

Abstract
Drug resistance represents a key aspect of human immunodeficiency virus (HIV) treatment failure. It is important to develop nonhuman primate models for studying issues of drug resistance and the persistence and transmission of drug-resistant viruses. However, relatively little work has been conducted using either simian immunodeficiency virus (SIV) or SIV/HIV recombinant viruses for studying resistance against integrase strand transfer inhibitors (INSTIs). Here, we used a T-cell-tropic SIV/HIV recombinant virus in which the capsid and vif regions of HIV-1 were replaced with their SIV counterparts (simian-tropic HIV-1 [stHIV-1](SCA,SVIF)) to study the impact of a number of drug resistance substitutions in the integrase coding region at positions E92Q, G118R, E138K, Y143R, S153Y, N155H, and R263K on drug resistance, viral infectivity, and viral replication capacity. Our results show that each of these substitutions exerted effects that were similar to their effects in HIV-1. Substitutions associated with primary resistance against dolutegravir were more detrimental to stHIV-1(SCA,SVIF) infectiousness than were resistance substitutions associated with raltegravir and elvitegravir, consistent with data that have been reported for HIV-1. These findings support the role of stHIV-1(SCA,SVIF) as a useful model with which to evaluate the role of INSTI resistance substitutions on viral persistence, transmissibility, and pathogenesis in a nonhuman primate model.
INTRODUCTION
The advent of highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV) infection represents a major accomplishment of modern medicine. Despite this, however, the occurrence of drug resistance mutations (DRMs) can reduce viral susceptibility to antiretroviral drugs (ARVs). Cell culture experiments have often been predictive of HIV drug resistance pathways (1, 2), but the development and persistence of DRMs is also governed by complex pharmacologic, viral, and host factors (1, 3, 4). Although animal models may allow for the direct study of DRMs and their effects on treatment success (1), there is no model that re-creates all aspects of HIV-1 infection in humans (5). This notwithstanding, infection of macaques with the simian immunodeficiency virus (SIV) or the simian-human immunodeficiency virus (SHIV) has provided important insights into HIV pathogenesis. The construction of simian-tropic HIV-1 (stHIV-1), with an 88% sequence homology to HIV-1 has been accomplished (5). By replacing the HIV-1 capsid and vif regions with the corresponding regions from SIV (2, 5, 6), this HIV-based chimera (stHIV-1(SCA,SVIF)) is capable of infecting both human and macaque cell lines by evading TRIM5α (tripartite motif-containing protein 5 alpha) and APOBEC3G (apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G) restrictions (5).
Integrase strand transfer inhibitors (INSTIs) represent the most recent class of ARVs and are effective, minimally toxic, and tolerable (7). Until now, all HIV drugs have been susceptible to the development of DRMs that often follows a similar pattern, whereby primary substitutions, which confer resistance while reducing viral fitness, are followed by compensatory substitutions that restore fitness while further increasing levels of resistance. The INSTIs raltegravir (RAL) and elvitegravir (EVG) are compromised by substitutions at positions Y143, Q148, and N155 (8, 9) and additionally at position E92 for EVG alone (10). In contrast, resistance to the newer INSTI dolutegravir (DTG) is associated with a substitution at position R263K, and no compensatory substitution for R263K has yet been identified (11). The lack of such compensation may help to explain the absence of detectable emerging resistance substitutions in treatment-naive individuals failing DTG-based therapy (11–14).
The G118R substitution was reported to emerge during selection studies with DTG as well as with the experimental integrase inhibitor MK-2048 (15, 16). Although this substitution has not been subsequently validated in clinical settings with DTG, it has been reported in an individual failing treatment with RAL (17). Similarly, other substitutions that have been shown to emerge in tissue culture under DTG pressure, i.e., L101I, T124A, and S153Y/F, have not been observed in clinical studies (18).
Previous studies have shown that HIV-infected humans and SIV-infected macaques share similar patterns of drug resistance substitutions (1, 19). Furthermore, tissue culture studies have demonstrated that the same substitutions that confer resistance against INSTIs in HIV also do so in SIV. Given the high cost of animal-based research and the need for additional models for the study of drug resistance, there is a need to study other viruses as well in order to validate and extend findings previously obtained with either HIV or SIV.
The active site of the integrase coding sequence of HIV-1 is conserved in stHIV-1. Thus, it is reasonable to speculate that stHIV-1(SCA,SVIF) may be a suitable animal model for the study of INSTI DRMs, and this may help in understanding the impact of resistance substitutions on treatment decisions.
Using site-directed mutagenesis, we therefore introduced key DRMs against dolutegravir (DTG), raltegravir (RAL), and elvitegravir (EVG) into the integrase (IN) region of stHIV-1 to determine if resistance was similar to that seen in HIV. Our results demonstrate that DRMs against INSTIs decrease stHIV-1 infectiousness and confer degrees of resistance against these drugs that are comparable to those reported with HIV-1. The DTG-specific DRMs have a greater negative effect on infectiousness compared to DRMs against RAL and EVG.
MATERIALS AND METHODS
Cells and antiviral compounds.
For the purpose of infectivity and resistance experiments, TZM-bl cells were obtained through the National Institutes of Health AIDS Reagent Program from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc. (catalogue no. 8129). 293T cells were used for transfection with replication-competent wild-type or mutant stHIV-1 and were obtained from the American Type Culture Collection (CRL-11268). Both cell lines were subcultured every 3 to 4 days in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin and kept at 37°C under 5% CO2. Umbilical cord blood mononuclear cells (CBMCs) were isolated by Ficoll-Hypaque (GE Healthcare) gradient centrifugation from blood obtained through the Department of Obstetrics, Jewish General Hospital, Montréal, Canada. The CBMCs were cultured as previously described (20).The stHIV-1(SCA,SVIF) plasmid was provided by Theodora Hatziioannou of the Aaron Diamond Research Center, New York, New York. DTG, RAL, and EVG were provided by GlaxoSmithKline/ViiV Healthcare, Merck Inc., and Gilead Sciences, respectively.
Mutagenesis of the integrase region of stHIV-1.
The stHIV-1(SCA,SVIF) plasmid was constructed (5) and provided by Theodora Hatziioannou of the Aaron Diamond Research Center. The QuikChange II XL site-directed mutagenesis kit (Stratagene) was used to generate desired mutations in the integrase region of stHIV-1 using the following primers: E92Q (sense, 5′-GCAGAAGTAATTCCAGCACAGACAGGGCAAGAAA-3′; antisense, 5′-TTTCTTGCCCTGTCTGTGCTGGAATTACTTCTGC-3′), E138K (sense, 5′-GGCGGGGATCAAGCAGAAATTTGGCATTCCCTA-3′; antisense, 5′-TAGGGAATGCCAAATTTCTGCTTGATCCCCGCC-3′), G118R (sense, 5′-CCAGTAAAAACAGTACATACAGACAATCGCAGCAATTTCACC-3′; antisense, 5′-GGTGAAATTGCTGCGATTGTCTGTATGTACTGTTTTTACTGG-3′), N155H (sense, 5′-AAAGTCAGGGGGTAATAGAATCTATGCATAAAGAATTAAAGAAAATTATAGGAC-3′; antisense, 5′-GTCCTATAATTTTCTTTAATTCTTTATGCATAGATTCTATTACCCCCTGACTTT-3′), R263K (sense, 5′-GTAGTGCCAAGAAAAAAAGCAAAGATCATCAGGG-3′; antisense, 5′-CCCTGATGATCTTTGCTTTTTTTCTTGGCACTAC-3′), S153Y (sense, 5′-TTCTTTAATTCTTTATTCATATATTCTATTACTCCTTGACTTTGGGGATTGTAG-3′; antisense, 5′-CTACAATCCCCAAAGTCAAGGAGTAATAGAATATATGAATAAAGAATTAAAGAA-3′), and Y143R (sense, 5′-CAGGAATTTGGCATTCCCCGCAATCCCCAAAGTCAGGG-3′; antisense, 5′-CCCTGACTTTGGGGATTGCGGGGAATGCCAAATTCCTG-3′). Following mutagenesis, the plasmids were digested with DpnI for 4 h at 37°C and transformed using Escherichia coli strain XL10-Gold ultracompetent cells, TetrΔ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44thi-1 recA1 gyrA96 relA1 lac Hte (F′ proAB lacIqZΔM15Tn10 [Tetr] Amy Camr) (Stratagene). Plasmid purification was accomplished using the QIAprep MiniPrep kit (Qiagen) and quantified using NanoDrop technology. The presence of the mutations in the integrase region of stHIV-1(SCA,SVIF) was confirmed by sequencing.
Generation of replication-competent stHIV-1.
The production of genetically homogeneous stHIV-1 wild-type (WT) or mutated viruses was accomplished as previously described for pNL4.3 plasmids (n = 2 for each virus) (21). Briefly, 12.5 μg of WT and mutated stHIV-1 proviral DNA were transfected into 293T cells using Lipofectamine 2000 (Invitrogen). Fresh medium was added 4 h following transfection. The culture supernatants were harvested, centrifuged, and passed through a 0.45-μm filter at 48 h posttransfection. The transfected stHIV-1 viruses were then stored at −80°C for future use. Since HIV-1 pol is conserved in stHIV-1, reverse transcriptase (RT) activity was measured as previously described (20).
stHIV-1 infectiousness in TZM-bl cells.
Noncompetitive short-term infectivity assays with TZM-bl cells (n = 6) were used to determine the infectivity of the stHIV-1 wild-type and mutant viruses as previously described for HIV-1 (16). Briefly, 30,000 TZM-bl cells were seeded into 96-well culture plates (Corning) and infected with virus normalized on the basis of RT activity. The luciferase assay system (Promega) and a MicroBeta2 luminometer (PerkinElmer) were used to determine luciferase activity at 48 h postinfection. The open-source statistics package OpenEpi (http://www.openepi.com) was used to evaluate significance using Student's t test. A significant difference was defined as a P value of <0.001.
Susceptibility of stHIV-1 to INSTIs in TZM-bl cells.
Short-term resistance assays with TZM-bl cells were used to determine stHIV-1 susceptibility to DTG, RAL, and EVG as previously described for HIV-1 (n = 9 for DTG, n = 6 each for RAL and EVG) (16). In brief, 30,000 cells per well were infected with stHIV-1 WT virus or viruses containing the E92Q, G118R, E138K, Y143R, S153Y, N155H, or R263K substitution in the presence of serial dilutions of DTG, RAL, or EVG in 96-well plates (Corning). The amount of stHIV-1 WT or mutant virus added was normalized based on RT activity (40,000 cpm/well). At 48 h postinfection, luciferase activity was measured using the luciferase assay system (Promega) and a MicroBeta2 luminometer (PerkinElmer) and was used to generate data on fold changes in effective inhibitory concentrations (EC50s) using the sigmoid-dose response function of GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA).
Animal and blood collection.
Whole monkey blood (obtained from Primus Bio-Ressources, Inc., Vaudreuil-Dorion, Québec) was collected from uninfected rhesus macaques (Macaca mulatta) and delivered in BD Vacutainer heparin tubes (Becton, Dickinson and Company). Peripheral blood mononuclear cells (PBMCs) were isolated from donor monkeys by Ficoll-Hypaque (GE Healthcare) gradient centrifugation from rhesus blood. The rhesus PBMCs were cultured as previously described (22).
stHIV-1 replication capacity in rhesus macaque PBMCs.
Long-term infection assays were used to quantify the stHIV-1 replication capacity in rhesus macaque PBMCs by measuring the levels of RT activity (counts per minute [cpm]) over time. One million cells per well were added into a 48-well culture plate (Becton, Dickinson and Company) in 1 ml of RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 20 U/ml interleukin 2 (IL-2). The cells were infected with the indicated amount of virus normalized on the basis of RT activity (40,000 cpm/well, n = 3). Infection was quantified by RT activity as previously described (22).
stHIV-1 replication capacity in human CBMCs.
The CBMCs were subcultured as described for the rhesus PBMCs. Long-term infection of the CBMCs was similar to that described for the rhesus PBMCs. The culture wells were refreshed with volumes of medium equivalent to the volumes of supernatants that were removed. stHIV-1 replication capacity was measured by the quantification of RT activity in the cell culture fluids of the CBMCs infected with stHIV-WT or mutant viruses over 21 days.
RESULTS
Effect of integrase substitutions on stHIV-1(SCA,SVIF) infectivity.
Single-cycle infection assays with TZM-bl cells were used to determine the infectivity of stHIV-1 wild-type (WT) and mutated viruses. The mutant viruses included each of the following: stHIV-1(E92Q), stHIV-1(G118R), stHIV-1(E138K), stHIV-1(Y143R), stHIV-1(S153Y), stHIV-1(N155H), and stHIV-1(R263K). The introduction of the DRMs reduced the infectivity of all mutated viruses compared to WT (Fig. 1). All differences were statistically significant (Student's t test, P < 0.001). The levels of infectiousness were calculated and expressed relative to WT, which was arbitrarily set as 1 (Fig. 2). Introduction of the N155H substitution had the smallest effect on stHIV-1 infectivity (3.3-fold decrease in infectiousness compared to WT), whereas R263K had the greatest effect (27.5-fold decrease in infectiousness). Introduction of the E92Q, G118R, E138K, Y143R, and S153Y DRMs resulted in intermediate reductions in stHIV-1 infectiousness (7.4-, 15.4-, 4.8-, 7.7-, and 11-fold, respectively).
FIG 1.

Effects of integrase substitutions on stHIV-1(SCA,SVIF) infectivity in TZM-bl cells. StHIV-1 infectiousness was measured by quantifying luciferase activity in terms of relative luminescent units (RLU) produced by TZM-bl cells at 48 h after infection with increasing concentrations of virus as measured by RT activity (as counts per minute [cpm], x axis). Means and standard error of the means (SEM) are presented. Lines are fits.
FIG 2.
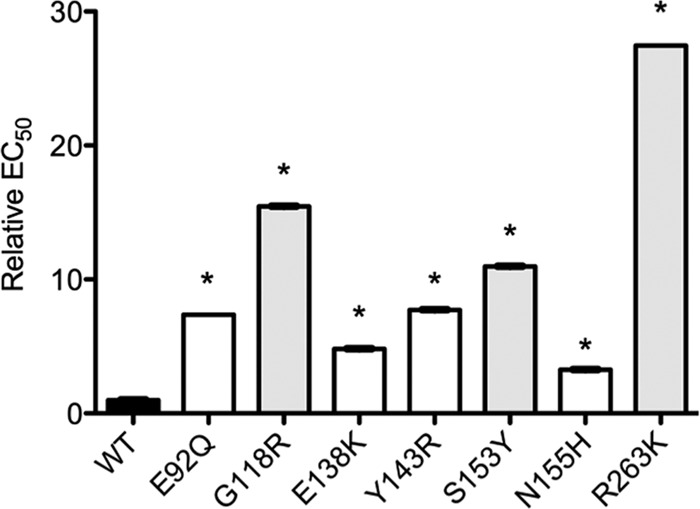
Impact of integrase substitutions on drug susceptibility. Relative half maximal effective virus concentrations (EC50s) were calculated and expressed relative to WT, which was arbitrarily set as 1. *, significant difference versus WT (Student's t test, P < 0.001).
Replication capacity of stHIV-1 mutant viruses in rhesus macaque PBMCs.
To determine whether the defect in infectiousness of the mutant viruses resulted in an impairment of stHIV-1 replication capacity, we measured the ability of these viruses to grow in rhesus macaque PBMCs over 24 days (Fig. 3). These experiments confirmed that all DRM viruses were impaired in replication capacity. Viruses bearing the G118R substitution were the most negatively affected. All substitutions that were tested significantly delayed early (<10 days) growth of stHIV-1. To exclude the possibility of cell-type-specific effects on viral growth, similar experiments were performed in the human CBMCs (Fig. 4). Similar to results in the macaque PBMCs, all DRM viruses negatively impacted stHIV-1 replication capacity in the CBMCs; in the macaque PBMCs, the greatest impact was observed with the G118R substitution. Overall, long-term replication studies in human CBMCs confirm the results obtained with the rhesus macaque PBMCs.
FIG 3.

Impact of integrase substitutions on stHIV-1 replication in rhesus macaque PBMCs. Replication was measured on the basis of RT activity in counts per minute (cpm) released by rhesus PBMCs into culture fluids following infection with stHIV-1 viruses. Error bars indicate means and SEM.
FIG 4.

Impact of integrase substitutions on stHIV-1 replication in human CBMCs. Reverse transcriptase (RT) activity was measured as counts per minute (cpm) in the culture fluids of CBMCs infected with stHIV viruses.
Susceptibility of stHIV-1 mutant viruses to INSTIs.
Previous studies with HIV revealed that the insertion of DRMs into the integrase region of pNL4.3 viruses resulted in reduced susceptibility to currently available INSTIs. In order to compare the levels of resistance between HIV and stHIV-1, we evaluated the replication capacity of mutated stHIV-1 viruses in the presence of increasing concentrations of RAL, EVG, or DTG using TZM-bl cells. Resistance to RAL was most prominent with the viruses containing E92Q, N155H, and Y143R, displaying increases in EC50s of 11.5-, 14.7-, and 35.9-fold, respectively, compared to the WT (Table 1 ). Viruses containing G118R conferred low-level resistance to RAL (5.8-fold), whereas the E138K, R263K, and S153Y viruses did not significantly confer resistance to this drug (0.41-, 0.83-, and 1.59-fold, respectively). All mutated stHIV-1 viruses conferred moderate to high levels of resistance to EVG, ranging from 16.6-fold for E138K to >300-fold for E92Q. With respect to DTG, the mutated viruses were associated with low levels of resistance, with viruses containing G118R conferring the highest level of resistance (6.4-fold). The R263K substitution did not confer significant levels of resistance against RAL but did confer low- and moderate-level resistance, respectively, to DTG (2.5-fold) and EVG (32.6-fold).
TABLE 1.
Effects of drug resistance substitutions on EC50s for DTG, RAL, and EVGa
| Genotype | Fold change EC50 (mean ± SEM) for: |
||
|---|---|---|---|
| DTG | RAL | EVG | |
| stHIV-1 | |||
| WT | 1 ± 0.1 | 1 ± 0.5 | 1 ± 0.6 |
| E92Q | 1.94 ± 0.3 | 11.5 ± 2 | >300 ± 7.6 |
| G118R | 6.44 ± 0.1 | 5.8 ± 0.6 | 20.1 ± 0.3 |
| E138K | 0.74 ± 0.4 | 0.41 ± 0.4 | 16.6 ± 0.3 |
| Y143R | 2.06 ± 0.1 | 35.9 ± 0.6 | 21.6 ± 0.2 |
| S153Y | 2.15 ± 0.2 | 1.59 ± 0.6 | 22.8 ± 0.4 |
| N155H | 0.76 ± 0.3 | 14.7 ± 0.6 | 55.8 ± 0.3 |
| R263K | 2.5 ± 0.3 | 0.83 ± 1.1 | 32.6 ± 0.4 |
| HIV-1 | |||
| WT | 1 | 1 | 1 |
| E92Q | 1.6b | 3.5b | >100c |
| G118R | 8.2d | 1d | 1d |
| E138K | 0.97b | 1b | 0.93b |
| Y143R | 1.4b | 16b | 2.3c |
| S153Y | 2.5b | 1.3b | 2.3b |
| N155H | 0.99b | 8.4b | 25b |
| R263K | 2.3e | 1e | 21.8e |
DISCUSSION
In an attempt to establish a model to study the effects of DRMs against INSTIs on viral replicative capacity and resistance, we introduced relevant resistance-associated substitutions into stHIV-1(SCA,SVIF). We had earlier performed similar studies with SIVmac239 and showed that SIV and HIV-1 share similar resistance profiles (22). The present study shows that stHIV-1 and HIV-1 also share similarities in regard to the impact of DRMs on resistance against INSTIs and on viral replicative capacity. Our study is important, because it expands the number of viral models that might be used to assess retroviral pathogenesis and antiviral effects in animal systems.
Our results show that stHIV-1 and HIV-1 share similar resistance pathway profiles (Table 1). As an example, the E92Q substitution, which has been shown to be prevalent in patients failing EVG-based therapy (23–28), was associated with the highest level of resistance against this drug when introduced into stHIV-1. In agreement with our studies on the role of the G118R substitution in HIV-1 (29), this substitution was the only one that conferred moderate-level resistance in stHIV-1 against DTG (change, 6.4-fold). This is in agreement with observations on the robustness of this drug against most resistance substitutions (7, 30). Similarly, the R263K substitution in integrase resulted in low-level resistance against DTG in stHIV-1 (Table 1) (16, 31). In contrast, introduction of the E138K substitution into stHIV-1 conferred 16.6-fold resistance against EVG, whereas this substitution had been shown to be innocuous against this drug in HIV-1 (18, 32). The resistance levels observed for stHIV-1 and HIV-1 were different for only some drugs. In particular, S153Y, which was observed during tissue culture selection experiments with DTG (16, 18) displayed similar levels of resistance against DTG in stHIV-1 and HIV, i.e., fold changes of 2.2- and 2.5-fold, respectively (Table 1), whereas S153Y conferred high-level resistance against EVG in stHIV-1 (22.8-fold) and low-level resistance against this same drug in HIV-1 (4- to 8-fold) (33, 34). Methodological differences may explain the observed disparities in resistance. However, given the similarities between stHIV(SCA,SVIF) and HIV-1, one cannot exclude the possibility that sequence variability in capsid (CA) coding regions may also contribute to the observed disparities. Although integrase and CA are not known to directly interact, the latter protein has been shown to contribute to the nuclear import of viral DNA (reviewed in reference 35) and may thus be indirectly important for integrase catalytic activity and integration. Since integrase also contributes to the nuclear import of the viral genome, viruses with integrase proteins that are deficient for DNA binding, such as R263K (16), may rely more on CA for nuclear import than viruses with WT integrase sequences.
The emergence of DRMs in vivo is contingent on the cost on viral replication capacity imparted by these substitutions (36). One example is the sequential emergence of the M184I and M184V resistance substitutions in individuals failing 3TC-based therapy (20, 37). Recently, our results suggested that the high replicative cost associated with the DTG-specific R263K resistance pathway could help to explain the rarity of DTG-resistance substitutions in treatment-naive individuals (11, 21, 30, 31). Therefore, we studied the impact of DRMs against INSTIs on viral infectiousness and replication capacity (Fig. 3). Our results show that stHIV-1 containing R263K reproduced the decrease in HIV-1 replicative capacity that has been reported with R263K-containing HIV-1. In particular, the G118R and R263K DTG-specific substitutions were the most detrimental to stHIV-1 viral infectivity (Fig. 1). S153Y was also associated with a high degree of impairment of viral replication. RAL- and EVG-specific DRMs, i.e., E92Q, E138K, Y143R, and N155H, were less detrimental to stHIV-1 replication capacity than were R263K and G118R, and N155H was the least disadvantageous for viral infectiousness.
The HIV-based chimeric virus stHIV-1 was derived by replacing the capsid and vif regions of HIV-1 with the corresponding regions of SIV and can infect both human primary cells and cell lines (5). We have now further investigated this by examining the impact of DRMs against INSTIs on the replication capacity of stHIV in human CBMCs. We found that the replication profiles of stHIV in CBMCs are similar to those seen in rhesus PBMCs (Fig. 3, 4). The G118R DTG-specific substitution was the most detrimental to stHIV-1 viral infectivity in CBMCs (Fig. 4), in agreement with what has been reported for G118R-containing HIV-1 and SIV (15, 22). stHIV-1 viruses bearing the Y143R substitution also displayed reduced replication capacity, as seen in HIV-1 and SIV (22, 38, 39). Similar to its effects in HIV-1 (32), E138K modestly diminished stHIV replication in the rhesus PBMCs and the human CBMCs (Fig. 3 and 4). Similarly, HIV-1 viruses bearing E92Q and N155H were modestly impaired in replication capacity. Importantly, the correlation between the results obtained with the human CBMCs and the macaque PBMCs suggests that potential reversions in the integrase region of the stHIV-1 variants did not influence the outcome of these experiments.
Together, this study demonstrates that DRMs in stHIV-1 resemble HIV-1 resistance substitutions in regard to diminished drug efficacy and viral replication defects associated with these substitutions. This suggests that stHIV-1 can be used to study the impact of DRMs against INSTIs on viral replicative capacity and expands the number of viral systems that can be used in animals to study the persistence, transmissibility, and pathogenicity of INSTI-resistant viruses. Hopefully, our findings will have relevance for the study of stHIV variants that display resistance against other classes of ARVs as well.
ACKNOWLEDGMENTS
We thank Theodora Hatziioannou of the Aaron Diamond AIDS Research Center, New York, for providing us with the stHIV-1 plasmid. We thank Maureen Oliveira for help with the rhesus PBMC isolation and activation.
S.H. is the recipient of a doctoral studentship from the Fonds de la Recherche du Québec en Santé (FRQS). This work was supported by the Canadian Institutes for Health Research.
REFERENCES
- 1.Van Rompay KK. 2010. Evaluation of antiretrovirals in animal models of HIV infection. Antiviral Res 85:159–175. doi: 10.1016/j.antiviral.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Na L, Tang YD, Liu JD, Yu CQ, Sun LK, Lin YZ, Wang XF, Wang X, Zhou JH. 2014. TRIMe7-CypA, an alternative splicing isoform of TRIMCyp in rhesus macaque, negatively modulates TRIM5alpha activity. Biochem Biophys Res Commun 446:470–474. doi: 10.1016/j.bbrc.2014.02.132. [DOI] [PubMed] [Google Scholar]
- 3.Borda JT, Alvarez X, Kondova I, Aye P, Simon MA, Desrosiers RC, Lackner AA. 2004. Cell tropism of simian immunodeficiency virus in culture is not predictive of in vivo tropism or pathogenesis. Am J Pathol 165:2111–2122. doi: 10.1016/S0002-9440(10)63261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohman BL, McChesney MB, Miller CJ, McGowan E, Joye SM, Van Rompay KK, Reay E, Antipa L, Pedersen NC, Marthas ML. 1994. A partially attenuated simian immunodeficiency virus induces host immunity that correlates with resistance to pathogenic virus challenge. J Virol 68:7021–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatziioannou T, Princiotta M, Piatak M Jr, Yuan F, Zhang F, Lifson JD, Bieniasz PD. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. doi: 10.1126/science.1130994. [DOI] [PubMed] [Google Scholar]
- 6.Kamada K, Igarashi T, Martin MA, Khamsri B, Hatcho K, Yamashita T, Fujita M, Uchiyama T, Adachi A. 2006. Generation of HIV-1 derivatives that productively infect macaque monkey lymphoid cells. Proc Natl Acad Sci U S A 103:16959–16964. doi: 10.1073/pnas.0608289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesplède T, Quashie PK, Zanichelli V, Wainberg MA. 2014. Integrase strand transfer inhibitors in the management of HIV-positive individuals. Ann Med 46:123–129. doi: 10.3109/07853890.2014.883169. [DOI] [PubMed] [Google Scholar]
- 8.Mouscadet JF, Delelis O, Marcelin AG, Tchertanov L. 2010. Resistance to HIV-1 integrase inhibitors: a structural perspective. Drug Resist Updat 13:139–150. doi: 10.1016/j.drup.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Huang W, Frantzell A, Fransen S, Petropoulos CJ. 2013. Multiple genetic pathways involving amino acid position 143 of HIV-1 integrase are preferentially associated with specific secondary amino acid substitutions and confer resistance to raltegravir and cross-resistance to elvitegravir. Antimicrob Agents Chemother 57:4105–4113. doi: 10.1128/AAC.00204-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quashie PK, Mesplède T, Wainberg MA. 2013. Evolution of HIV integrase resistance mutations. Curr Opin Infect Dis 26:43–49. [DOI] [PubMed] [Google Scholar]
- 11.Wainberg MA, Mesplède T, Raffi F. 2013. What if HIV were unable to develop resistance against a new therapeutic agent? BMC Med 11:249. doi: 10.1186/1741-7015-11-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walmsley SL, Antela A, Clumeck N, Duiculescu D, Eberhard A, Gutierrez F, Hocqueloux L, Maggiolo F, Sandkovsky U, Granier C, Pappa K, Wynne B, Min S, Nichols G. 2013. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med 369:1807–1818. doi: 10.1056/NEJMoa1215541. [DOI] [PubMed] [Google Scholar]
- 13.Raffi F, Rachlis A, Stellbrink HJ, Hardy WD, Torti C, Orkin C, Bloch M, Podzamczer D, Pokrovsky V, Pulido F, Almond S, Margolis D, Brennan C, Min S. 2013. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, noninferiority SPRING-2 study. Lancet 381:735–743. doi: 10.1016/S0140-6736(12)61853-4. [DOI] [PubMed] [Google Scholar]
- 14.Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, Baril JG, Domingo P, Brennan C, Almond S, Min S. 2013. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, noninferiority trial. Lancet Infect Dis 13:927–935. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PubMed] [Google Scholar]
- 15.Bar-Magen T, Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, Oliveira M, Hazuda DJ, Wainberg MA. 2010. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol 84:9210–9216. doi: 10.1128/JVI.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quashie PK, Mesplède T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malet I, Fourati S, Charpentier C, Morand-Joubert L, Armenia D, Wirden M, Sayon S, Van Houtte M, Ceccherini-Silberstein F, Brun-Vezinet F, Perno CF, Descamps D, Capt A, Calvez V, Marcelin AG. 2011. The HIV-1 integrase G118R mutation confers raltegravir resistance to the CRF02_AG HIV-1 subtype. J Antimicrob Chemother 66:2827–2830. doi: 10.1093/jac/dkr389. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother 55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. 2004. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 20.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol 85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wares M, Mesplède T, Quashie PK, Osman N, Han Y, Wainberg MA. 2014. The M50I polymorphic substitution in association with the R263K mutation in HIV-1 subtype B integrase increases drug resistance but does not restore viral replicative fitness. Retrovirology 11:7. doi: 10.1186/1742-4690-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassounah SA, Mesplède T, Quashie PK, Oliveira M, Sandstrom PA, Wainberg MA. 2014. Effect of HIV-1 integrase resistance mutations when introduced into SIVmac239 on susceptibility to integrase strand transfer inhibitors. J Virol 88:9683–9692. doi: 10.1128/JVI.00947-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molina JM, Lamarca A, Andrade-Villanueva J, Clotet B, Clumeck N, Liu YP, Zhong L, Margot N, Cheng AK, Chuck SL. 2012. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, noninferiority study. Lancet Infect Dis 12:27–35. doi: 10.1016/S1473-3099(11)70249-3. [DOI] [PubMed] [Google Scholar]
- 24.DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, Wei X, Yale K, Szwarcberg J, White K, Cheng AK, Kearney BP. 2012. Coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, noninferiority trial. Lancet 379:2429–2438. doi: 10.1016/S0140-6736(12)60918-0. [DOI] [PubMed] [Google Scholar]
- 25.Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, Gallant JE, Liu HC, Zhong L, Yale K, White K, Kearney BP, Szwarcberg J, Quirk E, Cheng AK. 2012. Coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus coformulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 379:2439–2448. doi: 10.1016/S0140-6736(12)60917-9. [DOI] [PubMed] [Google Scholar]
- 26.Rockstroh JK, DeJesus E, Henry K, Molina JM, Gathe J, Ramanathan S, Wei X, Plummer A, Abram M, Cheng AK, Fordyce MW, Szwarcberg J. 2013. A randomized, double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir DF versus ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr 62:483–486. doi: 10.1097/QAI.0b013e318286415c. [DOI] [PubMed] [Google Scholar]
- 27.Zolopa A, Sax PE, DeJesus E, Mills A, Cohen C, Wohl D, Gallant JE, Liu HC, Plummer A, White KL, Cheng AK, Rhee MS, Szwarcberg J. 2013. A randomized double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr 63:96–100. doi: 10.1097/QAI.0b013e318289545c. [DOI] [PubMed] [Google Scholar]
- 28.Elion R, Molina JM, Ramon Arribas Lopez J, Cooper D, Maggiolo F, Wilkins E, Conway B, Liu YP, Margot N, Rhee M, Chuck SL, Szwarcberg J. 2013. A randomized phase 3 study comparing once-daily elvitegravir with twice-daily raltegravir in treatment-experienced subjects with HIV-1 infection: 96-week results. J Acquir Immune Defic Syndr 63:494–497. doi: 10.1097/QAI.0b013e318298469c. [DOI] [PubMed] [Google Scholar]
- 29.Quashie PK, Mesplède T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase. Antimicrob Agents Chemother 57:6223–6235. doi: 10.1128/AAC.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mesplède T, Wainberg MA. 2014. Is resistance to dolutegravir possible when this drug is used in first-line therapy? Viruses 6:3377–3385. doi: 10.3390/v6093377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mesplède T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, Petropoulos CJ, Huang W, Wainberg MA. 2013. Viral fitness cost prevents HIV-1 from evading dolutegravir drug pressure. Retrovirology 10:22. doi: 10.1186/1742-4690-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesplède T, Osman N, Wares M, Quashie PK, Hassounah S, Anstett K, Han Y, Singhroy DN, Wainberg MA. 2014. Addition of E138K to R263K in HIV integrase increases resistance to dolutegravir, but fails to restore activity of the HIV integrase enzyme and viral replication capacity. J Antimicrob Chemother 69:2733–2740. doi: 10.1093/jac/dku199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. 2008. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, White KL, Miller MD. 2012. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antiviral Res 93:288–296. doi: 10.1016/j.antiviral.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Le Sage V, Mouland AJ, Valiente-Echeverria F. 2014. Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res 193:116–129. doi: 10.1016/j.virusres.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Wainberg MA, Zaharatos GJ, Brenner BG. 2011. Development of antiretroviral drug resistance. N Engl J Med 365:637–646. doi: 10.1056/NEJMra1004180. [DOI] [PubMed] [Google Scholar]
- 37.Hu Z, Kuritzkes DR. 2011. Interaction of reverse transcriptase (RT) mutations conferring resistance to lamivudine and etravirine: effects on fitness and RT activity of human immunodeficiency virus type 1. J Virol 85:11309–11314. doi: 10.1128/JVI.05578-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delelis O, Thierry S, Subra F, Simon F, Malet I, Alloui C, Sayon S, Calvez V, Deprez E, Marcelin AG, Tchertanov L, Mouscadet JF. 2010. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob Agents Chemother 54:491–501. doi: 10.1128/AAC.01075-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mbisa JL, Martin SA, Cane PA. 2011. Patterns of resistance development with integrase inhibitors in HIV. Infect Drug Resist 4:65–76. doi: 10.2147/IDR.S7775. [DOI] [PMC free article] [PubMed] [Google Scholar]