

Abstract
Naegleria fowleri is a pathogenic free-living amoeba (FLA) that causes an acute fatal disease known as primary amoebic meningoencephalitis (PAM). The major problem for infections with any pathogenic FLA is a lack of effective therapeutics, since PAM has a case mortality rate approaching 99%. Clearly, new drugs that are potent and have rapid onset of action are needed to enhance the treatment regimens for PAM. Diamidines have demonstrated potency against multiple pathogens, including FLA, and are known to cross the blood-brain barrier to cure other protozoan diseases of the central nervous system. Therefore, amidino derivatives serve as an important chemotype for discovery of new drugs. In this study, we validated two new in vitro assays suitable for medium- or high-throughput drug discovery and used these for N. fowleri. We next screened over 150 amidino derivatives of multiple structural classes and identified two hit series with nM potency that are suitable for further lead optimization as new drugs for this neglected disease. These include both mono- and diamidino derivatives, with the most potent compound (DB173) having a 50% inhibitory concentration (IC50) of 177 nM. Similarly, we identified 10 additional analogues with IC50s of <1 μM, with many of these having reasonable selectivity indices. The most potent hits were >500 times more potent than pentamidine. In summary, the mono- and diamidino derivatives offer potential for lead optimization to develop new drugs to treat central nervous system infections with N. fowleri.
INTRODUCTION
The first report of animal pathogenicity by free-living amoebae (FLA) was by Culbertson et al., who identified Acanthamoeba in necrotic lesions of monkeys and mice (1). He later suggested that FLA causes similar disease in humans. Fowler and Carter reported the first cases of an acute fatal disease caused by Naegleria fowleri in four victims in Australia in 1965 (2). Since the identification of primary amoebic meningoencephalitis (PAM), hundreds of cases have been reported worldwide, including 142 cases in the United States (3–5). Most infections with N. fowleri occur during the summer months, victims usually are symptomatic within 5 days, and the disease is almost always fatal. Infection usually occurs after the victim swam in warm, fresh, or brackish water or from exposure to contaminated tap water associated with the use of Neti pots for nasal irrigation (4–7). Amoebic invasion occurs via disruption of the olfactory mucosa, penetration of organisms into the submucosal nervous plexus, and ultimately passage through the cribriform plate to the frontal lobes of the brain (8, 9). Although most cases of PAM occur in warm climates, such as the southern tier states of the United States, pathogenic amoeba found in thermally polluted water sources, hot springs, and poorly chlorinated pools have been linked to infections (10, 11). Interestingly, most of the PAM cases have been diagnosed in developed countries, including the United States, Australia, and Europe. Although sporadic cases have been diagnosed in tropical regions of the world, it is likely that many cases in these areas go undetected or are misdiagnosed as bacterial or viral meningitis (5, 6). Recent evidence to support this hypothesis includes the large number of cases reported from Pakistan that appear to be related to poor chlorination of water used for ablution, a ritual cleansing that includes nasal passages (6). The prevalence of FLA in warm climates, the advent of global warming, and the use of immunosuppressive drugs combine to threaten expansion of the spatial distribution of pathogenic FLA with an increased incidence of disease.
The major problem for infections with any of the pathogenic FLA is the lack of effective therapeutics (5, 12, 13). PAM has a case fatality rate approaching 99%, even if the infection is diagnosed promptly and treated with the best available drug regimens. For N. fowleri infections in the United States, only three (3) cases have been successfully treated; all other documented cases were fatal (5). Treatment of PAM is empirical and based upon the first few cases of successful treatment. Amphotericin B is the drug of choice for PAM and is administered intravenously and intrathecally, usually in combination with rifampin, miltefosine, other antibiotics, or antifungals. A broader array of drugs has been used to treat systemic infections with Acanthamoeba spp. (12). For central nervous system (CNS), nasopharyngeal, and disseminated infections, a variety of azole antifungals (e.g., ketoconazole and itraconazole), pentamidine, and cotrimoxazole are used. Despite the slower, more insidious onset of disease in systemic Acanthamoeba infections, the outcome of systemic Acanthamoeba infections is death in these (usually) immunocompromised patients. Substantially more data are available on treatment of amoebic keratitis than any other FLA infection. Most of the regimens include the topical use of microbicides (chlorhexidine, polyhexamethylene biguanide [PHMB], and hexamidine) with or without neomycin, azoles, or antibiotics. Despite moderate success with chlorhexidine and PHMB for amoebic keratitis, both granulomatous amoebic encephalitis (GAE) and keratitis are difficult to treat due to the presence and persistence of both trophozoites and cysts of Acanthamoeba spp. Cysts are refractory to most drugs used to treat CNS infections; therefore, prolonged therapy is required due to this poor efficacy, prompting concerns for the emergence of drug resistance. Clearly, new drugs that can be administered parenterally for neurological disease or topically for keratitis are urgently needed.
Pentamidine and propamidine are diamidines with demonstrated potency against several pathogens, including pathogenic FLA (11, 12). Both of these are reported to be active against trophozoites of Naegleria fowleri and Acanthamoeba spp. in vitro (11, 14), and pentamidine has been used clinically in combination with other drugs for FLA infections. Brolene, a topical formulation of propamidine isothionate plus neomycin, is the first-line treatment for amoebic keratitis in Europe (47). These diamidino derivatives suffer from poor bioavailability, narrow therapeutic indices, and poor selectivity, yet they serve to validate the potential utility of the amidino chemotypes as anti-FLA drugs. In the past decade, an extensive series of amidino derivatives have been synthesized and assessed for activity as antiparasitic and antimicrobial agents. These include more than 2,000 derivatives that include dozens of structural classes with various linkers, prodrug moieties, and physiochemical properties. (15–31). Many of these compounds have potency in vitro and in vivo against a variety of parasitic protozoa, including Leishmania spp., Plasmodium falciparum, Trypanosoma cruzi, and T. brucei. Recent data demonstrate diamidines cross the blood-brain barrier (BBB) and have efficacy in late-stage animal models of human African trypanosomiasis (HAT) (32). Several of these pyridyl diamidine derivatives achieve steady-state levels of 10 to 40 μM in brain of rodents dosed orally, and in recent studies DB820 cured Vervet monkeys of late-stage HAT disease (15, 33). DB844, a prodrug that is rapidly converted to DB820, dosed at 5 mg/kg of body weight once a day for 5 days, cured monkeys infected with T. brucei rodiesense in the late-stage disease model (34). In yet another model of CNS disease, two arylimidamides were active in BALB/c mice infected with the apicomplexan parasite Neospora caninum and significantly reduced the cerebral parasite burden (35). Given the demonstrated susceptibility of FLA to diamidines and efficacy for CNS protozoal diseases, the amidino analogs are a very promising series for discovery of a new drug for the most severe forms of FLA-induced disease.
The focus of our work is to discover and develop a new drug to treat the usually fatal CNS infections caused by N. fowleri and Acanthamoeba spp. Here, we report the development and validation of new in vitro drug susceptibility assays for N. fowleri; we then used these models to screen over 150 amidino derivatives of multiple structural classes and have identified two hit series with nM potency that are suitable for further lead optimization as new drugs for these neglected diseases.
MATERIALS AND METHODS
Amoeba culture.
We used an isolate of Naegleria fowleri (ATCC 30215) originally collected from a child that died of PAM in South Australia in 1969. Trophozoites were grown axenically at 34°C in Nelsons complete medium (NCM). NCM is composed of 0.17% liver infusion broth (BD, Sparks, MD), 0.17% d-(+)-glucose, 0.0012% sodium chloride, 0.00136% potassium phosphate monobasic, 0.00142% sodium phosphate dibasic, 0.0004% calcium chloride, 0.0002% magnesium sulfate, supplemented with 10% fetal bovine serum (FBS) and 125 μg of penicillin-streptomycin. All reagents were obtained from Sigma-Aldrich (St. Louis, MO), and all experiments were performed using logarithmic-phase trophozoites.
Development of in vitro drug susceptibility assays.
Most previous drug susceptibility assays for N. fowleri are not amenable for medium or high throughput due to requirements for large culture volumes, long incubation periods with drug, and assay endpoints not easily optimized for automated, quantitative assessment. Therefore, we first assessed the potential for new in vitro assays with the goal of validating assays that could run in 96- and 384-well formats, which require no longer than 72 h of drug exposure and provided robust, quantitative endpoints for the assessment of drug response. The alamarBlue colorimetric microtiter plate assay was described first by McBride et al. for Acanthamoeba species (36) but has not been used previously for N. fowleri. The assay is based upon the reduction of resazurin, a nonfluorescent dye, to resorufin by metabolically active cells. The second assay we used was the CellTiter-Glo (CTG) 2.0 luminescent cell viability plate assay that was first used with amoeba by Debnath et al. to screen for compounds active against nonpathogenic Naegleria gruberi (37). The CTG assay is a quantitative assay that assesses the presence of ATP in lysed cells.
Since neither of these assays had been used with N. fowleri previously, we first established the optimal seeding density and length of assay (hours) for each assay in both 96- and 384-well microtiter plate formats. In brief, trophozoites of N. fowleri grown in NCM were seeded in triplicate at concentrations ranging from 1 × 105 cells/ml to 2 × 106 cells/ml for 96- and 384-well plates to determine the concentration of cells that best reduce alamarBlue at 24, 48, 72, and 96 h (see Fig. S1 in the supplemental material). From these studies, the alamarBlue assay period was established at 72 h with the reagent added at 48 h. The optimal seeding density for the 72-h assays was determined to be 100,000 cells/well and 3,000 cells/well for 96- and 384-well formats, respectively. We similarly optimized the seeding density for 72 h in the CTG assay, determining 4,000 and 3,000 cells/well for 96- and 384-well plate formats, respectively (see Fig. S1).
In vitro drug susceptibility methods.
All compounds were prepared as 5 mg/ml stock solutions in dimethylsulfoxide (DMSO). A Biomek 3000 automated liquid-handling workstation (Beckman Coulter) was used to serially dilute the compounds in 2-fold dilutions 6 or 11 times, yielding a concentration range of 24 ng/ml to 50 μg/ml in NCM with a final concentration of 1% DMSO. The Biomek workstation was used to transfer 10 μl or 6 μl of diluted compounds, followed by the addition of 100,000 or 3,000 N. fowleri trophozoites/well in 96- or 384-well screening plates, respectively, for alamarBlue colorimetric assays. For the CTG assays, 4,000 or 3,000 cells/well were used in white 96- or 384-well plates. The total volume for 96-well plates was 100 μl, and 60 μl was used for both 384-well screening plates. Positive-growth control wells in the screening plates contained N. fowleri trophozoites in NCM, and negative-growth control wells contained N. fowleri trophozoites in the presence of 135 μM amphotericin B (Sigma, St. Louis, MO). All assay plates were incubated for 72 h at 34°C. For the alamarBlue assays, 10% of the total well volume of reagent was added 24 h before the end of incubation, and the plates were incubated a final 24 h in complete darkness. At the end of incubation, all CellTiter-Glo 2.0 luminescent cell viability assay white plates were equilibrated to room temperature for 10 min, and 25 μl or 15 μl of CellTiter-Glo 2.0 reagent (Promega, Madison, WI) was added to all wells of the 96-well and 384-well plates, respectively, using a Biomek 3000 workstation. The plates were placed on an orbital shaker at room temperature for 2 min to induce cell lysis. After shaking, the plates were equilibrated at room temperature for 10 min to stabilize the luminescent signal. The resulting absorbance from the alamarBlue assay was determined at 570 nm and 600 nm using a Spectra Max M2é (Molecular Devices, Sunnyvale, CA). The ATP bioluminescence signal was measured at 490 nm using a Spectra Max L (Molecular Devices, Sunnyvale, CA). Curve fitting using nonlinear regression was carried out using DataAspects Plate Manager analysis software to determine 50 and 90% inhibitory concentrations (IC50 and IC90).
Cytotoxicity.
The cytotoxicity of compounds for J774 murine macrophages was determined by using the CellTiter 96 AQueous one-solution cell proliferation assay (Promega, Madison, WI). In brief, J774 cells were seeded at 5 × 105 cells/well in a 96-well tissue culture plate (Corning, NY) in the presence of the serial dilutions of active drugs against N. fowleri. Positive-control wells contained cells and media; negative-control wells contained media alone. Cells were grown in RPMI 1640 at a pH of 7.2 supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (all supplied by Invitrogen Corp., Carlsbad, CA). The inhibitor concentration started at 50 μg/ml and was diluted in doubling dilutions to assess cytotoxicity compared to that of Naegleria fowleri. The total volume of each well was 100 μl, and plates were incubated at 37°C, 5% CO2 for 72 h. Four h before the assessment time point, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Promega, Madison, WI) was added to each well. Inhibition of J774 growth was assessed at the 72-h time point, measuring the optical density (OD) values determined at 490 nm using a SpectraMax M2e (Molecular Devices, Sunnyvale, CA). Curve fitting using nonlinear regression was carried out using DataAspects Plate Manager analysis software to obtain IC50 and IC90 values.
Compounds.
Miltefosine was purchased from Cayman, whereas other standard drugs were obtained from Sigma-Aldrich. The amidino derivatives studied have been reported previously, and their purity was verified by elemental analysis and nuclear magnetic resonance spectroscopy. Representative references to their synthesis are the following: type I (29, 38), type II (23 to 25), type III (20), type IV (21), type V (26), and type VI (17). Hexamidine and octamidine were synthesized by using established methods.
RESULTS
Validation of drug susceptibility assays.
Two drug assays, alamarBlue and CTG, were assessed in both 96- and 384-well plate formats to enable medium- and high-throughput applications for N. fowleri drug discovery. Since the overall goal of our efforts was to discover new drugs that are potent and have a rapid onset of action, we validated the assays with 72 h of drug exposure. Quantitative dose-response data in the alamarBlue and CTG assays for standard drugs as well as new amidino inhibitors were similar regardless of the method used (Fig. 1; also see Fig. S1 in the supplemental material). Compared with previous methods, the efficacies of amphotericin B, pentamidine, and miltefosine in the alamarBlue and CTG assays were reduced (Table 1) yet consistent between the two new methods and in 96- or 384-well format.
FIG 1.
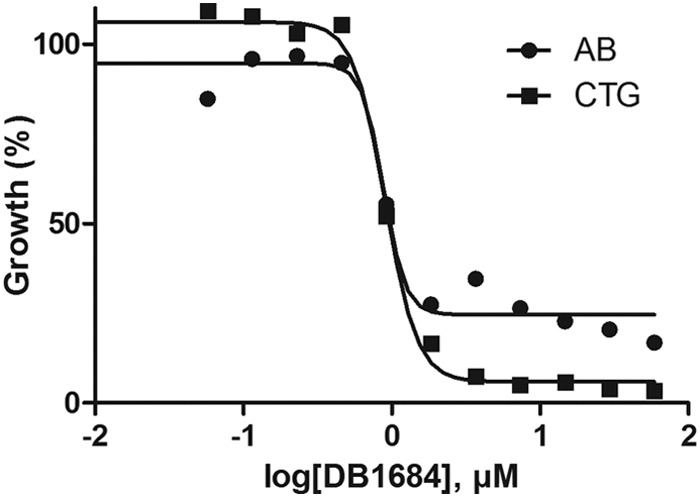
Quantitative dose-response data for Naegleria fowleri demonstrating similar activities of amidino compounds in the alamarBlue (AB) and CellTiter-Glo (CTG) assay formats.
TABLE 1.
Susceptibility of Naegleria fowleri to amphotericin B, miltefosine, and diamidines in vitro
| Drug and isolate | MIC (μM) | IC50 (μM) | Assay duration | Medium | Assay volume (ml) | No. of amoebas/ml | Assay endpoint | Reference or source |
|---|---|---|---|---|---|---|---|---|
| Amphotericin B | ||||||||
| Lee (M67) | 0.11 | 7 days | Mix | 30 | 104 | Cell counts | 45 | |
| ATCC 30808 | 0.84 | 60 h | Bacto Casitone | 40 | 25 × 106 | Cell counts | 11 | |
| ATCC 30215 | 1.6 | 2, 4, 6 days | NCM | 0.1 | 104 | Lactate dehydrogenase release | 44 | |
| ATCC 30215 | 47 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study | |
| Miltefosine | ||||||||
| CDC V511 | 40 | 1–4 days | NCM | 10 | 9.6 × 104 | Viability on recovery | 42 | |
| ATCC 30215 | 62 | 2, 4, 6 days | NCM | 0.1 | 104 | Lactate dehydrogenase release | 44 | |
| ATCC 30215 | >122.7 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study | |
| Pentamidine | ||||||||
| ATCC 30808 | >54 | 60 h | Bacto Casitone | 40 | 25 × 106 | Cell counts | 11 | |
| ATCC 30215 | >84.4 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study | |
| Propamidine | ||||||||
| ATCC 30215 | >160.1 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study | |
| Hexamidine | ||||||||
| ATCC 30215 | >117.3 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study | |
| Octamidine | ||||||||
| ATCC 30215 | 89.1 | 72 h | NCM | 9 | 106 | Absorbance/luminescence | Current study |
Screening results for amidino series.
Previous studies demonstrated activity of diamidino derivatives (e.g., pentamidine and propamidine) against pathogenic FLA (11, 14), with octamidine and hexamidine found to be the most active derivatives against Acanthamoeba spp. in vitro (14). Against N. fowleri in vitro, pentamidine, hexamidine, and propamadine were inactive, and octamidine was only moderately active (IC50 of 89.1 μM) (Table 1), suggesting an inherent difference in susceptibility to diamidines between different pathogenic free-living amoebae.
Multiple furamidine derivatives were shown to cross the blood-brain barrier and possess efficacy against several parasitic protozoa (15). Therefore, amidino derivatives possess potential for the discovery of novel, more potent derivatives as leads for the development of new drugs to treat CNS infections with N. fowleri. We evaluated over 150 amidino derivatives and related compounds that represent six major focused structural classes (summarized in Fig. 2) and two miscellaneous groups of cationic compounds (diamidines and diguanidines; structures not shown). Our objective was to identify compounds with IC50s of ≤1 μM and selectivity indices (SI) of >10 as hits for further development. Two of the six groups of compounds (types I and II) presented in Fig. 2 yielded compounds that met this standard. Three of the other four groups (IV to VI) yielded at least one compound that produced an IC50 near 10 μM. Consequently, we selected types I and II for further lead optimization, since several compounds were identified in each group with IC50s of <1 μM against N. fowleri and involved structures which could be readily modified in an effort to improve activity, selectivity, and physical properties.
FIG 2.

Structural classes of amidino compounds screened against Naegleria fowleri.
SAR studies of type I monoamidino compounds.
In the initial screens of amidino compounds, we identified the bis-benzimidazole monoamidines as hits with remarkable potency compared to pentamidine. Five of the type I series (DB173, DB183, DB210, HG17, and HG24 in Table 2 ) exhibited IC50s of <1 μM and provided reasonable SI. The most active compound was DB173, with an IC50 of 0.177 μM (Table 2). Six additional type I analogs produced IC50s between 1 and 10 μM. The structure-activity relationship (SAR) of the type I compounds was not obvious given the divergent results obtained with fairly minor changes in structures; however, the set represents a significant hit that merits further optimization.
TABLE 2.
Bis-benzimidazole amidines evaluated for activity against N. fowleri in vitro
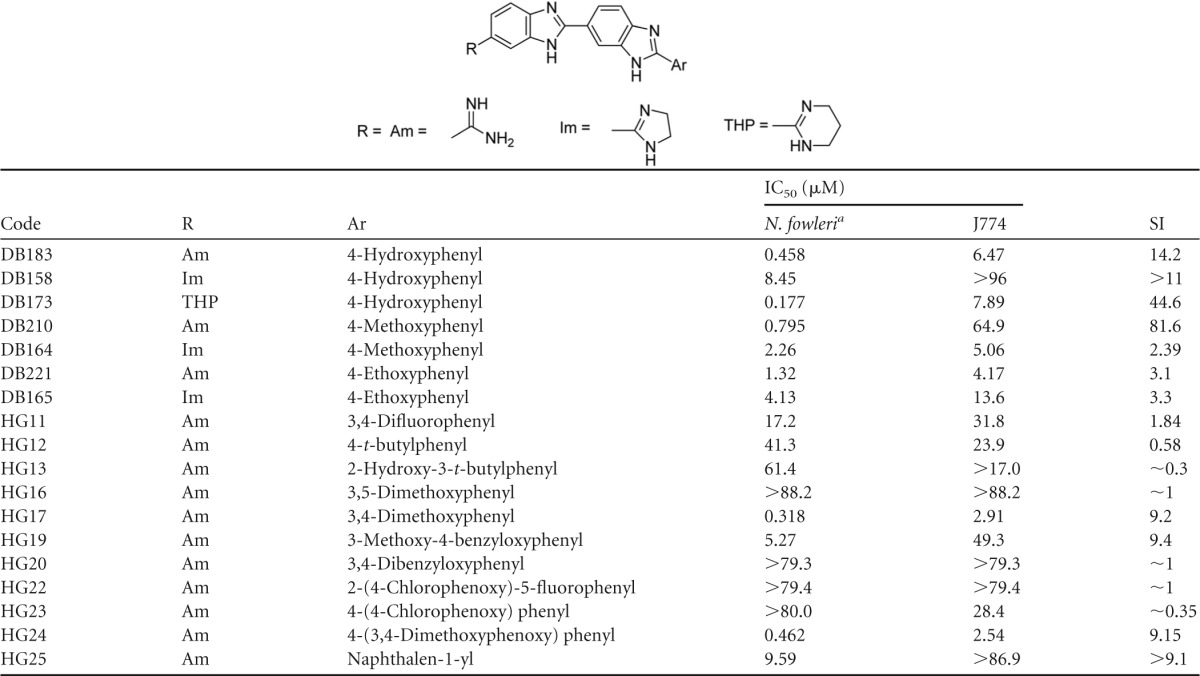
Means from at least two independent alamarBlue assays.
Optimization of type II diamidino compounds.
Five compounds of the type II series produced IC50s below 1 μM; however, only one of those (DB1766) exhibited an acceptable SI value (≥10). Three other active compounds (DB1684, DB1734, and DB1736) exhibit reasonable SI values (8.4 to 9.8) for early lead compounds. For the type II class, we have data for approximately 15 analogues (Table 3) that allow limited SAR conclusions to be drawn. Clearly, with type II compounds, we must diligently seek compounds with improved SI values. Often, cytotoxicity SARs are not obvious, and diamidines are no exception; however, we have found excellent SIs for numerous classes of diamidines (20, 22, 39), and we expect that we can successfully achieve similar results with type II compounds. Future studies will explore several approaches to determine the minimum size of the bis-benzimidazole diamidines (type II) that retain activity in an effort to identify a lead with physical properties that allow penetration of the BBB.
TABLE 3.
Bis-benzimidazole diamidines evaluated for activity against N. fowleri in vitro
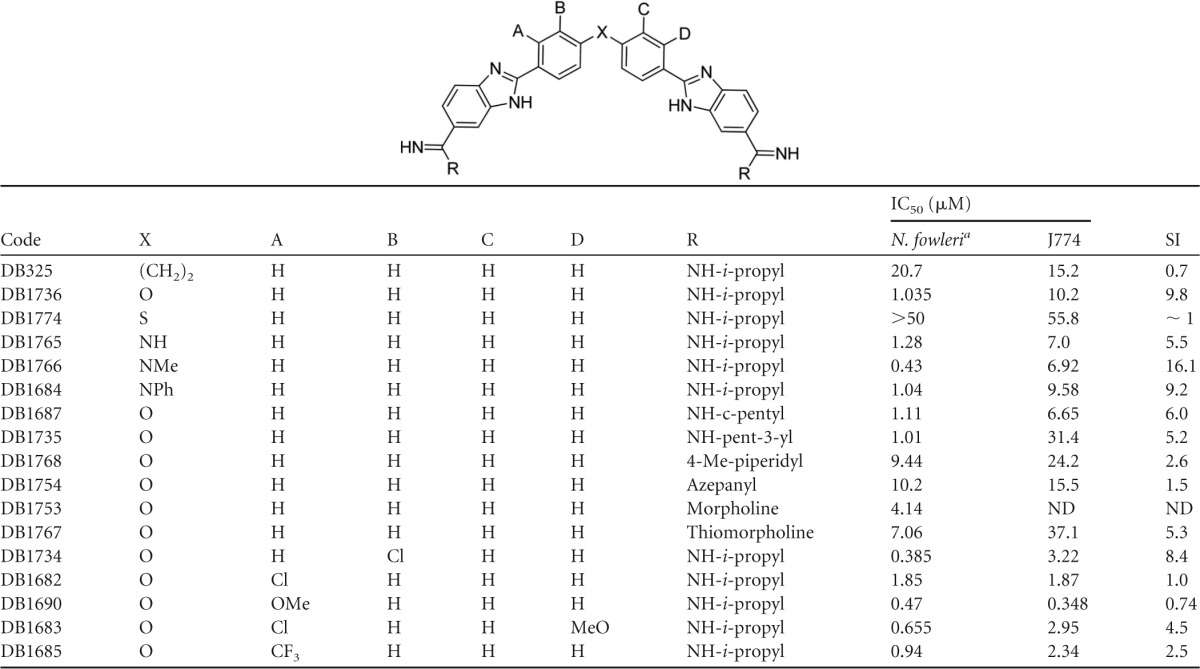
Mean of at least two independent alamarBlue assays.
Screening results for type III to VI amidino analogs.
The furamidine analogs (type III; see Table S1 in the supplemental material) produced IC50s ranging from 31 to 84 μM. The reduced potency of the furamidine analogs was disappointing, given this series contained four compounds known to penetrate the blood-brain barrier in both mice and monkey models for late-stage HAT disease. The linear benzimidazoles (type IV; see Table S2) and the indoles (type V; see Table S3) each yielded at least one compound with IC50s near 10 μM, although most compounds in these series were not potent against N. fowleri amoeba. Similarly, the arylimidamides (type VI; see Table S4) had only one compound with an IC50 near 10 μM. Finally, a series of miscellaneous amidino derivatives, mostly diamidines and diguanidines, were uniformly inactive against N. fowleri in vitro. Other miscellaneous amidino derivatives (>50) were found to be inactive and were not considered for lead optimization (data not shown).
DISCUSSION
The drugs that are currently used for treating pathogenic FLA infections have not been discovered and developed prospectively. Amphotericin B, antifungals, antibiotics, and microbicides available for other indications have been applied to the treatment of FLA in an empirical approach to find curative drugs (12). Amphotericin B, pentamidine, propamidine, miltefosine, and azoles are drugs that were developed originally against other pathogens but also were found to possess limited to moderate in vitro or vivo efficacy against pathogenic FLA infections. Since no large-scale drug-screening programs have been initiated for pathogenic FLA and there have not been any hit expansion or lead optimization of new chemotypes, a strategy for drug discovery for FLA had to be established. In this study, we aimed to develop a discovery paradigm to identify, evaluate, optimize, and advance new molecules toward the clinic for use as new therapies for pathogenic FLA. The primary enabling technology is new, quantitative in vitro methods for whole-cell phenotypic screens. This is the primary tool that enhances early drug discovery by supporting screens of large libraries of compounds and quantitative assessment of antiparasitic efficacy sufficient for structure-activity relationship studies. Secondarily, animal models of disease are required that mimic human disease, are amenable to infection with the human pathogen, and are validated by efficacy of known curative or partially efficacious drugs. These tools then are used to provide quantitative efficacy data required to fuel medicinal chemists' efforts to optimize structure-activity properties. Similar strategies and drug discovery pathways are used for novel antimalarial and antileishmanial drugs (40, 41).
Most previous in vitro assays for the assessment of anti-FLA drug screening are not amenable to high- or even medium-throughput screening. Endpoints for growth usually include morphology and visual counting of amoebae, assessment of viability that requires days to weeks, or release of lactate dehydrogenase (11, 42–45). In addition, these methods use large volumes of culture media (e.g., >10 ml) in flasks and require extended incubation periods of up to 6 days (11, 42–45). These methods are simply too cumbersome to support modern drug discovery requirements. Given the lack of in vitro assays for medium- to high-throughput drug susceptibility testing, we developed and validated two methods for HTS in 96- and 384-well plates and used the methods for the first time with pathogenic N. fowleri (rather than nonpathogenic N. gruberi) (37).
The potencies of amphotericin B and pentamidine were significantly lower in the two new assays used in this study than in previous experience (Table 1). These differences likely are due to a much longer exposure to drug, since some assays were up to 6 days compared to the 72-h assays used in this report. Second, the number of amoebae used per ml was higher in the alamarBlue assay than in some previously published methods (Table 1). Despite any differences, the reduced efficacy observed in the 72-h assay formats was similar regardless of the endpoint used (alamarBlue or CTG); therefore, it is likely that the new assays reflect the inherent lack of potency and slow onset of action of most of the drugs currently used to treat FLA infections. Another factor that may reduce potency in all assay formats is the requirement for highly complex media (e.g., NCM) with multiple components that can serve as a sink for drugs and reduce the free concentration needed to affect the parasite. Regardless, the 72-h assay format we used successfully enabled the discovery of compounds with potency more than 500 times that of previously described diamidines. The 72-h assays we used have multiple advantages, including reproducible, quantitative endpoints that are important for medicinal chemistry optimization, as well as enabling the detection of compounds with a more rapid onset of action. Given the rapidly progressive, fatal disease caused by N. fowleri, it is important to focus drug discovery efforts on compound series with rapid onset of action and to prioritize these over other scaffolds for lead optimization.
By using the new quantitative assays for assessing efficacy against N. fowleri in vitro, we have identified compounds that appear to be much more potent than currently used drugs for PAM. Our predetermined criteria for a hit was potency (IC50) of ≤1 μM and a selectivity index of >10. By using these criteria, we identified three monoamidino analogs (DB173, DB183, and DB210) and one diamidino derivative (DB1766) that are validated hits. In addition, we identified 5 derivatives deemed marginal hits, as defined by a potency of approximately 1 μM (DB1684 and DB1736) or selectivity indices of >8 (HG17, HG24, and DB1734). It is important to note that the hits we identified do not appear to be promiscuous inhibitors. Many of the hits and marginal hits have been screened previously for antiparasitic, antifungal, or antibacterial activities, and none appear to have indiscriminate activity across the spectrum of biological activities examined thus far (see Table S6 in the supplemental material). Furthermore, many of the compounds we tested that were not potent against N. fowleri in vitro, in particular type II derivatives, have broad activities in other biological systems (see Table S6). These results lead us to propose inhibitors for N. fowleri that have an encouraging degree of specificity that can be exploited in lead optimization. Therefore, from this study we identified nine compounds (hits and marginal hits in Table S6) that warrant further evaluation and optimization to identify lead compounds. For each of the hit compounds identified, the lead optimization process will require enhanced physiochemical properties to permit blood-brain barrier penetration and improved selectivity. A partial list of some of the properties to be optimized include lower molecular weight, log of distribution coefficient (logD) of approximately 2, and efficacy in vivo (N. fowler-infected mouse model).
While all the screened compounds initially were designed to be DNA minor groove binders, we have no evidence to suggest that the activity of the compounds against N. fowleri involves binding to DNA. Consequently, we consider the hit compounds as ones with an unknown target(s). Furthermore, the two classes with best potency, bis-benzimidazole amidines (type I) and bis-benzimidazole diamidines (type II) (Fig. 2), may not have a common mode of action, since the two structural types have significant differences. Type I is a monocation and type II is a dication; while the overall geometry of both is curved, the type II compounds are more acutely curved, and the two types may be unable to fit the same binding site. In addition, it is possible that each of the hits has more than one target and inhibits their targets with different potencies depending upon the structural modifications in the series. Once a lead compound is identified, specific mechanism-of-action studies will be warranted. Both type I and II cations are likely to be very hydrophilic (ACD estimated logD for DB183 of −0.65); therefore, to have a reasonable chance of passing the blood-brain barrier, it will be essential to focus on modifications that lead to compounds with a logD of 2 ± 1. Future studies should systematically vary the structures of types I and II to reduce their size and polarity in an attempt to optimize activity for N. fowleri, to develop reasonable selectivity, and to achieve the desired physiochemical properties. In addition, the type I and II derivatives should be assessed against multiple isolates of Acanthamoeba spp. Diamidino derivatives appear to be more potent against Acanthamoeba spp. than N. fowleri; therefore, the new hits identified in this study may be even more potent against Acanthamoeba and could be useful for developing new drugs to treat keratitis, GAE, or systemic infections with these important pathogens.
Advances in drug discovery for emerging infectious and tropical diseases have begun to deliver a pipeline of new chemotypes that are optimized for clinical development by academic and industrial scientists (40, 41). In comparison, pathogenic FLA have been ignored and early discovery studies have focused on a small number of drugs already approved for other indications or screens with nonpathogenic species of FLA. In this study, we have applied the tenets of modern drug discovery by using high-throughput phenotypic screens with pathogenic N. fowleri. To accomplish this goal, we validated two robust methods for quantitative dose-response and high-throughput screening of compounds for anti-FLA activity and applied these to conduct the first large-scale application of SAR to optimize new anti-FLA drugs. The mono- and diamidino compounds we discovered are synthetically tractable and highly attractive for hit-to-lead optimization.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from the National Institute of Allergy and Infectious Diseases (R21AI103664).
This work is dedicated to the memory of Martin John Rogers (NIH).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.05122-14.
REFERENCES
- 1.Culbertson CG, Smith JW, Minner JR. 1958. Acanthamoeba: observations on animal pathogenicity. Science 127:1506. [DOI] [PubMed] [Google Scholar]
- 2.Fowler M, Carter RF. 1965. Acute pyogenic meningitis probably due to Acanthamoeba sp.: a preliminary report. Br Med J 2:740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoder JS, Eddy BA, Visvesvara GS, Capewell L, Beach MJ. 2010. The epidemiology of primary amoebic meningoencephalitis in the U S A, 1962-2008. Epidemiol Infect 138:968–975. doi: 10.1017/S0950268809991014. [DOI] [PubMed] [Google Scholar]
- 4.Visvesvara GS, Stehr-Green JK. 1990. Epidemiology of free-living ameba infections. J Protozool 37:25S–33S. doi: 10.1111/j.1550-7408.1990.tb01142.x. [DOI] [PubMed] [Google Scholar]
- 5.Capewell LG, Harris AM, Yoder JS, Cope HR, Eddy BA, Roy SL, Visvesvara GS, Fox LM, Beach MJ. 23 October 2014. Diagnosis, clinical course, and treatment of primary amoebic meningoencephalitis in the United States, 1937-2013. J Pediatr Infect Dis Soc doi: 10.1093/jpids/piu103. [DOI] [PubMed] [Google Scholar]
- 6.Siddiqui R, Khan NA. 2014. Primary amoebic meningoencephalitis caused by Naegleria fowleri: an old enemy presenting new challenges. PLoS Negl Trop Dis 8:e3017. doi: 10.1371/journal.pntd.0003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadlec V, Skvarova J, Cerva L, Nebazniva D. 1980. Virulent Naegleria fowleri in indoor swimming pool. Folia Parasitol 27:11–17. [PubMed] [Google Scholar]
- 8.Martinez J, Duma RJ, Nelson EC, Moretta FL. 1973. Experimental naegleria meningoencephalitis in mice. Penetration of the olfactory mucosal epithelium by Naegleria and pathologic changes produced: a light and electron microscope study. Lab Investig 29:121–133. [PubMed] [Google Scholar]
- 9.Martinez AJ, Nelson EC, Duma RJ. 1973. Animal model of human disease. Primary amebic meningoencephalitis, Naegleria meningoencephalitis, CNS protozoal infection. Am J Pathol 73:545–548. [PMC free article] [PubMed] [Google Scholar]
- 10.Ondarza RN, Iturbe A, Hernandez E. 2007. The effects by neuroleptics, antimycotics and antibiotics on disulfide reducing enzymes from the human pathogens Acanthamoeba polyphaga and Naegleria fowleri. Exp Parasitol 115:41–47. doi: 10.1016/j.exppara.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Ondarza RN, Iturbe A, Hernandez E. 2006. In vitro antiproliferative effects of neuroleptics, antimycotics and antibiotics on the human pathogens Acanthamoeba polyphaga and Naegleria fowleri. Arch Med Res 37:723–729. doi: 10.1016/j.arcmed.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Visvesvara GS. 2010. Amebic meningoencephalitides and keratitis: challenges in diagnosis and treatment. Curr Opin Infect Dis 23:590–594. doi: 10.1097/QCO.0b013e32833ed78b. [DOI] [PubMed] [Google Scholar]
- 13.Schuster FL, Visvesvara GS. 2004. Opportunistic amoebae: challenges in prophylaxis and treatment. Drug Resist Updat 7:41–51. doi: 10.1016/j.drup.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Perrine D, Chenu JP, Georges P, Lancelot JC, Saturnino C, Robba M. 1995. Amoebicidal efficiencies of various diamidines against two strains of Acanthamoeba polyphaga. Antimicrob Agents Chemother 39:339–342. doi: 10.1128/AAC.39.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenzler T, Yang S, Patrick DA, Braissant O, Ismail MA, Tidwell RR, Boykin DW, Wang MZ, Brun R. 2014. In vitro and in vivo evaluation of 28DAP010, a novel diamidine for treatment of second-stage African sleeping sickness. Antimicrob Agents Chemother 58:4452–4463. doi: 10.1128/AAC.02309-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang MZ, Zhu X, Srivastava A, Liu Q, Sweat JM, Pandharkar T, Stephens CE, Riccio E, Parman T, Munde M, Mandal S, Madhubala R, Tidwell RR, Wilson WD, Boykin DW, Hall JE, Kyle DE, Werbovetz KA. 2010. Novel arylimidamides for treatment of visceral leishmaniasis. Antimicrob Agents Chemother 54:2507–2516. doi: 10.1128/AAC.00250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens CE, Tanious F, Kim S, Wilson WD, Schell WA, Perfect JR, Franzblau SG, Boykin DW. 2001. Diguanidino and “reversed” diamidino 2,5-diarylfurans as antimicrobial agents. J Med Chem 44:1741–1748. doi: 10.1021/jm000413a. [DOI] [PubMed] [Google Scholar]
- 18.Paine MF, Wang MZ, Generaux CN, Boykin DW, Wilson WD, De Koning HP, Olson CA, Pohlig G, Burri C, Brun R, Murilla GA, Thuita JK, Barrett MP, Tidwell RR. 2010. Diamidines for human African trypanosomiasis. Curr Opin Investig Drugs 11:876–883. [PubMed] [Google Scholar]
- 19.Lansiaux A, Tanious F, Mishal Z, Dassonneville L, Kumar A, Stephens CE, Hu Q, Wilson WD, Boykin DW, Bailly C. 2002. Distribution of furamidine analogues in tumor cells: targeting of the nucleus or mitochondria depending on the amidine substitution. Cancer Res 62:7219–7229. [PubMed] [Google Scholar]
- 20.Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. 2003. Synthesis and antiprotozoal activity of aza-analogues of furamidine. J Med Chem 46:4761–4769. doi: 10.1021/jm0302602. [DOI] [PubMed] [Google Scholar]
- 21.Ismail MA, Batista-Parra A, Miao Y, Wilson WD, Wenzler T, Brun R, Boykin DW. 2005. Dicationic near-linear biphenyl benzimidazole derivatives as DNA-targeted antiprotozoal agents. Bioorg Med Chem 13:6718–6726. doi: 10.1016/j.bmc.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 22.Ismail MA, Arafa RK, Brun R, Wenzler T, Miao Y, Wilson WD, Generaux C, Bridges A, Hall JE, Boykin DW. 2006. Synthesis, DNA affinity, and antiprotozoal activity of linear dications: terphenyl diamidines and analogues. J Med Chem 49:5324–5332. doi: 10.1021/jm060470p. [DOI] [PubMed] [Google Scholar]
- 23.Hu LX, Kully ML, Boykin DW, Abood N. 2009. Synthesis and structure-activity relationship of dicationic diaryl ethers as novel potent anti-MRSA and anti-VRE agents. Bioorg Med Chem Lett 19:4626–4629. doi: 10.1016/j.bmcl.2009.06.077. [DOI] [PubMed] [Google Scholar]
- 24.Hu LX, Kully ML, Boykin DW, Abood N. 2009. Synthesis and in vitro activity of dicationic bis-benzimidazoles as a new class of anti-MRSA and anti-VRE agents. Bioorg Med Chem Lett 19:1292–1295. doi: 10.1016/j.bmcl.2009.01.075. [DOI] [PubMed] [Google Scholar]
- 25.Hu L, Kully ML, Boykin DW, Abood N. 2009. Optimization of the central linker of dicationic bis-benzimidazole anti-MRSA and anti-VRE agents. Bioorg Med Chem Lett 19:3374–3377. doi: 10.1016/j.bmcl.2009.05.061. [DOI] [PubMed] [Google Scholar]
- 26.Farahat AA, Paliakov E, Kumar A, Barghash AE, Goda FE, Eisa HM, Wenzler T, Brun R, Liu Y, Wilson WD, Boykin DW. 2011. Exploration of larger central ring linkers in furamidine analogues: synthesis and evaluation of their DNA binding, antiparasitic and fluorescence properties. Bioorg Med Chem 19:2156–2167. doi: 10.1016/j.bmc.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 27.de Souza EM, Oliveira GM, Boykin DW, Kumar A, Hu Q, De Nazare CSM. 2006. Trypanocidal activity of the phenyl-substituted analogue of furamidine DB569 against Trypanosoma cruzi infection in vivo. J Antimicrob Chemother 58:610–614. doi: 10.1093/jac/dkl259. [DOI] [PubMed] [Google Scholar]
- 28.De Souza EM, Lansiaux A, Bailly C, Wilson WD, Hu Q, Boykin DW, Batista MM, Araujo-Jorge TC, Soeiro MN. 2004. Phenyl substitution of furamidine markedly potentiates its anti-parasitic activity against Trypanosoma cruzi and Leishmania amazonensis. Biochem Pharmacol 68:593–600. doi: 10.1016/j.bcp.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 29.Czarny A, Wilson WD, Boykin DW. 1996. Synthesis of mono-cationic and dicationic analogs of Hoechst 33258. J Heterocyclic Chem 33:1393–1397. doi: 10.1002/jhet.5570330463. [DOI] [Google Scholar]
- 30.Chaires JB, Ren J, Hamelberg D, Kumar A, Pandya V, Boykin DW, Wilson WD. 2004. Structural selectivity of aromatic diamidines. J Med Chem 47:5729–5742. doi: 10.1021/jm049491e. [DOI] [PubMed] [Google Scholar]
- 31.Brendle JJ, Outlaw A, Kumar A, Boykin DW, Patrick DA, Tidwell RR, Werbovetz KA. 2002. Antileishmanial activities of several classes of aromatic dications. Antimicrob Agents Chemother 46:797–807. doi: 10.1128/AAC.46.3.797-807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenzler T, Yang S, Braissant O, Boykin DW, Brun R, Wang MZ. 2013. Pharmacokinetics, Trypanosoma brucei gambiense efficacy, and time of drug action of DB829, a preclinical candidate for treatment of second-stage human African trypanosomiasis. Antimicrob Agents Chemother 57:5330–5343. doi: 10.1128/AAC.00398-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang S, Wenzler T, Miller PN, Wu H, Boykin DW, Brun R, Wang MZ. 2014. Pharmacokinetic comparison to determine the mechanisms underlying the differential efficacies of cationic diamidines against first- and second-stage human African trypanosomiasis. Antimicrob Agents Chemother 58:4064–4074. doi: 10.1128/AAC.02605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thuita JK, Wang MZ, Kagira JM, Denton CL, Paine MF, Mdachi RE, Murilla GA, Ching S, Boykin DW, Tidwell RR, Hall JE, Brun R. 2012. Pharmacology of DB844, an orally active aza analogue of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl Trop Dis 6:e1734. doi: 10.1371/journal.pntd.0001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schorer M, Debache K, Barna F, Monney T, Muller J, Boykin DW, Stephens CE, Hemphill A. 2012. Di-cationic arylimidamides act against Neospora caninum tachyzoites by interference in membrane structure and nucleolar integrity and are active against challenge infection in mice. Int J Parasitol Drugs Drug Resist 2:109–120. doi: 10.1016/j.ijpddr.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McBride J, Ingram PR, Henriquez FL, Roberts CW. 2005. Development of colorimetric microtiter plate assay for assessment of antimicrobials against Acanthamoeba. J Clin Microbiol 43:629–634. doi: 10.1128/JCM.43.2.629-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debnath A, Tunac JB, Galindo-Gomez S, Silva-Olivares A, Shibayama M, McKerrow JH. 2012. Corifungin, a new drug lead against Naegleria, identified from a high-throughput screen. Antimicrob Agents Chemother 56:5450–5457. doi: 10.1128/AAC.00643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alp M, Goker H, Brun R, Yildiz S. 2009. Synthesis and antiparasitic and antifungal evaluation of 2′-arylsubstituted-1H,1′H-[2,5′]bisbenzimidazolyl-5-carboxamidines. Eur J Med Chem 44:2002–2008. doi: 10.1016/j.ejmech.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Hu L, Patel A, Bondada L, Yang S, Wang MZ, Munde M, Wilson WD, Wenzler T, Brun R, Boykin DW. 2013. Synthesis and antiprotozoal activity of dicationic 2,6-diphenylpyrazines and aza-analogues. Bioorg Med Chem 21:6732–6741. doi: 10.1016/j.bmc.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, O'Loughlin KG, Shackleford DM, Justino de Almeida M, Carrillo AK, Clark JA, Dennis AS, Diep J, Deng X, Duffy S, Endsley AN, Fedewa G, Guiguemde WA, Gomez MG, Holbrook G, Horst J, Kim CC, Liu J, Lee MC, Matheny A, Martinez MS, Miller G, Rodriguez-Alejandre A, Sanz L, Sigal M, Spillman NJ, Stein PD, Wang Z, Zhu F, Waterson D, Knapp S, Shelat A, Avery VM, Fidock DA, Gamo FJ, Charman SA, Mirsalis JC, Ma H, Ferrer S, Kirk K, Angulo-Barturen I, Kyle DE, DeRisi JL, Floyd DM, Guy RK. 2014. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci U S A 111:E5455–5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuster FL, Guglielmo BJ, Visvesvara GS. 2006. In-vitro activity of miltefosine and voriconazole on clinical isolates of free-living amoebas: Balamuthia mandrillaris, Acanthamoeba spp., and Naegleria fowleri. J Eukaryot Microbiol 53:121–126. doi: 10.1111/j.1550-7408.2005.00082.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim JH, Lee YJ, Sohn HJ, Song KJ, Kwon D, Kwon MH, Im KI, Shin HJ. 2008. Therapeutic effect of rokitamycin in vitro and on experimental meningoencephalitis due to Naegleria fowleri. Int J. Antimicrob agents 32:411–417. doi: 10.1016/j.ijantimicag.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 44.Kim JH, Jung SY, Lee YJ, Song KJ, Kwon D, Kim K, Park S, Im KI, Shin HJ. 2008. Effect of therapeutic chemical agents in vitro and on experimental meningoencephalitis due to Naegleria fowleri. Antimicrob Agents Chemother 52:4010–4016. doi: 10.1128/AAC.00197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goswick SM, Brenner GM. 2003. Activities of azithromycin and amphotericin B against Naegleria fowleri in vitro and in a mouse model of primary amebic meningoencephalitis. Antimicrob Agents Chemother 47:524–528. doi: 10.1128/AAC.47.2.524-528.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Poeta M, Schell WA, Dykstra CC, Jones SK, Tidwell RR, Kumar A, Boykin DW, Perfect JR. 1998. In vitro antifungal activities of a series of dication-substituted carbazoles, furans, and benzimidazoles. Antimicrob Agents Chemother 42:2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Visvesvara GS, Moura H, Schuster FL. 2007. Pathogenic and opportunistic free-living amoebae: Acanthamoeba spp., Balamuthia mandrillaris, Naegleria fowleri, and Sappinia diploidea. FEMS Immunol Med Microbiol 50:1–26. doi: 10.1111/j.1574-695X.2007.00232.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.