

Abstract
We tested the antituberculosis drug SQ109, which is currently in advanced clinical trials for the treatment of drug-susceptible and drug-resistant tuberculosis, for its in vitro activity against the trypanosomatid parasite Trypanosoma cruzi, the causative agent of Chagas disease. SQ109 was found to be a potent inhibitor of the trypomastigote form of the parasite, with a 50% inhibitory concentration (IC50) for cell killing of 50 ± 8 nM, but it had little effect (50% effective concentration [EC50], ∼80 μM) in a red blood cell hemolysis assay. It also inhibited extracellular epimastigotes (IC50, 4.6 ± 1 μM) and the clinically relevant intracellular amastigotes (IC50, ∼0.5 to 1 μM), with a selectivity index of ∼10 to 20. SQ109 caused major ultrastructural changes in all three life cycle forms, as observed by light microscopy, scanning electron microscopy (SEM), and transmission electron microscopy (TEM). It rapidly collapsed the inner mitochondrial membrane potential (Δψm) in succinate-energized mitochondria, acting in the same manner as the uncoupler FCCP [carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone], and it caused the alkalinization of internal acidic compartments, effects that are likely to make major contributions to its mechanism of action. The compound also had activity against squalene synthase, binding to its active site; it inhibited sterol side-chain reduction and, in the amastigote assay, acted synergistically with the antifungal drug posaconazole, with a fractional inhibitory concentration index (FICI) of 0.48, but these effects are unlikely to account for the rapid effects seen on cell morphology and cell killing. SQ109 thus most likely acts, at least in part, by collapsing Δψ/ΔpH, one of the major mechanisms demonstrated previously for its action against Mycobacterium tuberculosis. Overall, the results suggest that SQ109, which is currently in advanced clinical trials for the treatment of drug-susceptible and drug-resistant tuberculosis, may also have potential as a drug lead against Chagas disease.
INTRODUCTION
Chagas disease is caused by the trypanosomatid parasite Trypanosoma cruzi and affects ∼8 to 10 million individuals, mostly in Latin America (1), with the U.S. Centers for Disease Control and Prevention estimating that ∼300,000 individuals are also infected in the United States (2). The drugs benznidazole and nifurtimox (Fig. 1, compounds 1 and 2, respectively) are used to treat the acute and early chronic forms of the disease, but their efficacies against established chronic infection, the most common presentation of the disease, are lower and variable. Chagas disease is calculated to result in a $7 billion annual global cost and represents the largest parasitic disease burden of the American continents (3). There is thus interest in developing new therapeutic options. Among these are inhibitors of ergosterol (Fig. 1, compound 3) biosynthesis. Ergosterol is an essential membrane sterol in yeasts, fungi, and many protozoa, and it is the functional equivalent of cholesterol (Fig. 1, compound 4) in humans; antifungal sterol biosynthesis inhibitors (such as posaconazole [Fig. 1, compound 5]) are also of interest as new anti-T. cruzi drugs, which we discuss in more detail below.
FIG 1.

Inhibitors and sterols of interest.
In previous work, we noticed reports (4–6) that the antiarrhythmic drug amiodarone (Fig. 1, compound 6) (used to treat arrhythmias in Chagas disease patients) also had activity against the yeast Saccharomyces cerevisiae and that amiodarone potentiated the effects of azole drugs. This suggested that amiodarone might also inhibit ergosterol (Fig. 1, compound 3) biosynthesis in T. cruzi because, at least in yeast, it acted synergistically with azoles (which inhibit lanosterol 14α-demethylase). This was found to be the case (7), with amiodarone inhibiting the enzyme oxidosqualene cyclase (lanosterol synthase) in T. cruzi (7), thereby decreasing ergosterol levels. In addition, it acted synergistically with posaconazole against T. cruzi in vitro and was active in vivo in a mouse model of infection (7). Similar results were later found with Leishmania spp. (8, 9), and amiodarone is now used clinically for the treatment of chronic Chagas disease (10) and disseminated cutaneous leishmaniasis (11), as discussed in a recent review (12). Similar results have also been obtained with a newer (and perhaps less toxic) analog of amiodarone, dronedarone (13) (Fig. 1, compound 7). What is interesting about amiodarone and dronedarone is that they also release Ca2+ from intracellular stores in both T. cruzi and Leishmania amazonensis, an activity probably related to the fact that these compounds are uncouplers (14–16), lipophilic bases that can transport H+.
In this work, we investigated another lipophilic base, SQ109 (17–21), an ethylene diamine containing N-geranyl and N-adamantyl groups (Fig. 1, compound 8). SQ109 is currently in phase II clinical trials for the treatment of drug-sensitive and drug-resistant tuberculosis (18–20). One major mode of action for SQ109 in Mycobacterium tuberculosis has been proposed (21) to be its inhibition of MmpL3 (mycobacterial membrane protein large 3), a trehalose monomycolate transporter that is used in cell wall biosynthesis in M. tuberculosis. Our interest in SQ109 arose since, in addition to inhibiting M. tuberculosis cell growth, it inhibits the growth of other bacteria, such as Helicobacter pylori (22), Clostridium difficile (18), Enterococcus spp. (18), Haemophilus influenzae (18), Neisseria gonorrhoeae (18), and Streptococcus pneumoniae (18); the fungi Candida albicans (23), Aspergillus fumigatus (18), and Cryptococcus neoformans (18); and the malaria parasite Plasmodium falciparum (24). Since none of these bacteria, fungi, or the malaria parasite contain bioinformatically identifiable mmpL3 orthologs, there must be an alternative site (or sites) of action in these organisms, and in recent work (24), we found that SQ109 can inhibit enzymes involved in quinone biosynthesis (MenA and MenG). In addition, it acts as an uncoupler, collapsing pH gradients (ΔpH) and membrane potentials (Δψ) in bacterial systems (24), thereby reducing ATP synthesis. In unrelated work, we also reported (25) that SQ109 was an inhibitor of dehydrosqualene synthase (from Staphylococcus aureus), a protein whose three-dimensional structure (26) and mechanism of action (27) are very similar to those of squalene synthase (SQS) (28), which catalyzes the first step of sterol biosynthesis in eukaryotes, such as T. cruzi, suggesting that SQ109 might also inhibit SQS.
Here, we report the antiproliferative and ultrastructural effects of SQ109 against the three life cycle stages of T. cruzi, i.e., trypomastigotes, epimastigotes, and amastigotes; its synergistic interaction with posaconazole; its uncoupling activity on T. cruzi mitochondria; its alkalinizing effects on acidic compartments; its effects on sterol biosynthesis; and the X-ray structures of SQ109 bound to T. cruzi and human squalene synthase.
MATERIALS AND METHODS
Parasites and host cell culture.
In most cases, the assays were performed using T. cruzi epimastigotes, trypomastigotes, or intracellular amastigotes of the Y strain (TcII) (29). The T. cruzi trypomastigotes were obtained from the supernatants of previously infected LLC-MK2 cells (ATCC [American Type Culture Collection], Rockville, MD) cultured in RPMI 1640 medium with garamycin (Gibco, Grand Island, NY) and 10% fetal bovine serum (FBS) (Cultilab, São Paulo, Brazil) at 37°C in a 5% CO2 atmosphere. Subconfluent cultures of LLC-MK2 cells were infected with 5 × 106 trypomastigotes. Extracellular parasites were removed after 2 h, the cells were washed, and the cultures were maintained in RPMI 1640 medium containing 10% FBS until trypomastigotes emerged from the infected cells (usually after 120 h). The epimastigotes were cultivated in liver infusion broth-tryptose (LIT) medium supplemented with 10% FBS (30) and were collected by centrifugation at 350 × g after 96 h of cultivation.
Drug solutions.
Stock solutions of SQ109 and analogs (0.01 mM) were prepared in dimethyl sulfoxide (DMSO) (Merck, Darmstadt, Germany), with the final concentration of DMSO in the experiments never being >0.05%.
Effects of SQ109 and analogs on LLC-MK2 cells.
The LLC-MK2 cells were treated with SQ109 (2.5 to 20 μM) and incubated for 96 h at 37°C. Fresh RPMI 1640 medium containing only 10% FBS was added to the untreated samples as a control. To determine toxicity, the MTS/PMS [3-(4,5-dimethyl-2-thiazolyl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt/5-methylphenazinium methyl sulfate] assay was performed as described by Henriques et al. (31). The selectivity index of SQ109 was determined based on its activities against the trypomastigote and intracellular amastigote forms of T. cruzi, calculated as the ratio of the 50% cytotoxic concentration (CC50) of mammalian cells to the 50% inhibitory concentration (IC50) or 50% lysing concentration (LC50) of T. cruzi. All experiments were performed in duplicate. The means were determined from ≥3 experiments.
Antitrypanosomal activity.
The trypomastigotes were obtained from the supernatant of a previously infected cell line (LLC-MK2). Between 5 and 7 days after infection, protozoa were collected from the supernatants of the infected LLC-MK2 cells and were then incubated with fresh RPMI 1640 medium supplemented with 0.5% FBS, with or without SQ109 (0.05 to 5 μM), for 24 h at 37°C under a 5% CO2 atmosphere. The concentration of SQ109 at which 50% of the parasites were lysed (LC50) was calculated by counting the cells in a Neubauer chamber. The experiment was performed in duplicate for each of the three different experiments.
For the antiproliferative assay involving epimastigotes, 106 parasites/ml were cultivated in LIT medium supplemented with 10% FBS. After 24 h of epimastigote growth, concentrations of 0.5 to 10 μM SQ109 (or analogues) were added to the culture and incubated for 120 h at 28°C. Cells were collected every 24 h for counting in a Neubauer chamber. Two controls were used and consisted of liver infusion broth-tryptose (LIT) supplemented with 10% FBS and LIT plus 0.05% DMSO. The cells were allowed to adhere to the coverslips with 0.1 mg/ml poly-l-lysine, fixed with Bouin's solution, and stained with Giemsa for morphological analysis using a Zeiss Axioplan 2 light microscope (Oberkochen, Germany) equipped with a Color View XS digital video camera.
To investigate the effect of SQ109 on amastigotes, LLC-MK2 cells were incubated for 2 h with T. cruzi trypomastigotes at a ratio of 10 parasites to 1 cell. The noninternalized parasites were removed by washing, and the host cells were incubated for 24 h at 37°C to allow full internalization and differentiation of trypomastigotes to amastigotes. Fresh 10% FBS-RPMI 1640 medium alone (control) or containing the inhibitors (0.5 to 6 μM) was added to the infected cells, which were then incubated for 96 h at 37°C. The infected cultures were fixed in Bouin's solution and stained with Giemsa. The number of parasites was determined using a Zeiss Axioplan (Jena, Germany) light microscope equipped with a 100× lens. The antiproliferative assay was performed as described by Veiga-Santos et al. (32).
The infection index (i.e., the percentage of infected host cells multiplied by the average number of intracellular amastigotes per infected host cell) was determined by counting a total of 500 host cells.
Inhibitor activity was calculated using the SigmaPlot (version 10) program (Systat Software, Inc., San Jose, CA, USA). The results are expressed as the mean values from three independent experiments.
We also carried out an additional set of amastigote assays using the T. cruzi CL strain overexpressing a tdTomato red fluorescent protein, performed basically as described earlier (33).
To evaluate antiproliferative synergism against intracellular amastigotes, fractional inhibitory concentration indices (FICIs) were calculated as described by Hallander et al. (34). An isobologram was constructed by plotting the concentrations of the drugs that alone or in combination induced an IC50 of that of intracellular amastigotes, as described above.
To compare the control and treated groups between all assays, paired t tests were applied using a 95% confidence interval (GraphPad Prism version 5.00 for Windows; GraphPad Software, Inc., San Diego, CA).
Scanning electron microscopy.
T. cruzi epimastigotes were treated with 4.6 μM SQ109 and incubated for 24 and 48 h at 28°C. The trypomastigotes were treated with 75 nM SQ109 and incubated for 6 and 12 h at 37°C. The samples were subsequently fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 1 h, washed, and adhered to a specimen support coated with poly-l-lysine. The cells were then postfixed in 1% OsO4 and 0.8% potassium ferrocyanide in 0.1 M sodium cacodylate buffer (pH 7.2) at room temperature for 20 min, washed, dehydrated in ethanol, and critical-point dried in CO2. In addition, the samples were ion sputtered with a chrome layer using a Leica EM SCD500 (Leica Microsystems, Nussloch, Germany) high-vacuum sputter coater and observed under a Quanta X50 scanning electron microscope. Images were obtained and processed using xTm version 4.1.1.1935 (FEI Company).
Transmission electron microscopy.
For transmission electron microscopy (TEM), T. cruzi epimastigotes were treated with SQ109 (4.6 μM) and were incubated for 24 and 48 h at 28°C. The trypomastigotes were incubated with 75 nM drug for 24 h. LLC-MK2 host cells infected with T. cruzi trypomastigotes were treated with 1.5 μM SQ109 and incubated for 96 h at 37°C. After the experimental procedure, the samples were processed as described by de Macedo-Silva et al. (9). Ultrathin sections were stained with uranyl acetate and lead citrate and were observed with a Jeol JEM-1200EX (Jeol, Akishima, Japan) transmission electron microscope operating at 80 kV.
Hemolysis assay.
The hemolytic activity of SQ109 was evaluated as described by Veiga-Santos et al. (35). A 4% suspension of fresh defibrinated human blood was prepared in a sterile 5% glucose solution and treated with SQ109 (10 to 80 μM) for 24 h at 37°C. Next, the cells were centrifuged at 250 × g for 5 min. The supernatant absorbance at 540 nm was calculated to determine the percent hemolysis. Amphotericin B (Cristália, São Paulo, Brazil) was used as the hemolytic reference drug, and Triton X-100 (Vetec, Rio de Janeiro, Brazil) was used as the positive control. Each experiment was conducted in duplicate and repeated ≥3 times.
Analysis of mitochondrial membrane potential.
We spectrofluorometrically monitored the mitochondrial membrane potential using safranine as the probe (36, 37). T. cruzi epimastigotes (108 cells) were added to 2.4 ml of buffer containing 2 mM succinate and 5 μM safranine, and the reaction was initiated with or without 60 μM digitonin and with or without the additives shown in Fig. 2. The incubations were conducted at 28°C. Fluorescence changes were monitored using a Hitachi 4500 spectrofluorometer (excitation wavelength, 496 nm; emission wavelength, 586 nm).
FIG 2.

Effects of SQ109 on the proton motive force (PMF) in digitonin-permeabilized T. cruzi. Epimastigotes (108 cells) were added to buffer (2.4 ml) containing 2 mM succinate and 5 μM safranine, and the reaction was initiated with or without (gray trace in panel A) 60 μM digitonin. (A) SQ109 (10 μM) and FCCP (8 μM) were added where indicated. (B) ADP (10 μM), oligomycin (Oligo) (2 μg/ml), CaCl2 (16 μM), EGTA (200 μM), SQ109 (10 μM), and FCCP (8 μM) were added where indicated. (C) CaCl2 (16 μM), EGTA (200 μM), various concentrations (0 to 40 μM) of SQ109, and FCCP (8 μM) were added where indicated. (D) Changes in safranine fluorescence after addition of SQ109 (4 to 40 μM) or FCCP (8 μM), as shown in panel C. The results are the means ± standard deviation (SD) values from three independent experiments.
Sterol biosynthesis inhibition.
T. cruzi epimastigotes (Y strain) were grown for 96 h at 28°C in LIT medium supplemented with 10% heat-inactivated fetal bovine serum in the absence or presence of 6 μM SQ109. The cells from the cultures were washed with buffer A with glucose (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM d-glucose, and 50 mM HEPES [pH 7.0]) and pelleted, and chloroform-methanol (2:1 [vol/vol]) was added (∼30 ml of solvent per g of cells [wet weight]), followed by vigorous stirring and overnight incubation at 4°C. The extract was then filtered using Whatman no. 2 paper, and the filtrate (a single phase) was dried under nitrogen. The extract was reextracted using chloroform-methanol (9:1 [vol/vol]) and dried under nitrogen. The extracted lipids were saponified with 5 ml of ethanolic-potassium hydroxide (5 g of KOH, 7 ml of water, 14 ml of ethanol) for 1 h at 80°C. After cooling, 2.5 ml of water and 1.5 ml of hexane were added and the phases allowed to separate. The aqueous phase was then reextracted twice with hexane. The hexane extracts were dried under nitrogen, dissolved in chloroform, and converted to the trimethylsilyl derivatives by using bis-trimethylsilyl acetamide. Both the standards and samples (1 μl) were injected in split mode (10:1) on a gas chromatography-mass spectrometry (GC/MS) system, which consisted of an Agilent 6890 (Agilent, Inc., Palo Alto, CA, USA) gas chromatograph, an Agilent 5973 mass selective detector, and an HP 7683B autosampler. The samples were analyzed on a 30-m DB5 column with a 0.32-mm inside diameter (i.d.) and 0.25-μm film thickness, with an injection port temperature of 300°C, interface set to 300°C, ion source adjusted to 230°C, and MS quadrupole at 150°C. The helium carrier gas was set at a constant flow rate of 2.7 ml min−1. The temperature program was 2 min at 250°C, followed by an oven temperature increase of 25°C min−1 to 320°C for 10 min. The mass spectrometer was operated in positive electron impact (EI) mode at a 69.9 eV ionization energy in the m/z 50 to 800 scan range. The spectra of all chromatogram peaks were analyzed using the HP ChemStation (Agilent) program. The identifications and quantifications were performed using the mass spectra obtained from authentic standards and with NIST08 and W8N08 libraries (John Wiley & Sons, Inc., USA).
Synthetic aspects.
SQ109 and its analogs were from the batches whose syntheses and characterization were described previously (24).
Expression and purification of T. cruzi squalene synthase.
T. cruzi squalene synthase (TcSQS) expression was induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 25°C for 18 h and human SQS (HsSQS) with 1.0 mM IPTG at 37°C for 6 h. The cell pastes were harvested by centrifugation at 4,000 × g for 15 min and resuspended in lysis buffer containing 25 mM Tris-HCl (pH 7.5), 20 mM imidazole, and 150 mM NaCl. Cell lysate was prepared with a French pressure cell press (AIM-AMINCO Spectronic Instruments) and then centrifuged at 17,000 × g for 30 min to remove the cell debris. The cell extract was loaded onto a lysis buffer-equilibrated nickel-nitrilotriacetic acid (Ni-NTA) column, followed by washing with 20 mM imidazole-containing buffer. The His-tagged enzyme was then eluted with an imidazole gradient (from 20 to 250 mM), dialyzed twice against 5 liters of 25 mM Tris-HCl (pH 7.5), and loaded onto a 20-ml DEAE Sepharose Fast Flow column (GE Healthcare Life Sciences). The buffer and gradient were 25 mM Tris (pH 7.5) and 0 to 500 mM NaCl. The purities of the recombinant proteins (>95%) were checked by SDS-PAGE analysis.
Crystallization, data collection, and structure determination of TcSQS and HsSQS.
We used the hanging-drop vapor diffusion method at room temperature for crystallization. In general, 2 μl of TcSQS protein solution (8 mg/ml TcSQS, 25 mM Tris-HCl [pH 7.5], 2.5 mM farnesyl-S-thiolodiphosphate [FSPP]) was mixed with 2 μl of reservoir solution. Single crystals were obtained in 0.1 M Tris (pH 8.5) and 21% polyethylene glycol 3350 (PEG 3350). Crystals of TcSQS were obtained only in the presence of FSPP. To obtain SQ109-bound crystals, we soaked TcSQS-FSPP crystals with cryoprotectant (0.15 M Tris [pH 8.5], 25% PEG 3350, and 8% glycerol) that contained 10 mM SQ109 for 3 h.
For HsSQS, 2 μl of protein solution (5 mg/ml HsSQS, 25 mM Tris-HCl [pH 7.5]) was mixed with 2 μl of reservoir solution. Single crystals were obtained in 0.2 M sodium citrate (pH 8.2) and 28% PEG 3350. Crystals of HsSQS in complex with SQ109 were obtained by cocrystallization (0.2 M sodium citrate [pH 8.2], 24 to 28% PEG 3350, 5 mM SQ109). Prior to data collection at 100 K, the crystals were mounted in a cryoloop and flash-frozen in liquid nitrogen with 0.3 M sodium citrate (pH 8.2), 30% PEG 3350, and 2% glycerol as a cryoprotectant. The diffraction data were collected at beamline BL13A1 of the National Synchrotron Radiation Research Center (NSRRC) (Hsinchu, Taiwan) and processed using the HKL-2000 program. Prior to structural refinements, 5% randomly selected reflections were set aside for calculating Rfree. The crystal structures of TcSQS and HsSQS were solved by using the molecular replacement method with the CNS program (38). The previously determined TcSQS-FSPP (PDB ID 3WCA) and HsSQS-FSPP (PDB ID 3WC9) structures were used as the search models. The TcSQS crystal belongs to the P212121 space group and contains four protein molecules in an asymmetric unit. The HsSQS crystal belongs to the P21 space group and contains six protein molecules in an asymmetric unit. The subsequent incorporation of FSPP, SQ109, and water molecules was at the 1.0 σ map level. All structural refinements were carried out using the CNS (38) and Coot (39) programs. The data collection and structure refinement statistics are summarized in Table S1 in the supplemental material. All of the structural diagrams were drawn using the PyMOL software (40).
Data deposition.
The crystallography, atomic coordinates, and structural factors of SQ109 have been deposited in the Protein Data Bank (www.pdb.org) under IDs 3WSB and 3WSA.
RESULTS AND DISCUSSION
SQ109 rapidly kills trypomastigotes but does not lyse red blood cells.
We first investigated whether SQ109 had any effect on survival of the infective nonproliferative trypomastigote stage of the T. cruzi life cycle, which is of potential interest in the context of the development of blood-sterilizing agents in areas with a high prevalence of Chagas disease, as well as for the treatment of both acute and chronic T. cruzi infections. We obtained trypomastigotes from the first-burst release of T. cruzi-infected LLC-MK2 cells and incubated them with SQ109 for 24 h at 37°C (Fig. 3). As can be seen in Fig. 3A, parasite survival was greatly decreased in the presence of SQ109, with an IC50 of 50 ± 8 nM. For comparison, crystal violet (previously used in some areas as a blood-sterilizing agent) had an IC50 of ∼12 μM in this assay, while benznidazole (the drug used to treat acute infections) is essentially inactive under the same conditions (IC50, >400 μM). SQ109 had little effect on red blood cell hemolysis (Fig. 3B), for which the 50% effective concentration (EC50) was >80 μM, leading to a selectivity index (EC50 of red cells/IC50 of T. cruzi trypomastigotes) of >1,600.
FIG 3.

Effects of SQ109 on trypomastigotes and red blood cells. (A) Effects of SQ109 on LLC-MK2 culture-derived trypomastigotes. Activity was evaluated after a 24-h treatment with SQ109 at 37°C. *, statistically significant at a P value of <0.001. (B) Hemolysis of human red blood cells treated with SQ109. A suspension of fresh defibrinated human blood was treated with SQ109 (10 to 80 μM) and incubated at 37°C and 5% CO2 for 24 h, and the absorbance at 540 nm was read. Amphotericin B (AM-B) was used as the hemolytic reference drug and Triton X-100 as the positive control. All experiments were conducted in triplicate, 3 times.
Scanning electron microscopy (SEM) of drug-treated trypomastigotes revealed the formation and release to the extracellular medium of vesicles (Fig. 4). Only a few vesicles were seen after a 6-h incubation with 75 nM SQ109, and most were associated with the cell surface of the parasite (Fig. 4A and B). However, after a 12-h treatment, the trypomastigotes displayed a very large number of vesicles having diameters of ∼0.5 nm (Fig. 4C and D), indicating that incubation with SQ109 leads to cell lysis.
FIG 4.

Effects of 75 nM SQ109 on T. cruzi trypomastigotes, as observed by scanning electron microscopy. (A) Untreated trypomastigotes exhibit a (typical) elongated shape and have a smooth cell surface. (B to D) Trypomastigotes treated with SQ109 for 6 h (B) and 12 h (C and D) show torsion of the cell body, the appearance of a large number of vesicles associated with the plasma membrane (arrows), and wrinkling of the parasite's surface. Scale bars = 2.5 μm. The observation of deranged morphologies at 6 to 12 h suggests that the depletion of sterols (which requires multiple cell divisions, and these cells are not dividing) is not involved.
We also studied drug-treated trypomastigotes by using transmission electron microscopy (TEM) (Fig. 5). The untreated parasites exhibited a characteristic ultrastructure with normal flagellum and kinetoplast organization (Fig. 5A). Incubation with 75 nM SQ109 for 12 h caused an intense alteration of the kinetoplast organization (Fig. 5B and C), as well as release to the extracellular medium of membranous material (Fig. 5C and D) resembling a membrane-shedding process. Such fast and potent activity suggests that the primary mechanism of action against this stage of the parasite may not be due to the inhibition of sterol biosynthesis, because in these nonproliferating cells, the sterol turnover rate is expected to be extremely low (several days). Likewise, it seems unlikely that the inhibition of quinone biosynthesis is involved (again because of the times needed to deplete quinone pools), suggesting, as with bacteria (24), that one mode of SQ109 action in trypomastigotes may be as an uncoupler, collapsing the inner mitochondrial membrane potential (and ΔpH), as reported elsewhere for dronedarone and amiodarone (12, 13) against T. cruzi and L. amazonensis. We examined this possibility and report the results below.
FIG 5.

Effects of 75 nM SQ109 on T. cruzi trypomastigotes treated for 12 h and visualized by TEM. (A) Untreated trypomastigotes. Kinetoplasts (k) and flagella (f) are indicated. (B to D) SQ109-treated trypomastigotes showing a loss of kinetoplast DNA (kDNA) organization (arrow) (B and C) and release to the extracellular medium of membrane sheets and vesicles (arrowheads) (C, D, and inset). Bars: 0.5 μm (A to D); 1 μm (D inset).
Effects of SQ109 on epimastigotes.
We next investigated the effects of SQ109 and the three analogs 9 to 11 (24) (Fig. 6) on the growth of T. cruzi epimastigotes (equivalent to the proliferative stages present in the hindgut of the insect vectors) grown axenically in LIT medium.
FIG 6.
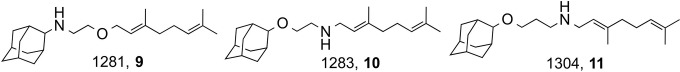
SQ109 analogs.
A representative set of cell proliferation curves as a function of time in the presence of various concentrations of SQ109 (500 nM to 10 μM) is shown in Fig. 7A, which yields an IC50 of 4.6 ± 1 μM. The IC50s for the three other compounds tested were similar (compound 9, 6.9 μM; compound 10, 9.3 μM; compound 11, 6.1 μM; data not shown). We investigated these three compounds since they have been reported to have activities against both M. tuberculosis and the malaria parasite P. falciparum, with compound 10 being 5× more active than was SQ109 against M. tuberculosis; however, with T. cruzi, all three compounds were slightly less active than was SQ109. As can be seen in the photomicrographs (obtained by using a light microscope) in Fig. 7B to D, treatment with 4.6 μM SQ109 (IC50) resulted in the rounding of many parasites, just as was observed with amiodarone treatment of T. cruzi (32). This can be seen even more clearly in the scanning electron microscopy (SEM) results shown in Fig. 7E to H, in which rounding of the cell body in the treated epimastigotes, as well as the appearance of large depressions (Fig. 7F to H), is apparent.
FIG 7.

SQ109 effects on epimastigotes. (A) Effects of various concentrations of SQ109, added at 24 h, on the growth of T. cruzi epimastigotes (Y strain, TcII) treated with SQ109 for 120 h at 28°C. The parasites were cultured in the absence (control) or presence of SQ109 (from 0.5 to 10 μM). The experiments were performed in triplicate, and the bars represent the standard deviations. (B to D) Light microscopy of epimastigotes treated with SQ109. (B) Untreated parasite with kinetoplast (k) shown. Epimastigotes were treated with 4.6 μM SQ109 for 24 h (C) and 48 h (D) and showed rounding of the cell body and the presence of vacuoles in the cytoplasm (arrows). Scale bars = 5 μm (B to D). (E to H) Effects of SQ109 on T. cruzi morphology, as revealed by SEM. SQ109 causes drastic morphology alterations in T. cruzi epimastigotes treated with 4.6 μM SQ109 after 24 and 48 h of treatment. (E) Untreated epimastigotes have a typical elongated shape with a smooth cell surface. (F and G) Epimastigotes treated for 24 h show plasma membrane alterations, rounding of the cell body (arrowhead), and depression of the cell surface (arrows). (H) Epimastigotes treated for 48 h with SQ109 show large plasma membrane depressions (arrows), which were seen also in some cells at shorter times. Scale bars = 2.5 μm (E to H). As with trypomastigotes, the observation of major changes in cell morphology within a short time span (24 h) suggests that the inhibition of enzymes involved in sterol biosynthesis may not be involved.
Thin sections of treated epimastigotes observed by transmission electron microscopy (TEM) also showed drastic changes in the Golgi complex cisternae, the appearance of mitochondrial swelling, the formation of myelin-like figures, and cytoplasmic vesiculation (see Fig. S1 and S2 in the supplemental material).
Effects of SQ109 on intracellular amastigotes, and synergy with posaconazole.
We next tested SQ109 (and the three analogs) against the proliferation of T. cruzi (Y strain) intracellular amastigotes in cultured murine peritoneal macrophages in a 96-hour assay. The typical dose-response results (for SQ109) are shown in Fig. 8A, from which we deduce an IC50 of 1.2 ± 0.4 μM. For the analogs, the IC50s were 1.6 μM for compound 9, 1.9 μM for compound 10, and 1.7 μM for compound 11 (data not shown). In a second series of experiments with SQ109 against the CL strain, we found an IC50 of 520 ± 70 nM under conditions for which the IC50 for benznidazole was 1.3 ± 0.5 μM (n = 3). As can be seen from the isobologram shown in Fig. 8B, SQ109 acts synergistically with posaconazole against amastigotes, with a fractional inhibitory concentration index (FICI) of 0.48. In comparison, the FICI for the amiodarone-posaconazole combination reported previously is slightly better, at 0.42 (7), but any FICI of <0.5 is regarded as demonstrating synergy. However, as can be seen in Fig. 8C, there is some host cell toxicity with SQ109 alone (CC50, ≈9 ± 2.5 μM), although, as noted, SQ109 is already in phase II clinical trials for tuberculosis.
FIG 8.

Effects of SQ109 on amastigotes, and synergy with posaconazole. (A) Effect of SQ109 on intracellular amastigotes. (B) Isobologram for posaconazole and SQ109 against intracellular amastigotes; the IC50s are plotted for the drugs acting alone or in combination. The FICI is 0.48 and indicates synergy. (C) Effects of SQ109 on LLC-MK2 cells. The bars represents the means ± standard deviations (n = 3).
We also observed the effects of SQ109 on intracellular amastigotes by TEM (Fig. 9). The untreated parasites exhibited a characteristic ultrastructure with a normal flagellar pocket, kinetoplast organization, nucleus, and Golgi complex (Fig. 9A). The incubation of intracellular amastigotes with 1.5 μM SQ109 for 96 h caused alterations in the Golgi complex cisternae, extreme accumulation of vesicles in the cytoplasm of the parasite, and the formation of structures similar to autophagosomes (Fig. 9B and C). The cells treated for 96 h with the combination of posaconazole (0.25 nM) plus SQ109 (0.4 μM), concentrations that were chosen because they correspond to those at which the two compounds given together had synergistic effects in vitro (Fig. 8B), showed extreme mitochondrial swelling, as well as the formation of numerous vesicles in the flagellar pocket (Fig. 9D). These vesicles resemble those previously described in amastigotes treated with 1 nM posaconazole for 96 h (32).
FIG 9.

TEM images of intracellular amastigotes treated with SQ109 or SQ109 plus posaconazole for 96 h. (A) Untreated intracellular amastigotes. Intracellular amastigotes treated with 1.5 μM SQ109 caused alterations in Golgi complex cisternae (arrows) (B) and formation of structures similar to autophagosomes (stars) (C). (D) Treatment of intracellular amastigotes with 0.25 nM posaconazole plus 0.4 μM SQ109 shows mitochondrial swelling and numerous vesicles in the flagellar pocket. k, kinetoplast; n, nucleus; G, Golgi; fp, flagellar pocket.
SQ109 collapses the inner mitochondrial membrane potential in T. cruzi and releases H+ from intracellular acidic compartments.
Since the effects of SQ109 on cell activity/growth/ultrastructure were quite rapid, this seemed to rule out the possibility that the inhibition of, e.g., quinone or sterol biosynthesis was a primary mechanism of action (and there is no mmpL3 in T. cruzi), so we next tested whether SQ109 had effects on the proton motive force (more specifically, the inner mitochondrial membrane potential [Δψm]) using the safranine method (36, 37) with epimastigotes. Figure 2A shows that addition of 10 μM SQ109 decreased the Δψm, which was further reduced by addition of the protonophore uncoupler FCCP [carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone] at 8 μM. Figure 2B shows that the T. cruzi mitochondria were able to phosphorylate ADP, as demonstrated by the small decrease in the Δψm after its addition. This activity was inhibited by the ATP synthase inhibitor oligomycin (2 μg/ml). In addition, the mitochondria were able to transport Ca2+, as shown by the decrease in the Δψm after the addition of CaCl2, and the Δψm returned to basal levels after the addition of the Ca2+-chelator EGTA. The further addition of SQ109, followed by FCCP, collapsed the Δψm. Figure 2C and D show that SQ109 collapsed the Δψm in a dose-dependent manner, and SQ109 alone (in the absence of digitonin; Fig. 2A, top trace) or solvent (0.2% DMSO) alone had no effect. These results show that mitochondria in permeabilized T. cruzi epimastigotes are able to develop an inner mitochondrial membrane potential, phosphorylate ATP (as shown by the transient collapse in the Δψm by ADP), and transport Ca2+ and that SQ109 collapses the Δψm in a dose-dependent manner. These effects on the proton motive force are very rapid, as expected, are very similar to those observed previously for SQ109 in bacterial systems (23, 24), and are likely to make a significant contribution to the inhibition of cell growth.
In addition to collapsing the Δψm in succinate-energized mitochondria, SQ109 released H+ from intracellular compartments in the same manner as that observed previously with another lipophilic base, amiodarone, acting on Leishmania mexicana promastigotes (8). As shown in Fig. S3 in the supplemental material, SQ109 (10 μM) caused a rapid (∼200 to 300 s) release of the fluorescent indicator acridine orange from intracellular compartments, an effect that was enhanced upon the addition of the antiporter nigericin. Similar effects are seen with the reverse addition (nigericin, and then SQ109), just as with dronedarone/amiodarone and nigericin in L. mexicana (8). These results indicate, therefore, that there is a release of H+ from acidic compartments (e.g., acidocalcisomes and reservosomes) in T. cruzi, in addition to the effects on succinate-energized mitochondria. Overall, these results show that the Δψm in T. cruzi mitochondria is collapsed by SQ109, that H+ is released from intracellular compartments, and that both effects occur rapidly (within a few minutes), effects that are expected to contribute to cell death.
Structure of SQ109 bound to squalene synthase.
As noted above, SQ109 acts as an uncoupler, in addition to releasing H+ from intracellular compartments such as acidocalcisomes. These effects are also seen with the lipophilic bases amiodarone and dronedarone (compound 7) (7), and with these two compounds, sterol biosynthesis is also inhibited. In addition, in previous work, we found that SQ109 was an inhibitor of the bacterial enzyme dehydrosqualene synthase, a squalene synthase homolog (26, 27), and we reported (25) two X-ray crystallographic structures of SQ109 bound to S. aureus dehydrosqualene synthase (SaCrtM; PDB IDs 4EA1 and 4EA2). We also found that SQ109 was a modest (IC50, ∼100 μM) inhibitor of T. cruzi squalene synthase (25), whose structure we recently reported (33). Since SQS inhibition is of interest as a T. cruzi drug target, we next sought to determine how it binds, since clearly, as with amiodarone, targeting both sterol biosynthesis and the proton motive force (PMF) should lead to a potent inhibition of cell growth. We used the recombinant protein construct described previously (41) and crystallized TcSQS in the presence of the farnesyl diphosphate (FPP) substrate analog FSPP (farnesyl-S-thiolodiphosphate), and then we soaked the crystals in an SQ109 cryoprotectant solution. In addition, we obtained the structure of human SQS (HsSQS) complexed with SQ109, this time by cocrystallization. Full data acquisition and refinement details are given in Table S1 in the supplemental material. The electron densities can be found in Fig. S4 in the supplemental material.
In Fig. 10A, we show a stereo view of the TcSQS + SQ109 + FSPP complex, and in Fig. 10B, we show a magnified view of the active site region with the bound ligands (SQ109 and FSPP). The TcSQS structure is similar to those reported for HsSQS and SaCrtM, and, as expected, TcSQS contains two farnesyl diphosphate (FPP) binding sites, S1 and S2. S1 is the allylic diphosphate-binding site in which FPP ionizes to form a farnesyl cation-diphosphate-Mg2+ species. This species then undergoes nucleophilic attack from the double bond in the FPP in the second site, S2, to generate (after rearrangement) presqualene diphosphate. As can be seen in Fig. 10A and B, SQ109 occupies both the S1 and the top part of the S2 site, perhaps mimicking a carbocation transition state/reactive intermediate, while the FPP analog FSPP occupies the bottom part of the S2 site and is tilted away at the top of the site. In the HsSQS + SQ109 structure (the active site shown in Fig. 10C), SQ109 binds in an essentially flipped-around manner to that seen with the TcSQS structure. It remains to be determined whether potent and selective TcSQS inhibitors can be developed based on these structures, but they do provide a starting point.
FIG 10.

SQ109 binds to the SQS active site. (A) T. cruzi SQS + FSPP + SQ109 complex (PDB ID 3WSB). Lime green, protein; purple, FSPP; cyan, SQ109. (B) Ligands in superimposed CrtM + FSPP and TcSQS + FSPP + SQ109 complexes. Green, FSPP in CrtM; purple, FSPP in TcSQS; cyan, SQ109 in TcSQS. (C) Ligands in superimposed human SQS + SQ109 (PDB ID 3WSA) and TcSQS + FSPP + SQ109 complexes. Orange, SQ109 in HsSQS; purple, FSPP in TcSQS; cyan, SQ109 in TcSQS. Although it is a very modest SQS inhibitor, SQ109 might be a lead for the development of inhibitors that target prenyltransferases and the Δψm, in basically the same way that amiodarone and dronedarone do.
Effects of SQ109 on sterol biosynthesis in T. cruzi.
We next sought to see if there were any significant effects of SQ109 on sterol biosynthesis, because, at least in amastigotes, there was synergy with the azole posaconazole, although clearly this synergy might also arise from effects on Ca2+ homeostasis (which is also seen with posaconazole [7]), or both effects might indeed be important. In untreated epimastigotes (see Fig. S5A in the supplemental material) we found that the major sterols (Fig. 11, compounds 13 and 14) had reduced side chains (compound 13, ergosta-5,7-dien-3-β-ol, and compound 14, 24-ethyl-cholesta-5,7-diene-3β-ol). Upon treatment with SQ109, three new species (compounds 15, 16, and 17) appeared: 24-methyl-cholesta-5,7,24-trien-3β-ol, 24-ethyl-cholesta-5,8,22-trien-3β-ol, and 24-ethyl-cholestadiene-5,8,24-trien-3β-ol (see Fig. S5B in the supplemental material). All have side-chain unsaturation that was not seen in the absence of SQ109, with compound 13 being the reduced form of compound 15 and compound 14 the reduced form of compound 17, suggesting that SQ109 may inhibit an NADPH-dependent reductase. Taken together with those of the previous section, these results are consistent with the observation that there are synergistic interactions between SQ109 and posaconazole, since both affect sterol biosynthesis and they can both affect cation homeostasis. There was, however, no decrease in the total endogenous sterol pool with SQ109, ruling out oxidosqualene cyclase and squalene synthase as primary targets, since their inhibition would block the formation of compounds 15 to 17.
FIG 11.

Structures of sterols of interest.
The SQ109 mechanism of action.
Answering the question “what is the mechanism of action of SQ109 against T. cruzi?” is clearly challenging, since we have data on three distinct life cycle forms of the parasite, the clinically relevant trypomastigotes and amastigotes, as well as the extracellular/insect form epimastigotes, to consider.
It is certainly theoretically possible that there are completely different mechanisms of action in each cell type. Alternatively, there might be very similar mechanisms of action, and we propose here that one major mechanism of action involves SQ109 acting as an uncoupler. SQ109 has been shown to act as an uncoupler in bacteria (24), and the same effect is seen here with epimastigotes, in which SQ109 collapses the inner mitochondrial membrane potential (Δψm) in addition to releasing H+ from intracellular acidic compartments, such as the acidocalcisomes. These effects occur on a very rapid time scale and are consistent with the deranged morphologies of trypomastigotes (at 6 to 12 h) and epimastigotes (at 12 to 24 h). We also find that SQ109 synergizes with posaconazole (in amastigotes). This might arise from both compounds targeting sterol biosynthesis, ion channels/uncoupling, both processes, or an as yet unidentified target(s). The effects on Δψm/ΔH+ are a likely mechanism, since as we show here and elsewhere, SQ109 is an uncoupler (24); plus, as shown elsewhere, posaconazole, in addition to its effects on sterol biosynthesis, acts to increase intracellular Ca2+ (Cai2+) (in epimastigotes). The synergistic effects seen here are very reminiscent of those found with the combination amiodarone plus posaconazole, in which amiodarone affects both Cai2+ and sterol biosynthesis, as does posaconazole (7). However, in that case, amiodarone inhibits sterol biosynthesis at the level of oxidosqualene cyclase, while the effects of SQ109 on sterol biosynthesis are more downstream and there is no block of endogenous sterol biosynthesis. Moreover, the rapid (<24-h) onset of growth arrest by SQ109 appears incompatible with the notion that its primary mechanism of action is the blockade of de novo synthesis of 24-alkyl sterols, since the results of many studies indicate that the full proliferation arrest is associated only with the complete disappearance of mature (24-alkyl and 14-desmethyl) endogenous sterols. This means that for T. cruzi, whose generation time is 24 to 30 h, complete growth inhibition is observed only after 72 to 120 h of drug exposure, while for Leishmania spp., for which the generation time is 12 to 18 h, complete growth arrest in the presence of optimal drug concentration is seen at 36 to 72 h (33, 42–50). For the same reason, T. cruzi trypomastigotes, which are nonproliferative cells, are refractory to bona fide ergosterol biosynthesis inhibitors such as ketoconazole, posaconazole, terbinafine, azasterols, and aryl-quinuclidines in the nanomolar range of concentrations at which they are active against epimastigotes and intracellular amastigotes. That is not what is seen with SQ109, for which there is potent (50 nM) activity at short (6-h) time periods.
Potential clinical relevance.
The results described above are of interest, since we find that the tuberculosis drug SQ109 is active against all three life cycle stages of the trypanosomatid parasite T. cruzi, the causative agent of Chagas disease. The most potent activity is found against the highly infective trypomastigote form, for which the IC50 is ∼50 nM. This result is of potential importance in the context of the sterilization of blood (for transfusions), given that there is very little effect on red cell hemolysis (EC50, >80 μM, selectivity index, >1,600). Activity is lower against extracellular epimastigotes (IC50, ∼5 μM) but better against the clinically relevant intracellular amastigote forms (IC50, ∼0.5 to 1 μM, depending on the strain). The IC50s obtained against the clinically relevant trypomastigotes (∼16.5 ng/ml) and intracellular amastigotes (∼0.33 μg/ml) are 50- and 2.5-fold lower than the MIC against drug-resistant M. tuberculosis (18).
Importantly, the IC50s against both vertebrate stages (trypomastigotes and amastigotes) fall in the range of the plasma and internal organ levels of SQ109 found in humans receiving well-tolerated doses of the drug. In preclinical studies, it has been shown that although SQ109 has a limited oral bioavailability, probably due to its highly lipophilic character, it strongly concentrates in mouse internal extravascular organs, such as the lung, spleen, liver, and heart, in which the tissue levels are 15- to >45-fold higher than those in plasma (51). In humans, the drug has a large volume of distribution and a long terminal half-life (61.1 h for a 300-mg oral dose) (18), two important pharmacokinetic parameters that in previous work have been linked to the remarkable in vivo anti-T. cruzi activities of sterol biosynthesis inhibitors, such as posaconazole (52, 53). Based on these results, it can be inferred that the plasma and expected internal organ levels of SQ109 in humans receiving well-tolerated doses (300 mg/day) of SQ109 (18) fall in the range of the IC50s found here against T. cruzi trypomastigotes and intracellular amastigotes, making SQ109, which is currently in advanced clinical trials for tuberculosis, of interest in the context of the development of drugs (either in monotherapy, or in combination therapies) for the etiological treatment of Chagas disease. Further work on animal models is in progress.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Synchrotron Research Center of Taiwan for beam time allocation and data collection assistance.
This work was supported in part by the U.S. Public Health Service (NIH grants GM065307, CA158191, and AI107663), the National Basic Research Program of China (grant 2011CBA00805), FONICIT-Venezuela grant 2011000884, the Brazilian funding agencies CNPq, CAPES, FINEP, and FAPERJ, the Junta de Andalucía (grants BIO-199 and P09-CVI-5367), and the Plan Nacional (grant SAF2010-20059).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03972-14.
REFERENCES
- 1.World Health Organization. 2014. Chagas disease (American trypanosomiasis). World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs340/en/. [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2013. Parasites—American trypanosomiasis (also known as Chagas disease). Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/parasites/chagas/epi.html. [Google Scholar]
- 3.Lee BY, Bacon KM, Bottazzi ME, Hotez PJ. 2013. Global economic burden of Chagas disease: a computational simulation model. Lancet Infect Dis 13:342–348. doi: 10.1016/S1473-3099(13)70002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellis C. 2003. Antifungals: doing the two step. Nat Rev Drug Discov 2:605. doi: 10.1038/nrd1167. [DOI] [Google Scholar]
- 5.Courchesne WE. 2002. Characterization of a novel, broad-based fungicidal activity for the antiarrhythmic drug amiodarone. J Pharmacol Exp Ther 300:195–199. doi: 10.1124/jpet.300.1.195. [DOI] [PubMed] [Google Scholar]
- 6.Gupta SS, Ton VK, Beaudry V, Rulli S, Cunningham K, Rao R. 2003. Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J Biol Chem 278:28831–28839. doi: 10.1074/jbc.M303300200. [DOI] [PubMed] [Google Scholar]
- 7.Benaim G, Sanders JM, Garcia-Marchan Y, Colina C, Lira R, Caldera AR, Payares G, Sanoja C, Burgos JM, Leon-Rossell A, Concepcion JL, Schijman AG, Levin M, Oldfield E, Urbina JA. 2006. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J Med Chem 49:892–899. doi: 10.1021/jm050691f. [DOI] [PubMed] [Google Scholar]
- 8.Serrano-Martín X, Garcia-Marchan Y, Fernandez A, Rodriguez N, Rojas H, Visbal G, Benaim G. 2009. Amiodarone destabilizes intracellular Ca2+ homeostasis and biosynthesis of sterols in Leishmania mexicana. Antimicrob Agents Chemother 53:1403–1410. doi: 10.1128/AAC.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Macedo-Silva ST, de Oliveira Silva TL, Urbina JA, de Souza W, Rodrigues JC. 2011. Antiproliferative, ultrastructural, and physiological effects of amiodarone on promastigote and amastigote forms of Leishmania amazonensis. Mol Biol Int 2011:876021. doi: 10.4061/2011/876021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paniz-Mondolfi AE, Perez-Alvarez AM, Lanza G, Marquez E, Concepcion JL. 2009. Amiodarone and itraconazole: a rational therapeutic approach for the treatment of chronic Chagas' disease. Chemotherapy 55:228–233. doi: 10.1159/000219436. [DOI] [PubMed] [Google Scholar]
- 11.Paniz-Mondolfi AE, Perez-Alvarez AM, Reyes-Jaimes O, Socorro G, Zerpa O, Slova D, Concepcion JL. 2008. Concurrent Chagas' disease and borderline disseminated cutaneous leishmaniasis: the role of amiodarone as an antitrypanosomatidae drug. Ther Clin Risk Manag 4:659–663. doi: 10.2147/TCRM.S2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benaim G, Paniz Mondolfi AE. 2012. The emerging role of amiodarone and dronedarone in Chagas disease. Nat Rev Cardiol 9:605–609. doi: 10.1038/nrcardio.2012.108. [DOI] [PubMed] [Google Scholar]
- 13.Benaim G, Hernandez-Rodriguez V, Mujica-Gonzalez S, Plaza-Rojas L, Silva ML, Parra-Gimenez N, Garcia-Marchan Y, Paniz-Mondolfi A, Uzcanga G. 2012. In vitro anti-Trypanosoma cruzi activity of dronedarone, a novel amiodarone derivative with an improved safety profile. Antimicrob Agents Chemother 56:3720–3725. doi: 10.1128/AAC.00207-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spaniol M, Bracher R, Ha HR, Follath F, Krahenbuhl S. 2001. Toxicity of amiodarone and amiodarone analogues on isolated rat liver mitochondria. J Hepatol 35:628–636. doi: 10.1016/S0168-8278(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 15.Peña A, Calahorra M, Michel B, Ramirez J, Sanchez NS. 2009. Effects of amiodarone on K+, internal pH and Ca2+ homeostasis in Saccharomyces cerevisiae. FEMS Yeast Res 9:832–848. doi: 10.1111/j.1567-1364.2009.00538.x. [DOI] [PubMed] [Google Scholar]
- 16.Felser A, Blum K, Lindinger PW, Bouitbir J, Krahenbuhl S. 2013. Mechanisms of hepatocellular toxicity associated with dronedarone–a comparison to amiodarone. Toxicol Sci 131:480–490. doi: 10.1093/toxsci/kfs298. [DOI] [PubMed] [Google Scholar]
- 17.Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, Einck L, Nacy CA. 2005. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother 56:968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- 18.Sacksteder KA, Protopopova M, Barry CE III, Andries K, Nacy CA. 2012. Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol 7:823–837. doi: 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 20.Stehr M, Elamin AA, Singh M. 2014. Filling the pipeline—new drugs for an old disease. Curr Top Med Chem 14:110–129. doi: 10.2174/1568026613666131113152908. [DOI] [PubMed] [Google Scholar]
- 21.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE III, Boshoff HI. 2012. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makobongo MO, Einck L, Peek RM Jr, Merrell DS. 2013. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS One 8:e68917. doi: 10.1371/journal.pone.0068917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anonymous. 2008. Sq109. Tuberculosis (Edinb) 88:159–161. doi: 10.1016/S1472-9792(08)70026-X. [DOI] [PubMed] [Google Scholar]
- 24.Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E. 2014. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem 57:3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin FY, Liu YL, Li K, Cao R, Zhu W, Axelson J, Pang R, Oldfield E. 2012. Head-to-head prenyl tranferases: anti-infective drug targets. J Med Chem 55:4367–4372. doi: 10.1021/jm300208p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. 2008. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin FY, Liu CI, Liu YL, Zhang Y, Wang K, Jeng WY, Ko TP, Cao R, Wang AH, Oldfield E. 2010. Mechanism of action and inhibition of dehydrosqualene synthase. Proc Natl Acad Sci U S A 107:21337–21342. doi: 10.1073/pnas.1010907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ Jr. 2000. Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J Biol Chem 275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 29.Zingales B, Andrade SG, Briones MR, Campbell DA, Chiari E, Fernandes O, Guhl F, Lages-Silva E, Macedo AM, Machado CR, Miles MA, Romanha AJ, Sturm NR, Tibayrenc M, Schijman AG, Second Satellite Meeting . 2009. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz 104:1051–1054. doi: 10.1590/S0074-02762009000700021. [DOI] [PubMed] [Google Scholar]
- 30.Camargo EP. 1964. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev Inst Med Trop Sao Paulo 6:93–100. [PubMed] [Google Scholar]
- 31.Henriques C, Moreira TLB, Maia-Brigagao C, Henriques-Pons A, Carvalho TMU, de Souza W. 2011. Tetrazolium salt based methods for high-throughput evaluation of anti-parasite chemotherapy. Anal Methods 3:2148–2155. doi: 10.1039/c1ay05219e. [DOI] [Google Scholar]
- 32.Veiga-Santos P, Barrias ES, Santos JF, de Barros Moreira TL, de Carvalho TM, Urbina JA, de Souza W. 2012. Effects of amiodarone and posaconazole on the growth and ultrastructure of Trypanosoma cruzi. Int J Antimicrob Agents 40:61–71. doi: 10.1016/j.ijantimicag.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Shang N, Li Q, Ko TP, Chan HC, Li J, Zheng Y, Huang CH, Ren F, Chen CC, Zhu Z, Galizzi M, Li ZH, Rodrigues-Poveda CA, Gonzalez-Pacanowska D, Veiga-Santos P, de Carvalho TM, de Souza W, Urbina JA, Wang AH, Docampo R, Li K, Liu YL, Oldfield E, Guo RT. 2014. Squalene synthase as a target for Chagas disease therapeutics. PLoS Pathog 10:e1004114. doi: 10.1371/journal.ppat.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hallander HO, Dornbusch K, Gezelius L, Jacobson K, Karlsson I. 1982. Synergism between aminoglycosides and cephalosporins with antipseudomonal activity: interaction index and killing curve method. Antimicrob Agents Chemother 22:743–752. doi: 10.1128/AAC.22.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veiga-Santos P, Pelizzaro-Rocha KJ, Santos AO, Ueda-Nakamura T, Dias Filho BP, Silva SO, Sudatti DB, Bianco EM, Pereira RC, Nakamura CV. 2010. In vitro anti-trypanosomal activity of elatol isolated from red seaweed Laurencia dendroidea. Parasitology 137:1661–1670. doi: 10.1017/S003118201000034X. [DOI] [PubMed] [Google Scholar]
- 36.Vercesi AE, Bernardes CF, Hoffmann ME, Gadelha FR, Docampo R. 1991. Digitonin permeabilization does not affect mitochondrial function and allows the determination of the mitochondrial membrane potential of Trypanosoma cruzi in situ. J Biol Chem 266:14431–14434. [PubMed] [Google Scholar]
- 37.Huang G, Vercesi AE, Docampo R. 2013. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun 4:2865. doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54(Pt 5):905–921. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrödinger. 2014. PyMOL, a molecular visualization system on an open source foundation, maintained and distributed by Schrödinger. Schrödinger, New York, NY. [Google Scholar]
- 41.Sealey-Cardona M, Cammerer S, Jones S, Ruiz-Perez LM, Brun R, Gilbert IH, Urbina JA, Gonzalez-Pacanowska D. 2007. Kinetic characterization of squalene synthase from Trypanosoma cruzi: selective inhibition by quinuclidine derivatives. Antimicrob Agents Chemother 51:2123–2129. doi: 10.1128/AAC.01454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larralde G, Vivas J, Urbina JA. 1988. Concentration and time dependence of the effects of ketoconazole on growth and sterol synthesis by Trypanosoma (Schizotrypanum) cruzi epimastigotes. Acta Cient Venez 39:140–146. [PubMed] [Google Scholar]
- 43.Urbina JA, Lazardi K, Aguirre T, Piras MM, Piras R. 1988. Antiproliferative synergism of the allylamine SF86-327 and ketoconazole on epimastigotes and amastigotes of Trypanosoma (Schizotrypanum) cruzi. Antimicrob Agents Chemother 32:1237–1242. doi: 10.1128/AAC.32.8.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urbina JA, Vivas J, Visbal G, Contreras LM. 1995. Modification of the sterol composition of Trypanosoma (Schizotrypanum) cruzi apimastigotes by delta 24(25)-sterol methyl transferase inhibitors and their combinations with ketoconazole. Mol Biochem Parasitol 73:199–210. doi: 10.1016/0166-6851(95)00117-J. [DOI] [PubMed] [Google Scholar]
- 45.Rodrigues JCF, Attias M, Rodriguez C, Urbina JA, de Souza W. 2002. Ultrastructural and biochemical alterations induced by 22,26-azasterol, a Δ24(25)-sterol methyltransferase inhibitor, on promastigote and amastigote forms of Leishmania amazonensis. Antimicrob Agents Chemother 46:487–499. doi: 10.1128/AAC.46.2.487-499.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liendo A, Visbal G, Piras MM, Piras R, Urbina JA. 1999. Sterol composition and biosynthesis in Trypanosoma cruzi amastigotes. Mol Biochem Parasit 104:81–91. doi: 10.1016/S0166-6851(99)00129-2. [DOI] [PubMed] [Google Scholar]
- 47.Urbina JA, Payares G, Contreras LM, Liendo A, Sanoja C, Molina J, Piras M, Piras R, Perez N, Wincker P, Loebenberg D. 1998. Antiproliferative effects and mechanism of action of SCH56592 against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob Agents Chemother 42:1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbina JA, Concepcion JL, Rangel S, Visbal G, Lira R. 2002. Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol Biochem Parasitol 125:35–45. doi: 10.1016/S0166-6851(02)00206-2. [DOI] [PubMed] [Google Scholar]
- 49.Urbina JA, Concepcion JL, Caldera A, Payares G, Sanoja C, Otomo T, Hiyoshi H. 2004. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob Agents Chemother 48:2379–2387. doi: 10.1128/AAC.48.7.2379-2387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandes Rodrigues JC, Concepcion JL, Rodrigues C, Caldera A, Urbina JA, de Souza W. 2008. In vitro activities of ER-119884 and E5700, two potent squalene synthase inhibitors, against Leishmania amazonensis: antiproliferative, biochemical, and ultrastructural effects. Antimicrob Agents Chemother 52:4098–4114. doi: 10.1128/AAC.01616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia L, Tomaszewski JE, Hanrahan C, Coward L, Noker P, Gorman G, Nikonenko B, Protopopova M. 2005. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br J Pharmacol 144:80–87. doi: 10.1038/sj.bjp.0705984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urbina JA. 2010. New insights in Chagas disease treatment. Drugs Future 35:409–419. doi: 10.1358/dof.2010.035.05.1484391. [DOI] [Google Scholar]
- 53.Buckner FS, Urbina JA. 2012. Recent developments in sterol 14-demethylase inhibitors for Chagas disease. Int J Parasitol Drugs Drug Resist 2:236–242. doi: 10.1016/j.ijpddr.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.