | 1.7 | 320 | 190 | 3.5 | 2.0 |
ATV, atovaquone; CQ, chloroquine.
IC50 values were averaged from ≥3 independent experiments run in triplicate. The standard deviations were <10%.
In vitro Qo versus Qi preference of ELQs.
To explore how chemical structure contributed to Qi versus Qo site preference within the ELQ series, we conducted a large-scale assessment of our ELQ library using the P. falciparum D1 and Tm90-C2B drug-resistant clones. We used the fluorescence-based SYBR green I assay (27) to determine the IC50 for each compound and created cross-resistance indices by normalizing D1 and Tm90-C2B activities against that of the Dd2 parental strain. Because the D1 and Tm90-C2B clones contain Qi and Qo site point mutations, respectively, we used D1 cross-resistance as an indicator of preferential Qi site activity and Tm90-C2B cross-resistance as an indicator of preferential Qo site activity.
ELQ 6 and 7 positions.
We assessed the roles of the 6-position and 7-position groups (Fig. 1) in Qo/Qi preference by first evaluating a set of ELQs containing the 3-position alkyl chain of endochin (Table 2). At the 7 position, all chemical groups were associated with an increase in Tm90-C2B cross-resistance relative to that of the undecorated compound ELQ-127. Electron-withdrawing substituents had the most dramatic effect, especially for the 7-Cl compound ELQ-109, which was >25 times less potent against the Tm90-C2B strain. In contrast, the effect of 6-position groups was dependent on electrostatic character. Electron-donating groups (e.g., SCH3, OH, and OCH3) dramatically reduced potency and increased cross-resistance in Tm90-C2B parasites, while electron-withdrawing groups (e.g., halogens and NO2) were universally associated with D1 cross-resistance and Qi targeting. Overall, the most Qi-selective compound was ELQ-130, which contained a 6-chloro substituent and was undecorated at the 7 position.
FIG 1.
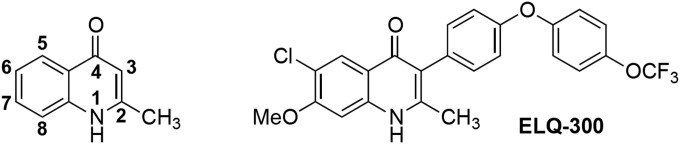
Left, general structure of 4(1H)-quinolones with numbered chemical positions. Right, structure of ELQ-300, the compound used to generate the Qi mutant D1 clone.
TABLE 2.
Comparative activities of 3-alkyl ELQs with various 6- and 7-position substituents
| ELQ | Substituent at position: |
IC50 value (nM) or ratio fora: |
||||
|---|---|---|---|---|---|---|
| 3 | 6 | 7 | Dd2 | C2B/Dd2 | D1/Dd2 | |
| 131 | C7H15 | F | H | 47 | 1.9 | 2.8 |
| 130 | C7H15 | Cl | H | 29 | 0.9 | 9.7 |
| 162 | C7H15 | NO2 | H | 260 | 1.4 | 4.6 |
| 150 | C7H15 | OCH3 | H | 440 | 3.4 | 0.9 |
| 133 | C7H15 | SCH3 | H | 280 | 5.9 | 1.5 |
| 127 | C7H15 | H | H | 39 | 1.7 | 2.4 |
| 120 | C7H15 | H | F | 9.6 | 19 | 1.0 |
| 109 | C7H15 | H | Cl | 7.1 | 26 | 1.4 |
| 118 | C7H15 | H | CN | 38 | 14 | 0.8 |
| 110 | C7H15 | H | NO2 | 27 | 15 | 1.7 |
| 100 | C7H15 | H | OCH3 | 5.2 | 6.1 | 0.9 |
IC50 values were averaged from ≥3 independent experiments run in triplicate. The standard deviations were <10%.
In a second set of compounds consisting of 3-diarylether ELQs, 6-position halogens had an even more dramatic effect on D1 cross-resistance and Qi site preference (Table 3). While ELQ-130 was approximately 10-fold less active against the D1 clone, the corresponding 6-chloro 3-diarylether ELQ-296 was 15-fold less active, and a similar loss of activity was observed for the 6-fluoro comparators ELQ-131 and ELQ-314. Intriguingly, the combination of 6-halogen and 7-methoxy groups produced an unanticipated increase in Qi site preference among the 3-diarylether ELQs (Table 3). Compared to the 6-halogen compounds ELQ-314, ELQ-296, and ELQ-339, the corresponding 7-methoxy analogs (ELQ-316, ELQ-300, and ELQ-340, respectively) demonstrated both increased potency and more pronounced D1 cross-resistance, which correlated with halogen size.
TABLE 3.
Comparative activities of 3-diarylether ELQs containing 6-position halogens and 7-position OCH3 groups
| ELQ | Substituent at position: |
IC50 value (nM) or ratio fora: |
||||
|---|---|---|---|---|---|---|
| 3 | 6 | 7 | Dd2 | C2B/Dd2 | D1/Dd2 | |
| 271 | DAEb | H | H | 12 | 2.8 | 0.2 |
| 298 | DAE | H | OCH3 | 5.5 | 0.7 | 0.8 |
| 314 | DAE | F | H | 33 | 1.3 | 3.4 |
| 316 | DAE | F | OCH3 | 3.1 | 1.1 | 3.6 |
| 296 | DAE | Cl | H | 29 | 0.9 | 16 |
| 300 | DAE | Cl | OCH3 | 6.6 | 0.7 | 24 |
| 339 | DAE | Br | H | 170 | 1.1 | 14 |
| 340 | DAE | Br | OCH3 | 25 | 0.4 | 65 |
IC50 values were averaged from ≥3 independent experiments run in triplicate. The standard deviations were <10%.
DAE, para-OCF3 diaryl ether side chain, as present in ELQ-300.
ELQ 3-position side chains.
The difference in D1 cross-resistance observed between the 3-alkyl and the 3-diarylether ELQs suggested that the 3-position side chain might play an integral role in Qo versus Qi targeting for the ELQs. To assess this relationship, we next analyzed a series of 6-chloro ELQs containing various 3-position side chains (Table 4). Among these compounds, the most dramatic D1 cross-resistance was observed for the 3-biphenyl compound ELQ-269, while the lowest degree of cross-resistance was associated with ELQ-200, which contained a truncated 3-alkyl side chain. Generally, D1 cross-resistance correlated with side-chain length and rigidity but was not noticeably altered by the presence of additional charged groups or heterocycles, as was demonstrated by the comparison between ELQ-296 and ELQ-317.
TABLE 4.
Comparative activities of 6-chloro ELQs with various 3-position side chains
| ELQ | Substituent at positiona: |
IC50 value (nM) or ratio forb: |
||||
|---|---|---|---|---|---|---|
| 3 | 6 | 7 | Dd2 | C2B/Dd2 | D1/Dd2 | |
| 220 | Benzyl | Cl | H | 510 | 4.1 | 2.7 |
| 200 | C4H9 | Cl | H | 121 | 1.6 | 5.1 |
| 130 | C7H15 | Cl | H | 29 | 0.9 | 9.7 |
| 296 | DAE | Cl | H | 29 | 0.9 | 16 |
| 317 | Het- DAE | Cl | H | 58 | 0.5 | 15 |
| 269 | Biphenyl | Cl | H | 42 | 0.3 | 110 |
DAE, para-OCF3 diaryl ether side chain, as present in ELQ-300. Het-DAE, para-OCF3 diaryl ether analog, containing a pyridyl inner ring.
IC50 values were averaged from ≥3 independent experiments run in triplicate. The standard deviations were <10%.
ELQ 5 and 7 positions.
While 6-position halogens and rigid 3-position side chains were associated with the highest degree of Qi site selectivity, Qo site inhibition was strongly favored in ELQs containing 5,7-dihalogen groups (Table 5). The 5,7-difluoro compound ELQ-121 was >600 times less potent against Tm90-C2B parasites and was the most Qo-selective compound within our ELQ library. Structurally, the 5-position fluorine contributed most strongly to this Qo site preference; Tm90-C2B parasites were more cross-resistant to the 5-fluoro analog ELQ-136 than to the 7-fluoro analog ELQ-120. In contrast to the effect of isolated 7-position halogens, cross-resistance did not increase with 5,7-dihalogen size, and the 5,7-dichloro compound ELQ-124 was less Qo selective than was the difluoro compound ELQ-121.
TABLE 5.
Comparative activities of ELQs containing 5- or 7-position halogens, with various Qi-directing substituents
| ELQ | Substituent at positiona: |
IC50 value (nM) or ratio forb: |
|||||
|---|---|---|---|---|---|---|---|
| 3 | 5 | 6 | 7 | Dd2 | C2B/Dd2 | D1/Dd2 | |
| 136 | C7H15 | F | H | H | 3.1 | 76 | 1.3 |
| 121 | C7H15 | F | H | F | 0.5 | 610 | 0.6 |
| 124 | C7H15 | Cl | H | Cl | 24 | 15 | 0.9 |
| 141 | Benzyl | F | H | F | 38 | 120 | 1.4 |
| 400 | DAE | F | H | F | 1.5 | 23 | 1.1 |
| 140 | C7H15 | F | F | F | 2.8 | 34 | 1.1 |
| 404 | DAE | F | F | F | 4.9 | 5.8 | 1.4 |
| 428 | DAE | F | Cl | F | 11 | 3.4 | 3.3 |
| 429 | DAE | F | OCH3 | F | 46 | 26 | 2.0 |
DAE, para-OCF3 diaryl ether side chain, as present in ELQ-300.
IC50 values were averaged from ≥3 independent experiments run in triplicate. The standard deviations were <10%.
Intriguingly, the Qo-directing effects of the 5,7-difluoro configuration were effectively modulated by the addition of Qi-directing chemical substituents (Table 5). The combination of a 3-position diarylether chain with 5,7-difluoro groups produced ELQ-400, which was associated with diminished Tm90-C2B cross-resistance relative to that of the 3-alkyl compound ELQ-121. Tm90-C2B cross-resistance was further reduced by the incorporation of halogens at the 6 position, and low levels of both D1 and Tm90-C2B cross-resistance were observed for the 5,7-difluoro 6-chloro analog ELQ-428. Conversely, the addition of a 6-methoxy group between the flanking fluorine atoms increased Tm90-C2B cross-resistance relative to that of ELQ-400, which was consistent with the predicted effect of an electron-donating 6-position substituent.
DISCUSSION
Using a set of mutant P. falciparum strains, including the novel D1 clone, we have identified several key ELQ features associated with preferential Qo or Qi site inhibition of Plasmodium cyt bc1. The primary contributors to Qi site preference included (i) electron-withdrawing 6-position substituents (including halogens), (ii) combined 6-halogen 7-methoxy groups, and (iii) aryl 3-position side chains. Alternately, 7-position groups were broadly associated with Qo site targeting, especially in combination with 5-fluorine moieties, as was observed for the 5,7-difluoro ELQs.
The dramatic impact of subtle chemical changes on site specificity suggests that ELQ substituents may divergently interact with key residues of cyt bc1. Although exact binding interactions cannot be identified without a crystal structure of the Plasmodium enzyme, we were able to explain several of our observed structural effects using homology modeling in S. cerevisiae. In Qo site models, we found that the 1-NH group of the quinolone core made a predicted hydrogen bond with the conserved E272 residue, which is important for binding of the natural cyt bc1 substrate ubiquinol and the potent Qo site inhibitor stigmatellin (30). In this conformation, 7-position substituents were well tolerated and frequently formed additional predicted bonds with backbone atoms of the nearby M139 and G143 residues (Fig. 2A). Interestingly, this predicted binding alignment was similar to that recently reported for atovaquone (16), which may explain the reduced sensitivity of the ATV-resistant TM90-C2B clinical isolate to several members of the ELQ library, including 5,7-difluoro compounds.
FIG 2.
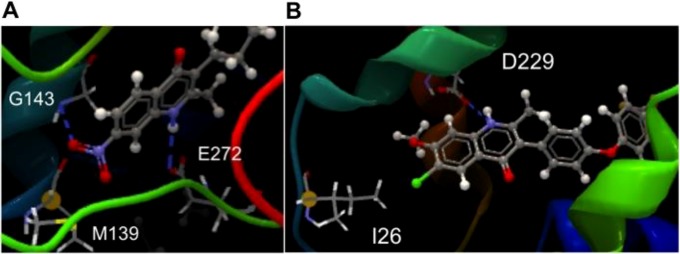
S. cerevisiae homology models depicting possible interactions between ELQs and cyt bc1. Shown are the predicted interactions between the 1-NH group of ELQ-110 and the E272 residue at the Qo site (A) and hydrogen bonding of the 1-NH group of ELQ-300 to the Qi site D229 residue (B), showing the 6-position chlorine in close proximity to the isoleucine residue mutated in the P. falciparum D1 clone. The dashed blue lines represent predicted hydrogen bonds.
With respect to the Qi site, we identified a potential hydrogen bond interaction between the 1-NH group of the quinolone core and the nearby D229 residue of the Qi binding pocket (Fig. 2B). In this conformation, the 6-position chlorine atom of ELQ-300 was in close proximity to I26, which is homologous to the I22 residue mutated in ELQ-300-resistant D1 parasites. While the involvement of the conserved quinolone core suggests that all ELQs are likely capable of Qi site interaction, the specific bond with the 1-NH group also provides a potential explanation for the increased Qi site selectivity of ELQs containing 6-position electron-withdrawing groups. Because the quinolone ring is aromatic in nature, electron-withdrawing groups located para to the 1-position nitrogen (e.g., at the 6 position) should draw electron density away from the 1-NH group, making the associated hydrogen more acidic and available for hydrogen bond formation. Alternately, 6-position electron-donating groups would be expected to decrease the potential for hydrogen bonding, which is consistent with our finding that such compounds demonstrate both decreased potency and reduced Qi site preference.
The predicted binding conformation of ELQ-300 at the Qi site also suggests that observed D1 cross-resistance is likely a direct reflection of the steric interference between 6-position substituents and the interfacing I22L residue (Fig. 2B). In addition to justifying the correlation between 6-halogen size and D1 cross-resistance, this steric hypothesis provides a compelling explanation for the lack of cross-resistance observed for the known Qi site inhibitor ELQ-271 (23), which is undecorated at the 6 position. While a more general Qi site P. falciparum mutant may be desirable from a screening perspective, our efforts to generate resistance under ELQ-271 pressure have thus far been unsuccessful. Furthermore, constant exposure to ELQ-300 at 10× its IC50 fails to select for resistant parasites (12), suggesting that a significant fitness cost may be associated with mutations that confer high-level resistance to ELQs or that there may simply be limited genetic space for mutations within the cyt b Qi site.
In conclusion, our studies have shown that the P. falciparum D1 clone has considerable potential for use in comparative assessments and large-scale structure-activity analyses. Intriguingly, this model demonstrated that the 4(1H)-quinolone scaffold was compatible with inhibition at either the Qo or Qi site of cyt bc1. Although it is unknown whether ELQs can simultaneously inhibit both catalytic sites, a dual-site interaction may explain why even the most Qo-selective compounds in the ELQ library still function as nanomolar inhibitors of Tm90-C2B parasites, and it might also explain the remarkable in vivo efficacies of the ELQs relative to those of comparison compounds, such as ATV (12, 31). In theory, dual-site inhibitors of the parasite cyt bc1 complex may be highly desirable, because high-level drug resistance would require the simultaneous appearance of two independent mutations within a single protein target. Ultimately, it will be necessary to parse the effects of isolated Qi site inhibition from those of a dual-site interaction so that cyt bc1 inhibitors can be optimized for clinical use. Until then, the structural insights obtained from the ELQ library and the D1 clone have the potential to guide drug development efforts for several chemical subseries and advance our understanding of how these compounds interact with P. falciparum cyt bc1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Medicines for Malaria Venture (Geneva, Switzerland) for their funding and project support. This project was also supported by the National Institutes of Health, grants AI079182 and AI100569 (to M.K.R) and AI028398 (to A.B.V.), the U.S. Department of Veterans Affairs (to M.K.R.), and the Stanley Medical Research Institute (to M.K.R.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04149-14.
REFERENCES
- 1.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. 2014. Malaria. Lancet 383:723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 2.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TN. 2013. Designing the next generation of medicines for malaria control and eradication. Malar J 12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. 2007. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 5.Vaidya AB. 2012. Naphthoquinones: atovaquone, and other antimalarials targeting mitochondrial functions, p 127–139. In Staines HM, Krishna S. (ed), Treatment and prevention of malaria: antimalarial drug chemistry, action and use. Springer, Basel, Switzerland. [Google Scholar]
- 6.Yeates CL, Batchelor JF, Capon EC, Cheesman NJ, Fry M, Hudson AT, Pudney M, Trimming H, Woolven J, Bueno JM, Chicharro J, Fernandez E, Fiandor JM, Gargallo-Viola D, Gomez de las Heras F, Herreros E, Leon ML. 2008. Synthesis and structure-activity relationships of 4-pyridones as potential antimalarials. J Med Chem 51:2845–2852. doi: 10.1021/jm0705760. [DOI] [PubMed] [Google Scholar]
- 7.Bueno JM, Herreros E, Angulo-Barturen I, Ferrer S, Fiandor JM, Gamo FJ, Gargallo-Viola D, Derimanov G. 2012. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med Chem 4:2311–2323. doi: 10.4155/fmc.12.177. [DOI] [PubMed] [Google Scholar]
- 8.Hudson AT, Randall AW, Fry M, Ginger CD, Hill B, Latter VS, McHardy N, Williams RB. 1985. Novel anti-malarial hydroxynaphthoquinones with potent broad spectrum anti-protozoal activity. Parasitology 90(Pt 1):45–55. [DOI] [PubMed] [Google Scholar]
- 9.Nixon GL, Moss DM, Shone AE, Lalloo DG, Fisher N, O'Neill PM, Ward SA, Biagini GA. 2013. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother 68:977–985. doi: 10.1093/jac/dks504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Bagby GC, Rathbun RK, Levin JI, Hinrichs D, Riscoe MK. 2006. Evaluation and lead optimization of anti-malarial acridones. Exp Parasitol 114:47–56. doi: 10.1016/j.exppara.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Nilsen A, Miley GP, Forquer IP, Mather MW, Katneni K, Li Y, Pou S, Pershing AM, Stickles AM, Ryan E, Kelly JX, Doggett JS, White KL, Hinrichs DJ, Winter RW, Charman SA, Zakharov LN, Bathurst I, Burrows JN, Vaidya AB, Riscoe MK. 2014. Discovery, synthesis, and optimization of antimalarial 4(1H)-quinolone-3-diarylethers. J Med Chem 57:3818–3834. doi: 10.1021/jm500147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, Morrisey JM, Steuten J, Mutka T, Li Y, Wirjanata G, Ryan E, Duffy S, Kelly JX, Sebayang BF, Zeeman AM, Noviyanti R, Sinden RE, Kocken CH, Price RN, Avery VM, Angulo-Barturen I, Jimenez-Diaz MB, Ferrer S, Herreros E, Sanz LM, Gamo FJ, Bathurst I, Burrows JN, Siegl P, Guy RK, Winter RW, Vaidya AB, Charman SA, Kyle DE, Manetsch R, Riscoe MK. 2013. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 5:177ra137. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biagini GA, Fisher N, Shone AE, Mubaraki MA, Srivastava A, Hill A, Antoine T, Warman AJ, Davies J, Pidathala C, Amewu RK, Leung SC, Sharma R, Gibbons P, Hong DW, Pacorel B, Lawrenson AS, Charoensutthivarakul S, Taylor L, Berger O, Mbekeani A, Stocks PA, Nixon GL, Chadwick J, Hemingway J, Delves MJ, Sinden RE, Zeeman AM, Kocken CH, Berry NG, O'Neill PM, Ward SA. 2012. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc Natl Acad Sci U S A 109:8298–8303. doi: 10.1073/pnas.1205651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukens AK, Heidebrecht RW Jr, Mulrooney C, Beaudoin JA, Comer E, Duvall JR, Fitzgerald ME, Masi D, Galinsky K, Scherer CA, Palmer M, Munoz B, Foley M, Schreiber SL, Wiegand RC, Wirth DF. Diversity-oriented synthesis probe targets Plasmodium falciparum cytochrome b ubiquinone reduction site and synergizes with oxidation site inhibitors. J Infect Dis, in press. doi: 10.1093/infdis/jiu565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stocks PA, Barton V, Antoine T, Biagini GA, Ward SA, O'Neill PM. 2014. Novel inhibitors of the Plasmodium falciparum electron transport chain. Parasitology 141:50–65. doi: 10.1017/S0031182013001571. [DOI] [PubMed] [Google Scholar]
- 16.Birth D, Kao WC, Hunte C. 2014. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 5:4029. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 17.Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. 1996. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54:62–66. [DOI] [PubMed] [Google Scholar]
- 18.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. 2008. Antimalarial quinolones: synthesis, potency, and mechanistic studies. Exp Parasitol 118:487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winter R, Kelly JX, Smilkstein MJ, Hinrichs D, Koop DR, Riscoe MK. 2011. Optimization of endochin-like quinolones for antimalarial activity. Exp Parasitol 127:545–551. doi: 10.1016/j.exppara.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaidya AB, Lashgari MS, Pologe LG, Morrisey J. 1993. Structural features of Plasmodium cytochrome b that may underlie susceptibility to 8-aminoquinolines and hydroxynaphthoquinones. Mol Biochem Parasitol 58:33–42. doi: 10.1016/0166-6851(93)90088-F. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Zhu XL, Yang WC, Yang GF. 2014. Comparative kinetics of Qi site inhibitors of cytochrome bc1 complex: picomolar antimycin and micromolar cyazofamid. Chem Biol Drug Des 83:71–80. doi: 10.1111/cbdd.12199. [DOI] [PubMed] [Google Scholar]
- 22.Smilkstein MJ, Forquer I, Kanazawa A, Kelly JX, Winter RW, Hinrichs DJ, Kramer DM, Riscoe MK. 2008. A drug-selected Plasmodium falciparum lacking the need for conventional electron transport. Mol Biochem Parasitol 159:64–68. doi: 10.1016/j.molbiopara.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doggett JS, Nilsen A, Forquer I, Wegmann KW, Jones-Brando L, Yolken RH, Bordon C, Charman SA, Katneni K, Schultz T, Burrows JN, Hinrichs DJ, Meunier B, Carruthers VB, Riscoe MK. 2012. Endochin-like quinolones are highly efficacious against acute latent experimental toxoplasmosis. Proc Natl Acad Sci U S A 109:15936–15941. doi: 10.1073/pnas.1208069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallieres C, Fisher N, Antoine T, Al-Helal M, Stocks P, Berry NG, Lawrenson AS, Ward SA, O'Neill PM, Biagini GA, Meunier B. 2012. HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob Agents Chemother 56:3739–3747. doi: 10.1128/AAC.00486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riscoe MK, Kelly JX, Winter RW, Hinrichs DJ, Smilkstein MJ, Nilsen A, Burrows JN, Kyle DE, Manetsch M, Cross RM, Monastyrskyi A, Flanigan DL. December 2013. Compounds having antiparasitic or anti-infectious activity. (Oregon Health & Science University, USA; Medicines for Malaria Venture, Switzerland; University of South Florida, USA). US patent US8598354 B2. [Google Scholar]
- 26.Ekland EH, Schneider J, Fidock DA. 2011. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J 25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallieres C, Fisher N, Meunier B. 2013. Reconstructing the Qo site of Plasmodium falciparum bc1 complex in the yeast enzyme. PLoS One 8:e71726. doi: 10.1371/journal.pone.0071726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodyer ID, Taraschi TF. 1997. Plasmodium falciparum: a simple, rapid method for detecting parasite clones in microtiter plates. Exp Parasitol 86:158–160. doi: 10.1006/expr.1997.4156. [DOI] [PubMed] [Google Scholar]
- 30.Wenz T, Hellwig P, MacMillan F, Meunier B, Hunte C. 2006. Probing the role of E272 in quinol oxidation of mitochondrial complex III. Biochemistry 45:9042–9052. doi: 10.1021/bi060280g. [DOI] [PubMed] [Google Scholar]
- 31.Davies CS, Pudney M, Matthews PJ, Sinden RE. 1989. The causal prophylactic activity of the novel hydroxynaphthoquinone 566C80 against Plasmodium berghei infections in rats. Acta Leiden 58:115–128. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.