

Abstract
The cellular entry of HIV-1 into CD4+ T cells requires ordered interactions of HIV-1 envelope glycoprotein with C-X-C chemokine receptor type 4 (CXCR4) receptors. However, such interactions, which should be critical for rational structure-based discovery of new CXCR4 inhibitors, remain poorly understood. Here we first determined the effects of amino acid substitutions in CXCR4 on HIV-1NL4-3 glycoprotein-elicited fusion events using site-directed mutagenesis-based fusion assays and identified 11 potentially key amino acid substitutions, including D97A and E288A, which caused >30% reductions in fusion. We subsequently carried out a computational search of a screening library containing ∼604,000 compounds, in order to identify potential CXCR4 inhibitors. The computational search used the shape of IT1t, a known CXCR4 inhibitor, as a reference and employed various algorithms, including shape similarity, isomer generation, and docking against a CXCR4 crystal structure. Sixteen small molecules were identified for biological assays based on their high shape similarity to IT1t, and their putative binding modes formed hydrogen bond interactions with the amino acids identified above. Three compounds with piperidinylethanamine cores showed activity and were resynthesized. One molecule, designated CX6, was shown to significantly inhibit fusion elicited by X4 HIV-1NL4-3 glycoprotein (50% inhibitory concentration [IC50], 1.9 μM), to inhibit Ca2+ flux elicited by stromal cell-derived factor 1α (SDF-1α) (IC50, 92 nM), and to exert anti-HIV-1 activity (IC50, 1.5 μM). Structural modeling demonstrated that CX6 bound to CXCR4 through hydrogen bond interactions with Asp97 and Glu288. Our study suggests that targeting CXCR4 residues important for fusion elicited by HIV-1 envelope glycoprotein should be a useful and feasible approach to identifying novel CXCR4 inhibitors, and it provides important insights into the mechanism by which small-molecule CXCR4 inhibitors exert their anti-HIV-1 activities.
INTRODUCTION
Over the last 30 years, human immunodeficiency virus 1 (HIV-1) has become responsible for more than 30 million deaths worldwide, and approximately 35 million people are estimated to be currently infected with the virus (1). Major innovations and advancements have led to the current availability of many anti-HIV-1 inhibitors; however, continued discovery and development of novel inhibitors against existing and newly discovered targets are needed to overcome a number of inherent problems in current antiretroviral therapy (ART), including toxicities and the acquisition of drug resistance by HIV-1 (2).
C-X-C chemokine receptor type 4 (CXCR4) and C-C chemokine receptor type 5 (CCR5) are essential coreceptors for the entry of HIV-1 into host cells. Both CXCR4 and CCR5 are G-protein-coupled receptors (GPCRs) with structures containing seven transmembrane (TM) helices. Maraviroc is the only small-molecule, FDA-approved, therapeutic agent targeting CCR5. Compared to CCR5 inhibitors, fewer CXCR4 inhibitors have been reported as potential therapeutic agents for treating HIV-1 infections. In fact, to date no CXCR4 inhibitor has been approved for clinical use as an anti-HIV-1 agent, and there is an urgent need for novel small-molecule inhibitors targeting CXCR4. Such a molecule, by itself or particularly in combination with a CCR5 antagonist, should greatly improve the treatment options available for patients predominantly infected with X4 or dual-tropic HIV-1 strains.
Initial reports identified several peptides (such as T140) and macrocycles (such as AMD3100) that targeted CXCR4 (3–5). To improve oral bioavailability, attempts to replace or to decrease the size of the macrocycles while retaining anti-HIV-1 potency were made. One such effort led to the discovery of AMD070, a molecule with benzoimidazole and tetrahydroquinoline groups (6, 7). AMD070 is orally bioavailable and has good safety and pharmacokinetic profiles (8, 9). Jenkinson et al. reported on the anti-HIV-1 and pharmacological profiles of GSK812397, a molecule with some structural similarity to AMD070 (10). Thoma et al. identified several isothiourea derivatives that bind to CXCR4 and inhibit HIV-1 infection (11). The crystal structures of CXCR4 in complex with a small molecule (IT1t) and with a 16-residue cyclic peptide (CVX15) were determined (12). The structures demonstrated important features of CXCR4, but further understanding of the mechanisms of antiviral activity exerted by small-molecule inhibitors is required for rational structure-based design of new CXCR4 inhibitors. Moreover, only a limited number of studies have utilized the recently determined crystal structures of various GPCRs in the discovery of novel chemotypes or in the optimization of existing candidates. This might be partly because inhibitors may bind to the binding sites of GPCRs in an orthosteric or allosteric fashion. The orthosteric inhibitors directly bind to the active site and competitively inhibit the natural substrate or ligand, while the allosteric modulators show their effects distal from their binding locations (13). Thus, the functional significance of each binding site residue and which residues need to be selectively targeted, based on the mechanism of action, need to be elucidated for design and discovery of new inhibitors.
The interaction of the HIV-1 envelope glycoprotein gp120 with CXCR4 enables the virus to gain entry into cells. We wanted to better understand the structural and functional importance of CXCR4 residues implicated in gp120-elicited fusion and to determine whether preferential interactions of an inhibitor with such residues may give rise to inhibition of the fusion event and anti-HIV-1 activity. In the current study, we first introduced a variety of amino acid substitutions in CXCR4 to determine residues that are important for the interaction of CXCR4 with the gp120 envelope protein. We then hypothesized that molecules that formed critical polar interactions with such potentially key residues would be likely to interfere with the binding and interactions of CXCR4 with gp120, exerting antiviral activity. To identify such molecules, we utilized the crystal structure of CXCR4 in complex with IT1t (12), as follows. First, we carried out a computational search that identified molecules that had high shape similarity with IT1t. The putative interactions of such molecules with CXCR4 were examined by docking simulations with the crystal structure of CXCR4. Sixteen molecules that had hydrogen bond interactions with at least two CXCR4 amino acid residues determined to be important for gp120-elicited fusion were selected for assays. Three piperidinylethanamine (PEA) derivatives were identified as exhibiting binding to CXCR4 in Ca2+ flux assays with MOLT-4 cells and showed anti-HIV-1 activity against X4 HIV-1NL4-3. The PEA derivatives also inhibited the fusion of HIV-1NL4-3 envelope gp120 with CXCR4. We subsequently synthesized several PEA derivatives, and assays confirmed their activity and specificity as anti-HIV-1 inhibitors targeting CXCR4. Our study provides important insights regarding the mechanism of inhibition and suggests that study of the interactions of molecules with CXCR4 residues that are important for gp120-elicited fusion might be a worthwhile strategy for discovering new inhibitors of CXCR4. The present study should provide a useful platform for structure-based discovery of inhibitors of CXCR4. Similar strategies, based on residue-specific interactions responsible for a given mechanism, may be used for the discovery of novel chemotypes for other GPCRs.
MATERIALS AND METHODS
Cells and viruses.
MT-4 cells were grown in RPMI 1640 culture medium supplemented with 10% fetal calf serum (FCS) (Gemini Bio-Products). The multinuclear activation of the galactosidase indicator (MAGI) cell line (14) and the U373-MAGI cell line (15) were provided by the NIH AIDS Research and Reference Reagent Program and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS, 200 μg/ml G418, and 100 μg/ml hygromycin B. The 293T cells were cultured in DMEM with 10% FCS. Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats from HIV-1-seronegative individuals and were activated with 10 μg/ml phytohemagglutinin (PHA) prior to use, as described previously (16). Two HIV-1 strains, i.e., HIV-1NL4-3 (17) and HIV-1BaL (18), were employed for drug susceptibility assays.
HIV-1 gp120-elicited cell-cell fusion assay.
The HIV-1-gp120-elicited cell-cell fusion assays using a panel of CXCR4 mutants were conducted as reported previously (19), with minor modifications. The CXCR4 expression vector pcDNA3.1-CXCR4 (Missouri University of Science and Technology cDNA Resource Center, Rolla, MO) was employed, and a variety of plasmids carrying a mutant CXCR4-encoding gene (pcDNA3.1-CXCR4MT) were subsequently generated by employing the site-directed mutagenesis technique. The HIV-1 envelope expression vector pCXN-NL4-3env was generated by replacing the JRFL envelope gene of pCXN-JRenv (20) with the NL4-3 envelope gene.
The envelope expression vector (pCXN-NL4-3env) and Tat expression vector (19) (0.5 μg each) were cotransfected into 293T cells (2 × 105 cells, 3 ml in 6-well microculture plates) using Lipofectamine 2000 (Invitrogen), while the wild-type or mutant CXCR4 expression vector and pLTR-LucE (0.5 μg each) were cotransfected into U373-MAGI cells (2 × 105 cells, 3 ml in 6-well microculture plates), since they do not endogenously express CXCR4. On the next day, the two types of cotransfected cells were harvested and mixed in wells of 96-well plates (2 × 104 cells each). The cotransfected cells were incubated further for 6 h, and the luciferase activity in each well was detected using the Bright-Glo luciferase assay system (Promega); luminescence levels were measured using a Veritas microplate luminometer (Turner BioSystems, Sunnyvale, CA). Nonspecific luciferase activity was determined in wells containing control Tat+ Env− 293T cells and Luc+ CXCR4+ U373-MAGI cells, and the value of the nonspecific luminescence was subtracted from each experimental luminescence value. The inhibition of cell-cell fusion by the test compounds was determined under the same conditions but with various drug concentrations, and the 50% inhibitory concentration (IC50) values were determined.
Computational screening.
We searched the screening libraries from ChemBridge (http://www.chembridge.com) to identify potential small-molecule inhibitors of CXCR4. In brief, the computational search identified potential molecules with the following two simulations: (i) initial identification of molecules with high shape similarity to IT1t (12), a known CXCR4 inhibitor, and (ii) subsequent determination of the putative binding mode and interactions of these molecules with potentially key CXCR4 amino acid residues. A flow diagram of the screening protocol is shown in Fig. 1. Initially, known aggregators, molecules with more than 20 rotatable bonds, and molecules of <350 Da or >750 Da were eliminated from consideration. In order to determine the molecules with high shape similarity to IT1t, the possible stereoisomers and three-dimensional conformations were generated using Omega (version 2.3.2; OpenEye Scientific Software, Inc., Santa Fe, NM), and the shape overlay program ROCS (version 3.0.0; OpenEye Scientific Software) was used (21, 22). The crystal structure conformation of IT1t (12) was used as the query template against which molecules from the chemical database were aligned. Molecules whose Tanimoto coefficients for IT1t shape similarity were at least 0.7 were retained for molecular docking. In order to eliminate lack of compatibility between software suites from different sources, the possible ionization states at pH 7 ± 2, tautomers, stereoisomers, and ring conformations of these molecules were generated again with LigPrep (version 2.4; Schrödinger, LLC). These structures were docked into CXCR4 using Glide (version 5.6; Schrödinger, LLC) (23, 24). The crystal water molecules were removed, the docking grid around the binding site of IT1t binding to CXCR4 was generated, and molecules were docked. Molecules that did not form hydrogen bond interactions with at least two of the following residues determined to be important for the fusion of gp120 envelope protein to CXCR4 were eliminated: Asp97, Tyr116, Phe174, Ala175, Asp182, Asp187, Arg188, Tyr190, Asp262, Glu288, and Phe292. Selection of the candidate molecules was made by analyzing the putative binding mode and interactions with CXCR4 residues, without explicit consideration of the Glide docking scores.
FIG 1.

Flow diagram of the computational screening protocol. Starting from over 600,000 compounds from a general screening library, molecules that were too flexible or did not meet our desired molecular weight range were eliminated. The three-dimensional shape similarities of molecules with respect to IT1t, a known inhibitor, were determined, and molecules with Tanimoto coefficients of at least 0.7 were retained. The interactions of these molecules with CXCR4 were determined by molecular docking. Sixteen molecules were selected based on their ability to form hydrogen bonds with at least two critical residues, as determined by fusion assays with mutant CXCR4 and HIV-1 envelope protein and by their binding modes and locations in the active site. Ca2+ binding, cell-cell fusion, and antiviral assays identified three compounds with PEA cores that bound to CXCR4 and were active against X4 HIV-1.
Determination of inhibition of chemokine binding to CXCR4.
In order to determine whether the selected compounds bound to CXCR4, calcium flux binding inhibition assays were conducted using the Fluo-4 Direct calcium reagent (Invitrogen), according to the manufacturer's protocol. In brief, MOLT-4 cells (5 × 105 cells) were exposed to Fluo-4 Direct calcium reagent for 60 min at 37°C in RPMI 1640 medium containing 5% FCS. A test compound was added at various concentrations, the mixture was incubated for 30 min, and the cells were exposed to stromal cell-derived factor 1α (SDF-1α) at a concentration of 1 nM. Relative increases in cytosolic Ca2+ levels after SDF-1α exposure were determined by fluorescence-activated cell sorting (FACS) analysis with a FACSCalibur system (BD Biosciences), and IC50s for cytosolic Ca2+ mobilization (Ca2+ flux) by test compounds were determined by comparison with the Ca2+ flux levels in drug-free control samples.
Drug susceptibility and cytotoxicity assays.
The susceptibility of HIV-1 strains to various drugs was determined as described previously (16, 25), with minor modifications. In the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, MT-4 cells (5 × 104 cells/ml) were exposed to 100 times the 50% tissue culture infective dose (TCID50) of HIV-1 in the presence of various concentrations of drugs, in 96-well microculture plates, and were incubated at 37°C for 5 days. After culture, 10 μl of cell-staining solution (Cell Counting Kit-8; Dojindo Molecular Technologies, Inc., Japan) was added to each well in the plate, followed by incubation at 37°C for 1 to 2 h, and the optical density at 450 nm was measured in a microplate reader (model 3550; Bio-Rad). All assays were performed in duplicate, and data shown represent mean ± 1 standard deviation values derived from the results of at least two independent experiments.
PHA-activated PBMC (1 × 106 cells/ml) were exposed to 50 times the TCID50 of HIV-1NL4-3 in the presence or absence of various concentrations of drugs in 10-fold serial dilutions, in 96-well microculture plates. All assays were performed in triplicate. The amounts of p24 antigen produced by the cells were determined on day 7 in culture using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (PerkinElmer). Drug concentrations that resulted in 50% inhibition (IC50) of p24 antigen production were determined by comparison with the p24 production levels in drug-free control cell cultures. Antiviral assays using MAGI cells (MAGI cell assay) were also conducted, as reported previously (16).
The cytotoxicity of compounds in MT-4 cells was also determined using the MTT assay. Cells were plated in 96-well plates, exposed to various concentrations of the compound, and cultured under the same conditions as for the anti-HIV-1 assay. The number of viable cells in each well was determined using the Cell Counting Kit-8.
Synthesis of piperidinylethanamine derivatives.
The synthesis of a PEA derivative (inhibitor CX6) is shown in Fig. 2. Reductive amination of ethylpiperdine-3-carboxylate with cyclopentanone provided amine 1 in 81% yield. Dibal-H reduction at −78°C afforded aldehyde 2 in 75% yield. Amine 4 was prepared by alkylation of piperidine with commercially available bromide 3. Reductive amination of aldehyde 2 with amine 4 in the presence of titanium tetraisopropoxide furnished secondary amine 5. Amine 5 was then subjected to reductive amination with 3-hydroxybenzaldehyde to provide compound 6 (CX6) in 78% yield and 97% purity, as determined by high-performance liquid chromatography (HPLC). The detailed experimental procedure and characterization are described in the supplemental material. The characterization of other synthesized PEA derivatives (CX6, CX21, CX22, CX23, CX24, CX25, and CX27) is described in the supplemental material.
FIG 2.
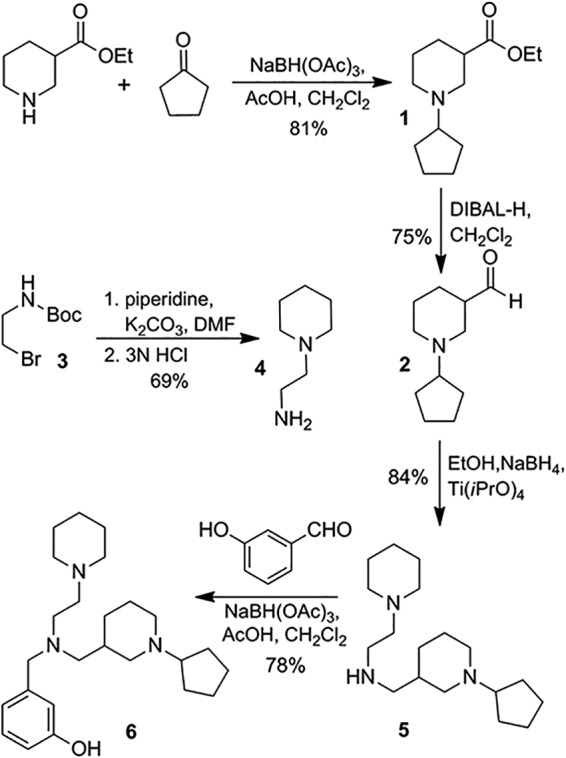
Synthesis of inhibitor 6 (CX6). AcOH, acetic acid; DMF, dimethylformamide; EtOH, ethanol.
RESULTS
Effects of amino acid substitutions in CXCR4 on cell fusion induced by HIV-1 envelope glycoprotein.
The HIV-1 envelope glycoprotein gp120 interacts with CXCR4 for entry to CD4+ T cells. The gp120-CXCR4 interaction is described by a two-site model in which the N terminus of CXCR4 interacts with the base of the hypervariable region 3 (V3) loop of gp120 (site 1) and CXCR4 residues in transmembrane (TM) helices and extracellular loop 2 (ECL2) interact with the tip of V3 (site 2) (12). The two-site model suggests that certain transmembrane and extracellular residues are important for gp120 binding, and we hypothesized that small molecules that bind to such residues in the orthosteric binding site would likely inhibit the CXCR4-gp120 interactions. To determine these residues, 56 amino acid residues in the extracellular and transmembrane regions of CXCR4 were selected for introduction of amino acid substitutions and the HIV-1 gp120-elicited cell fusion levels were determined and compared to those of wild-type CXCR4. The intracellular residues are important for signal transduction but do not seem to directly affect anti-HIV-1 inhibitor binding or gp120-elicited fusion; hence, they were not considered. The significance of some of the residues selected for substitution is as follows. Asp133, Arg134, and Tyr135 in TM3 are the conserved Asp-Arg-Tyr motif in various GPCRs and are reportedly important in triggering ligand-induced conformational changes that lead to receptor activation (26). Asp171 (TM4), Gly207 (TM5), Asp262 (TM6), Asp187 (ECL2), Arg188 (ECL2), Tyr190 (ECL2), and His281 (ECL3) are important in the binding of T140, AMD3100, or AMD3465 to CXCR4 (27–29). The second extracellular loop (ECL2) is known to be important for the structure and function of CXCR4, CCR5, and other GPCRs (30–32); therefore, several residues in ECL2 were selected. Cys109 located in the extracellular region of TM3 forms a disulfide bond with Cys186 in ECL2. This disulfide linkage is conserved for class A GPCRs and is known to be important for their structures and functions (12, 32). The effects of substitution of some of these residues on HIV-1 glycoprotein-elicited fusion were reported previously (33); the coverage was partial, however, and we carried out a comprehensive analysis of the effects of CXCR4 amino acid residues on virally induced fusion.
Figure 3A shows the changes in cell-cell fusion levels with wild-type versus mutant CXCR4. Eleven amino acid substitutions (D97A, Y116A, F174A, A175F, D182A, D187A, R188A, Y190A, D262A, E288A, and F292A) resulted in substantial reductions (more than 30%) in the levels of fusion elicited by HIV-1 envelope protein. Amino acid substitutions C109A, D133A, C186A, and D193A drastically reduced CXCR4-gp120 fusion; however, they also caused critical decreases in the levels of CXCR4 expression on the cell surface, as determined by FACS analysis. Therefore, we could not determine whether these reductions were because of functional importance or because of decreased expression levels. Substitutions of Asn176 (in ECL2) had no effects on the fusion levels, in agreement with the data of Brelot et al. (33). It is noteworthy that there are a number of negatively charged acidic residues (D97A, D182A, D187A, D262A, and E288A) whose substitutions significantly decreased gp120-elicited fusion (Fig. 3A). These acidic residues may interact with the basic residues of gp120 to affect coreceptor selectivity. Negatively charged acidic residues have also been shown to be important for the binding of SDF-1 and for CXCR4 coreceptor function in HIV-1 entry (33).
FIG 3.

(A) Effects of CXCR4 amino acid substitutions on cell fusion elicited by the HIV-1 envelope glycoprotein. Tat+ X4 Env+ 293T cells were cocultured with U373-MAGI cells expressing long terminal repeat (LTR)-luciferase and CXCR4 (wild-type [WT] or mutant) in a 96-well plate for 6 h. The luciferase activity (y axis), which indicated cell-cell fusion activity, was measured. The value in wells with U373-MAGI cells with wild-type CXCR4 is shown as 100% (black bar). For U373-MAGI cells with mutant CXCR4, activities of less than 70%, compared to that of the wild-type control, are shown by magenta bars, whereas activities of more than 70% are shown by blue bars. The white bars indicate substitutions that had both low cell expression and low luciferase activity. The data shown represent mean ± 1 standard deviation values derived from results of at least two independent experiments. (B) Locations of CXCR4 amino acids whose substitution decreased cell fusion elicited by HIV-1 envelope glycoprotein. A ribbon and van der Waals surface diagram of CXCR4 with the locations of residues whose substitution resulted in more than 30% decreases (magenta bars in panel A) in cell fusion elicited by HIV-1 envelope glycoprotein is shown. Red spheres, alpha carbons of the residues. The diagram was generated using Maestro (version 9.3; Schrödinger, LLC).
We analyzed the crystal structures of CXCR4 (12) to understand the interactions and orientations of the residues whose substitutions adversely affected the interactions of CXCR4 with the HIV-1 envelope protein (Fig. 3B). Six amino acid substitutions (F174A, A175F, D182A, D187A, R188A, and Y190A) that affected the fusion event were identified in or near ECL2 (Fig. 3B), strongly suggesting that this region as a whole probably affects fusion more than any other loop or transmembrane region of CXCR4. Asp187 and Arg188 have hydrogen bond interactions with each other through their side chains and form part of the binding cavity. However, Phe174, Ala175, and Tyr190 are not part of the ligand binding pocket of IT1t. Asp262, located in TM6, is not close enough to the ligand binding pocket of IT1t but forms hydrogen bond interactions with the much larger 16-residue peptide CVX15 (12). Site-directed mutagenesis studies have suggested that Asp262 is important for the binding of AMD3100 (27). Glu288 (in TM7) has a hydrogen bond with the side chain of Tyr116 (in TM3) and an intrahelical hydrogen bond with Phe292 (in TM7), and these three residues form part of the binding pocket within the transmembrane domain. The side chain of Asp97 forms part of the binding pocket for small molecules, whereas the carboxylate side chain of Asp182 is oriented toward the extracellular region and away from the binding pocket located inside the transmembrane domain. Substitutions of residues in TM5 or ECL3 did not affect the fusion event in the assay (Fig. 3A), indicating that these regions are not involved in the interaction of CXCR4 with the HIV-1 envelope protein.
Computational examination of molecules for identification of CXCR4 inhibitors.
We searched a general screening library from ChemBridge, which contains more than 600,000 molecules, to select molecules for assays as potential inhibitors of CXCR4. Known aggregators and molecules that did not have molecular weights between 350 and 750 were eliminated (Fig. 1), and the possible stereoisomers and conformations of the rest were determined with Omega (version 2.3.2; OpenEye Scientific Software, Inc., Santa Fe, NM) (21). Previous studies showed that determination of molecular similarity plays a critical role in the analysis of large databases of compounds in chemical and pharmaceutical research (34). Therefore, using the software tool ROCS (22), we determined the shape similarity of the molecules from the ChemBridge library with respect to IT1t, a molecule that binds with high affinity to CXCR4 and demonstrates anti-HIV-1 activity (11, 12). The highest Tanimoto coefficient for IT1t shape similarity from the database was 0.85. A total of 753 unique molecules with 1,005 configurations/conformations had shape overlap Tanimoto coefficients of at least 0.70. The binding modes and interactions of these molecules with CXCR4 were determined by molecular docking using Glide (version 5.6; Schrödinger, LLC) (23, 24). To avoid any issues that might arise through the use of conformations generated by Omega with Glide docking, LigPrep was used to generate molecular configurations and conformations for docking, and it generated 14,226. These configurations/conformations were docked to the crystal structure of CXCR4 to determine their possible binding modes. Since it had been determined that CXCR4 transmembrane residues Asp97, Tyr116, Asp262, Glu288, and Phe292 and ECL2 residues Phe174, Ala175, Asp182, Asp187, Arg188, and Tyr190 appeared to be important for the cell fusion event (Fig. 3A), we hypothesized that molecules that bound around the active site, as determined in the crystal structure, and formed hydrogen bond interactions with at least two of these residues would be likely to competitively inhibit the interactions of CXCR4 with gp120. Our candidate molecule selection was based on shape similarity to a known inhibitor (IT1t) and putative binding modes and interactions with residues that were determined to be important for the HIV-1 gp120-elicited cell fusion event with CXCR4. Of note, we did not use any energy-based or empirical scoring functions for estimation of relative affinities for the selection of compounds. Based on the hypothesis described above, we selected 16 compounds (named CX1 to CX16) from the ChemBridge general screening library for biological assays. Three compounds were piperidinylethanamine derivatives, and four compounds were tetrahydro-β-carboline derivatives. The other compounds contained quinoxaline, indole, indane, imidazole, imidazopyridine, imidazothiazole, piperazine, morpholine, and diazepane moieties. In computational examinations of screening libraries, completely different cores are selected for assays in some instances, but we chose multiple piperidinylethanamine and tetrahydro-β-carboline derivatives, as their binding modes were thought to be most substantive. While there are certain advantages to selecting only one compound for each core, we decided not to take that approach because we did not want to eliminate an active core if the only compound we chose happened to be inactive.
Inhibition of chemokine binding to CXCR4 by piperidinylethanamine derivatives.
We investigated whether the 16 selected compounds bound to CXCR4 by blocking the intracellular Ca2+ mobilization induced by SDF-1α, whose primary receptor is CXCR4. One of the compounds, described as CX6 (Fig. 4), blocked the SDF-1α-induced Ca2+ mobilization in MOLT-4 cells with an IC50 of 92 nM (Table 1; also see Fig. S1 in the supplemental material). In the same assay, AMD3100 showed inhibition of SDF-1α-induced Ca2+ mobilization with an IC50 of ∼10 nM (see Fig. S1 in the supplemental material). Two other compounds, namely, CX11 and CX13, both of which are piperidinylethanamine (PEA) derivatives like CX6 (Fig. 4), blocked SDF-1α-induced Ca2+ mobilization in MOLT-4 cells with IC50s of 161 and 149 nM, respectively (Table 1). None of the other compounds (see Fig. S2 in the supplemental material) bound to CXCR4. Of note, all three PEA derivatives (CX6, CX11, and CX13) failed to block the Ca2+ mobilization induced with RANTES, whose receptor is CCR5, indicating that these compounds did not bind to CCR5. When we investigated whether these three compounds had agonistic effects to induce Ca2+ mobilization in CXCR4+ cells, none induced Ca2+ mobilization, suggesting that CX6, CX11, and CX13 were antagonists of CXCR4 (see Fig. S3 in the supplemental material). In summary, the data presented above strongly suggested that the PEA derivatives bound to CXCR4 with specificity and were antagonists of CXCR4. Subsequently, we newly synthesized CX6 (Fig. 2; also see the descriptions in the supplemental material). Other PEA derivatives were also synthesized in high purity, and their characterization is described in the supplemental material.
FIG 4.

Structures of CXCR4 inhibitors. The structures of IT1t (11), a known inhibitor, and CX6 are shown. The newly identified inhibitors have a cyclopentylpiperidinylmethylpiperidinylethylamine (CpPMPEA) pharmacophore. The R-groups and anti-HIV-1 activities of the other derivatives are shown.
TABLE 1.
Anti-HIV activities, SDF-1α binding inhibition, and cytotoxicity of selected CXCR4 inhibitors
| Compounda | Anti-HIV activity IC50 (mean ± SD) (μM) |
IC50 (mean ± SD) (µM) in fusion assay with HIV-1NL4-3 Env (X4) | Binding IC50 (mean ± SD) (nM)b | CC50 (mean ± SD) (μM)c | |||
|---|---|---|---|---|---|---|---|
| MTT assay with HIV-1NL4-3 (X4) | p24 assay with HIV-1NL4-3 (X4) | MAGI cell assay with HIV-1LAI (X4) | MAGI cell assay with HIV-1BaL (R5) | ||||
| CX6 | 1.5 ± 0.4 | 3.0 ± 0.8 | 3.0 ± 1.5 | >10 | 1.9 ± 0.4 | 92 ± 20 | 58 ± 25 |
| CX11 | 2.6 ± 0.3 | 4.6 ± 2.0 | 5.2 ± 3.0 | >10 | 7.9 ± 1.1 | 161 ± 6 | 96 ± 22 |
| CX13 | 2.6 ± 1.0 | 6.7 ± 2.2 | 7.8 ± 1.3 | >10 | >10 | 149 ± 19 | >100 |
| CX20 | 0.6 ± 0.5 | 1.0 ± 0.5 | 1.2 ± 0.5 | >10 | 1.5 ± 0.2 | 76 ± 10 | 40 ± 3 |
| CX26 | 2.6 ± 1.3 | 2.6 ± 1.1 | 7.3 ± 1.1 | >10 | 2.9 ± 0.5 | 159 ± 20 | 36 ± 3 |
| AMD3100 | 0.02 ± 0.01 | NDd | 0.011 ± 0.008 | >10 | ND | 12 ± 1 | >10 |
| AMD11070 | 0.026 ± 0.01 | ND | ND | ND | ND | 32 ± 4 | >10 |
CX6, CX11, and CX13 were among the 16 compounds initially selected from the general screening library. CX20 and CX26, containing the CpPMPEA pharmacophore, were subsequently analyzed. Anti-HIV activity and binding data for the known CXCR4 inhibitors AMD3100 and AMD11070 are included for comparison. All assays were conducted in duplicate, and data shown represent mean ± 1 standard deviation (SD) values derived from the results of at least two independent experiments.
Binding IC50 values were determined in Ca2+ flux assays with MOLT-4 cells.
CC50 values were determined in MTT assays with MT4 cells.
ND, not determined.
Inhibition of fusion elicited by HIV-1NL4-3 envelope protein interactions with CXCR4 by piperidinylethanamine derivatives.
The fusion event elicited by the interactions of HIV envelope glycoprotein with CXCR4 enables the virus to gain entry into cells, eventually leading to viral replication. The 16 compounds were selected through docking simulations that suggested that they bound to a potential orthosteric binding site of CXCR4. These compounds formed hydrogen bonds with at least two amino acid residues most likely to be important for the fusion event and thus have the potential to competitively inhibit the fusion event. We thus assessed whether the compounds were actually able to inhibit the interactions of the HIV-1NL4-3 envelope protein with CXCR4 and to block the fusion event in the HIV-1 gp120-elicited cell-cell fusion assays with wild-type CXCR4. Both CX6 and CX11 blocked the fusion, with IC50s of 1.9 μM and 7.9 μM, respectively (Table 1). Moreover, none of the compounds, including CX6 and CX11, inhibited the fusion event, as examined with the HIV-1 gp120-elicited cell-cell fusion assay, with cells expressing wild-type CCR5 derived from HIV-1BaL (R5 HIV-1), as described previously (19, 30). This finding suggested that CX6 and CX11 inhibited the fusion event associated with CXCR4 but not that associated with CCR5.
Interactions of piperidinylethanamine derivatives with CXCR4.
The three-dimensional shape overlay of CX6 (shown in gray sticks) with IT1t (shown in green sticks), as determined by ROCS (version 3.0.0; OpenEye Scientific Software), is shown in Fig. 5A. The molecules have an excellent Tanimoto coefficient of 0.73 for shape overlap. The imidazothiazole ring of IT1t overlays the cyclopentylpiperidinyl group of CX6. The interactions of the identified compound (CX6) with CXCR4 were deduced by molecular docking analysis. The interactions of CX6 with CXCR4 are illustrated in Fig. 5B to E. The following amino acid residues of CXCR4 were seen to form the active site (<4 Å from the inhibitor) for the binding of CX6: Glu32 of the N terminus of CXCR4; Phe36, Asn37, Leu41, and Tyr45 of TM1; Trp94, Asp97, and Ala98 of TM2; Trp102 of ECL1; Val112, His113, and Tyr116 of TM3; Cys186 and Arg188 of ECL2; and His281, Ile284, Ser285, and Glu288 of TM7. Thus, the molecule CX6 bound in the active site predominantly formed by residues from TM1, TM2, TM3, TM7, and ECL2. As expected, no amino acid residues of TM4, TM5, or TM6 had interactions with CX6. The nitrogens of both piperidine groups were determined to be protonated (LigPrep version 2.4; Schrödinger, LLC). For CX6, the protonated nitrogen of cyclopentylpiperidinyl had hydrogen bond interactions with Glu288, and the protonated nitrogen of piperidinylethanamine had hydrogen bond interactions with Asp97. The phenol group of CX6 interacted with Glu32 located in the N terminus of CXCR4. Asp97 has been shown to be important for the binding of AMD070 as well as for CXCR4-gp120 fusion. Glu288 is important for the fusion event, as it has been shown that substitution of Glu288 with alanine results in loss of the CXCR4-gp120 fusion. Sequence alignment indicated that the residue corresponding to Glu288 of CXCR4 is Glu283 of CCR5. Therefore, E283 in CCR5 should be important for the binding of the CCR5 antagonist aplaviroc and its analogs, in line with previously published results (19, 30). Indeed, substitutions of E283 in CCR5 resulted in loss of the CCR5-gp120 fusion event, as described previously (19, 30).
FIG 5.

(A) ROCS shape overlap and similarity of the query molecule (IT1t) and an identified lead molecule (CX6). The figure shows a representation of the three-dimensional shape (volume) overlap and similarity between IT1t (green carbons) and the identified lead molecule CX6 (gray carbons). The surface of IT1t is shown in gray and that of the identified inhibitor CX6 is shown in orange. The molecules are structurally different but have a shape overlap Tanimoto coefficient of 0.73. (B and C) Model of the interaction of CX6 with CXCR4. CXCR4 TM helices are shown by a ribbon representation in panel B. CX6 is represented by tubes with gray (B) and green (C) carbons. The model suggests that CX6 forms polar interactions with Asp97 and Glu288, residues important for the fusion elicited by HIV envelope protein interactions with CXCR4. (D and E) Side view (D) and view from the top (E) of the molecular surface of CX6 bound in the active site of CXCR4. The diagrams were generated using Maestro (version 9.3; Schrödinger, LLC).
E283 of CCR5 and E288 of CXCR4 represent the 6th residue of TM7 in both chemokine receptors. Glutamic acid at position 6 in TM7 is a highly conserved amino acid residue in chemokine receptors and has been demonstrated to be important for the binding of nonpeptidic ligands to chemokine receptors CCR1 and CCR2 (35). Our current study suggests that Glu288 is an important residue to target for rational structure-based design and discovery of CXCR4 inhibitors.
Binding of PEA derivatives to CXCR4 and antiviral activity against X4 HIV-1.
Finally, we determined the anti-HIV-1 activity and cytotoxicity of CX6 with the MTT assay using X4 HIV-1NL4-3 and MT4 cells as target cells. CX6 suppressed the infectivity and replication of HIV-1NL4-3 with a good dose-response relationship (Fig. 6). As shown in Table 1, CX6 exerted substantial activity against HIV-1NL4-3 and HIV-1LAI, both of which are X4 HIV-1 strains, with IC50s of 1.5 and 3.0 μM, respectively. The 50% cytotoxic concentration (CC50) of CX6 was 58 μM, with a therapeutic index of 39. CX6 blocked the fusion event, as examined in the fusion inhibition assay using HIV-1NL4-3-derived envelope protein. In the Ca2+ flux inhibition assay, in which SDF-1α-elicited Ca2+ flux levels are determined in the presence or absence of a potential small-molecule inhibitor, CX6 was found to block the flux with an IC50 of 92 nM (Table 1). As expected, however, CX6 failed to block the infectivity and replication of R5 HIV-1 (HIV-1BaL; IC50, >10 μM), as examined in an anti-HIV assay using HIV-1BaL and MAGI cells. CX11 and CX13 similarly exerted activity against X4 HIV-1NL4-3 and X4 HIV-1LAI but failed to block the infectivity and replication of R5 HIV-1BaL (Table 1). Of note, CX13 was least active among the three PEA derivatives initially examined, and it only marginally suppressed the fusion event (25 to 27% reduction at 10 μM), as examined using the HIV-1NL4-3-derived envelope protein. Cyclopentylpiperidinylmethylpiperidinylethylamine (CpPMPEA) is the pharmacophore of these PEA derivatives (Fig. 4). Substitution of the cyclopentanepiperidinyl group with an m-methoxybenzene moiety abolished activity, confirming that CpPMPEA is critical for the observed activities.
FIG 6.
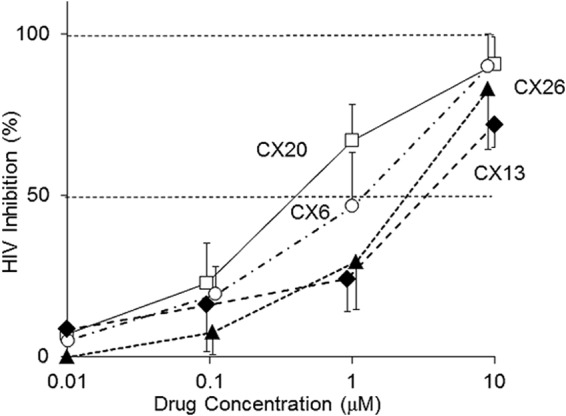
Antiviral activities of PEA derivatives against HIV-1NL4-3 (X4 HIV-1). The dose-response curves for the antiviral activities determined by MTT assays using MT4 cells and HIV-1NL4-3 are shown. All assays were conducted in duplicate, and the data shown represent mean ± 1 standard deviation values derived from the results of at least two independent experiments.
After establishing the activity and specificity of the PEA derivatives as described, we examined the activity of other PEA derivatives with the CpPMPEA pharmacophore (Fig. 4). CX20 and CX26 similarly exerted substantial activity against X4 HIV-1NL4-3 and X4 HIV-1LAI but failed to block the infectivity and replication of R5 HIV-1BaL (Fig. 4 and Table 1). Of note, CX20 exerted the most potent activity against X4 HIV-1NL4-3 and X4 HIV-1LAI, had a therapeutic index of 67, and most potently inhibited CXCR4-associated fusion and SDF-1-induced Ca2+ flux among the eight PEA derivatives examined. In this regard, a docking analysis demonstrated that the larger benzimidazole group of CX20, compared to the phenol moiety of CX6, forms a better hydrophobic contact with CXCR4, thus probably giving rise to its more potent anti-HIV-1 activity and greater binding affinity. It is also of note that CX26 and CX6 are isomers and differ in the position of the alcohol group in the phenol (CX26 has o-phenol, while CX6 has m-phenol). CX26 had comparable anti-HIV-1 activity, with an IC50 of 2.6 μM, with respect to CX6, suggesting that the phenol moiety should have good interactions with CXCR4 receptors. We synthesized six other PEA derivatives (CX21, CX22, CX23, CX24, CX25, and CX27) and determined their anti-HIV-1 activities. As assessed in the MTT assay, the compounds had activity against X4 HIV-1NL4-3, with IC50s between 0.6 and 2.9 μM (Fig. 4), confirming that the CpPMPEA pharmacophore plays significant roles in the interactions and activities of these derivatives with CXCR4.
DISCUSSION
As of this writing, maraviroc, which is active against R5 HIV-1, is the only CCR5 inhibitor in clinical use. Many infected patients carry both R5 HIV-1 and X4 HIV-1 strains, and no small-molecule inhibitor specifically targeting CXCR4 is in clinical use. Moreover, X4 HIV-1 may become predominant when HIV-1 disease progresses. Thus, the availability of a CXCR4 inhibitor would greatly increase the treatment options for patients infected with X4 and dual-tropic X4/R5 HIV-1 (2). In vitro studies suggest that CCR5 and CXCR4 inhibitors together provide greater synergistic effects (36). Therefore, the use of CCR5 and CXCR4 inhibitors together should have significant potential to provide greater benefits to patients, particularly over the use of maraviroc alone.
CCR5 has been actively pursued as a promising target, as some patients with a natural deletion of the CCR5 gene are apparently physiologically normal. CXCR4 belongs to the family of chemoattractant cytokines and is reportedly functionally important for angiogenesis, angiostasis, embryogenesis, cancer, and inflammatory diseases (37). These putative functions indicate the challenges involved in developing safe CXCR4 antagonists for clinical use. In fact, clinical trials of AMD3100 for HIV-1 treatment were discontinued due to toxicity, although the compound was later approved as a stem cell mobilizer. An ideal solution to CXCR4 inhibitor-induced toxicity would be to create a molecule that, when bound to CXCR4, could prevent HIV-1 fusion without interfering with normal CXCR4 functions. Besides discovering a molecule with a high selectivity index, an optimal dosage might need to be determined. It is quite possible that determining a specific dose of a CXCR4 inhibitor could lead to effective HIV interventions while minimizing toxicity. If these challenges are overcome, then CXCR4 inhibitors are likely to play significant roles in ART regimens.
Besides inhibitors targeting CCR5 and CXCR4, inhibitors such as AR177 (Zintevir) that target gp120 and demonstrate anti-HIV activity have been reported (38). Virtually all inhibitors of CXCR4-gp120 interactions previously reported in the literature were discovered and optimized before the crystal structure of CXCR4 became available. The CXCR4 crystal structure reported by Wu et al. (12), showing an orthosteric binding site, opened the possibility of the discovery of novel inhibitor chemotypes and rational optimization of inhibitors. We performed a comprehensive examination of the effects of amino acid substitutions in the transmembrane and extracellular domains of CXCR4 on HIV-1 gp120-elicited cell fusion and identified 11 residues important for preserving the interaction of CXCR4 with the gp120 protein. We hypothesized that molecules that formed hydrogen bond interactions with at least two of these residues might competitively inhibit the interaction of CXCR4 and gp120 and exert antiviral activity. A general screening library was searched to identify molecules that exhibited high three-dimensional shape similarity to a known CXCR4 inhibitor and formed hydrogen bond interactions with at least two of those 11 residues. Using a computational search that took into account the residue-by-residue interaction analysis of CXCR4, we identified piperidinylethanamine derivatives as a novel CXCR4 antagonist family. The most promising PEA derivative (CX20) inhibited SDF-1α-induced Ca2+ influx with CXCR4 in MOLT-4 cells with an IC50 of 76 nM and exerted anti-HIV-1 activity with an IC50 of 600 nM. AMD3100 and AMD11070 inhibited SDF-1α-induced Ca2+ influx with CXCR4 with IC50s of 12 nM and 32 nM, respectively (Table 1). The PEA derivatives did not have any activity against R5 HIV-1BaL. In summary, our Ca2+ flux, fusion, antiviral activity, and cytotoxicity assays demonstrate that PEA derivatives exert their activities through strong specific interactions with CXCR4 and not with CCR5. The current study provides a platform for structure-based discovery of CXCR4 inhibitors. The study also provides important insights into the mechanism of the antiviral activity of inhibitors targeting CXCR4, and our approach may be useful in exploring antagonists against other novel targets through understanding the mechanisms of action.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a grant from the Global Education and Research Center Aiming at the Control of AIDS (Global Center of Excellence supported by Monbu-Kagakusho), a grant for promotion of AIDS research from the Ministry of Health, Welfare, and Labor of Japan, the grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re-emerging Infectious Diseases from Monbu-Kagakusho (H.M.), and a grant from the National Institutes of Health (grant GM53386 [A.K.G.]). The study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (http://biowulf.nih.gov).
We are thankful to David Hoover for help in configuring software from OpenEye and Schrödinger for use with the NIH Biowulf Linux cluster and to Joseph Adelsberger of Leidos Biomedical Research, Inc., for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04654-14.
REFERENCES
- 1.Joint United Nations Programme on HIV/AIDS. 2013. Global report: UNAIDS report on the global AIDS epidemic 2013. UNAIDS, Geneva, Switzerland. [Google Scholar]
- 2.Maeda K, Das D, Nakata H, Mitsuya H. 2012. CCR5 inhibitors: emergence, success, and challenges. Expert Opin Emerg Drugs 17:135–145. doi: 10.1517/14728214.2012.673584. [DOI] [PubMed] [Google Scholar]
- 3.Tamamura H, Xu Y, Hattori T, Zhang X, Arakaki R, Kanbara K, Omagari A, Otaka A, Ibuka T, Yamamoto N, Nakashima H, Fujii N. 1998. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: a strong anti-HIV peptide T140. Biochem Biophys Res Commun 253:877–882. doi: 10.1006/bbrc.1998.9871. [DOI] [PubMed] [Google Scholar]
- 4.Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP. 1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 4:72–77. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Narumi T, Ozaki T, Sohma A, Ohashi N, Hashimoto C, Itotani K, Nomura W, Murakami T, Naoki YB, Tamamura H. 2011. Azamacrocyclic metal complexes as CXCR4 antagonists. ChemMedChem 6:834–839. doi: 10.1002/cmdc.201000548. [DOI] [PubMed] [Google Scholar]
- 6.Bridger GJ, Skerlj RT, Hernandez-Abad PE, Bogucki DE, Wang Z, Zhou Y, Nan S, Boehringer EM, Wilson T, Crawford J, Metz M, Hatse S, Princen K, De Clercq E, Schols D. 2010. Synthesis and structure-activity relationships of azamacrocyclic C-X-C chemokine receptor 4 antagonists: analogues containing a single azamacrocyclic ring are potent inhibitors of T-cell tropic (X4) HIV-1 replication. J Med Chem 53:1250–1260. doi: 10.1021/jm901530b. [DOI] [PubMed] [Google Scholar]
- 7.Skerlj RT, Bridger GJ, Kaller A, McEachern EJ, Crawford JB, Zhou Y, Atsma B, Langille J, Nan S, Veale D, Wilson T, Harwig C, Hatse S, Princen K, De Clercq E, Schols D. 2010. Discovery of novel small molecule orally bioavailable C-X-C chemokine receptor 4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. J Med Chem 53:3376–3388. doi: 10.1021/jm100073m. [DOI] [PubMed] [Google Scholar]
- 8.Moyle G, DeJesus E, Boffito M, Wong RS, Gibney C, Badel K, MacFarland R, Calandra G, Bridger G, Becker S. 2009. Proof of activity with AMD11070, an orally bioavailable inhibitor of CXCR4-tropic HIV type 1. Clin Infect Dis 48:798–805. doi: 10.1086/597097. [DOI] [PubMed] [Google Scholar]
- 9.Stone ND, Dunaway SB, Flexner C, Tierney C, Calandra GB, Becker S, Cao Y-J, Wiggins IP, Conley J, MacFarland RT, Park J-G, Lalama C, Snyder S, Kallungal B, Klingman KL, Hendrix CW. 2007. Multiple-dose escalation study of the safety, pharmacokinetics, and biologic activity of oral AMD070, a selective CXCR4 receptor inhibitor, in human subjects. Antimicrob Agents Chemother 51:2351–2358. doi: 10.1128/AAC.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkinson S, Thomson M, McCoy D, Edelstein M, Danehower S, Lawrence W, Wheelan P, Spaltenstein A, Gudmundsson K. 2010. Blockade of X4-tropic HIV-1 cellular entry by GSK812397, a potent noncompetitive CXCR4 receptor antagonist. Antimicrob Agents Chemother 54:817–824. doi: 10.1128/AAC.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thoma G, Streiff MB, Kovarik J, Glickman F, Wagner T, Beerli C, Zerwes H-G. 2008. Orally bioavailable isothioureas block function of the chemokine receptor CXCR4 in vitro and in vivo. J Med Chem 51:7915–7920. doi: 10.1021/jm801065q. [DOI] [PubMed] [Google Scholar]
- 12.Wu BL, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. 2010. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nussinov R, Tsai CJ. 2012. The different ways through which specificity works in orthosteric and allosteric drugs. Curr Pharm Des 18:1311–1316. doi: 10.2174/138161212799436377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimpton J, Emerman M. 1992. Detection of replication-competent and pseudotyped human-immunodeficiency-virus with a sensitive cell-line on the basis of activation of an integrated β-galactosidase gene. J Virol 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vodicka MA, Goh WC, Wu LI, Rogel ME, Bartz SR, Schweickart L, Raport CJ, Emerman M. 1997. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 233:193–198. doi: 10.1006/viro.1997.8606. [DOI] [PubMed] [Google Scholar]
- 16.Maeda K, Yoshimura K, Shibayama S, Habashita H, Tada H, Sagawa K, Miyakawa T, Aoki M, Fukushima D, Mitsuya H. 2001. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J Biol Chem 276:35194–35200. doi: 10.1074/jbc.M105670200. [DOI] [PubMed] [Google Scholar]
- 17.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clavel F, Guetard D, Brunvezinet F, Chamaret S, Rey MA, Santosferreira MO, Laurent AG, Dauguet C, Katlama C, Rouzioux C, Klatzmann D, Champalimaud JL, Montagnier L. 1986. Isolation of a new human retrovirus from west-African patients with AIDS. Science 233:343–346. doi: 10.1126/science.2425430. [DOI] [PubMed] [Google Scholar]
- 19.Maeda K, Das D, Yin PD, Tsuchiya K, Ogata-Aoki H, Nakata H, Norman RB, Hackney LA, Takaoka Y, Mitsuya H. 2008. Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV-1 fusion: insights into the mechanism of allosteric inhibition. J Mol Biol 381:956–974. doi: 10.1016/j.jmb.2008.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda Y, Foda M, Matsushita S, Harada S. 2000. Involvement of both the V2 and V3 regions of the CCR5-tropic human immunodeficiency virus type 1 envelope in reduced sensitivity to macrophage inflammatory protein 1α. J Virol 74:1787–1793. doi: 10.1128/JVI.74.4.1787-1793.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. 2010. Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and Cambridge structural database. J Chem Inf Model 50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawkins PCD, Skillman AG, Nicholls A. 2007. Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 23.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 24.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. 2006. Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 25.Ide K, Aoki M, Amano M, Koh Y, Yedidi RS, Das D, Leschenko S, Chapsal B, Ghosh AK, Mitsuya H. 2011. Novel HIV-1 protease inhibitors (PIs) containing a bicyclic P2 functional moiety, tetrahydropyrano-tetrahydrofuran, that are potent against multi-PI-resistant HIV-1 variants. Antimicrob Agents Chemother 55:1717–1727. doi: 10.1128/AAC.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berchiche YA, Chow KY, Lagane B, Leduc M, Percherancier Y, Fujii N, Tamamura H, Bachelerie F, Heveker N. 2007. Direct assessment of CXCR4 mutant conformations reveals complex link between receptor structure and Gαi activation. J Biol Chem 282:5111–5115. doi: 10.1074/jbc.C600270200. [DOI] [PubMed] [Google Scholar]
- 27.Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW. 2001. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem 276:14153–14160. doi: 10.1074/jbc.M704739200. [DOI] [PubMed] [Google Scholar]
- 28.Boulais PE, Dulude D, Cabana J, Heveker N, Escher E, Lavigne P, Leduc R. 2009. Photolabeling identifies transmembrane domain 4 of CXCR4 as a T140 binding site. Biochem Pharmacol 78:1382–1390. doi: 10.1016/j.bcp.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Rosenkilde MM, Gerlach LO, Hatse S, Skerlj RT, Schols D, Bridger GJ, Schwartz TW. 2007. Molecular mechanism of action of monocyclam versus bicyclam non-peptide antagonists in the CXCR4 chemokine receptor. J Biol Chem 282:27354–27365. doi: 10.1074/jbc.M704739200. [DOI] [PubMed] [Google Scholar]
- 30.Maeda K, Das D, Ogata-Aoki H, Nakata H, Miyakawa T, Tojo Y, Norman R, Takaoka Y, Ding JP, Arnold GF, Arnold E, Mitsuya H. 2006. Structural and molecular interactions of CCR5 inhibitors with CCR5. J Biol Chem 281:12688–12698. doi: 10.1074/jbc.M512688200. [DOI] [PubMed] [Google Scholar]
- 31.Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, Christopoulos A. 2007. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem 282:25677–25686. doi: 10.1074/jbc.M702311200. [DOI] [PubMed] [Google Scholar]
- 32.Wheatley M, Wootten D, Conner MT, Simms J, Kendrick R, Logan RT, Poyner DR, Barwell J. 2012. Lifting the lid on GPCRs: the role of extracellular loops. Br J Pharmacol 165:1688–1703. doi: 10.1111/j.1476-5381.2011.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brelot A, Heveker N, Montes M, Alizon M. 2000. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem 275:23736–23744. doi: 10.1074/jbc.M000776200. [DOI] [PubMed] [Google Scholar]
- 34.Nicholls A, McGaughey GB, Sheridan RP, Good AC, Warren G, Mathieu M, Muchmore SW, Brown SP, Grant JA, Haigh JA, Nevins N, Jain AN, Kelley B. 2010. Molecular shape and medicinal chemistry: a perspective. J Med Chem 53:3862–3886. doi: 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenkilde MM, Schwartz TW. 2006. GluVII:06—a highly conserved and selective anchor point for non-peptide ligands in chemokine receptors. Curr Top Med Chem 6:1319–1333. doi: 10.2174/15680266106061319. [DOI] [PubMed] [Google Scholar]
- 36.Nakata H, Steinberg SM, Koh Y, Maeda K, Takaoka Y, Tarnarnura H, Fujii N, Mitsuya H. 2008. Potent synergistic anti-human immunodeficiency virus (HIV) effects using combinations of the CCR5 inhibitor aplaviroc with other anti-HIV drugs. Antimicrob Agents Chemother 52:2111–2119. doi: 10.1128/AAC.01299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peled A, Wald O, Burger J. 2012. Development of novel CXCR4-based therapeutics. Expert Opin Investig Drugs 21:341–353. doi: 10.1517/13543784.2012.656197. [DOI] [PubMed] [Google Scholar]
- 38.Este JA, Cabrera C, Schols D, Cherepanov P, Gutierrez A, Witvrouw M, Pannecouque C, Debyser Z, Rando RF, Clotet B, Desmyter J, De Clercq E. 1998. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol Pharmacol 53:340–345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.