

Abstract
In a loss-of-viability screen using small molecules against methicillin-resistant Staphylococcus aureus (MRSA) strain USA300 with a sub-MIC of a β-lactam, we found a small molecule, designated DNAC-1, which potentiated the effect of oxacillin (i.e., the MIC of oxacillin decreased from 64 to 0.25 μg/ml). Fluorescence microscopy indicated a disruption in the membrane structures within 15 min of exposure to DNAC-1 at 2× MIC. This permeabilization was accompanied by a rapid loss of membrane potential, as monitored by use of the DiOC2 (3,3′-diethyloxacarbocyanine iodide) dye. Macromolecular analysis showed the inhibition of staphylococcal cell wall synthesis by DNAC-1. Transmission electron microscopy of treated MRSA USA300 cells revealed a slightly thicker cell wall, together with mesosome-like projections into the cytosol. The exposure of USA300 cells to DNAC-1 was associated with the mislocalization of FtsZ accompanied by the localization of penicillin-binding protein 2 (PBP2) and PBP4 away from the septum, as well as mild activation of the vraRS-mediated cell wall stress response. However, DNAC-1 does not have any generalized toxicity toward mammalian host cells. DNAC-1 in combination with ceftriaxone is also effective against an assortment of Gram-negative pathogens. Using a murine subcutaneous coinjection model with 108 CFU of USA300 as a challenge inoculum, DNAC-1 alone or DNAC-1 with a sub-MIC of oxacillin resulted in a 6-log reduction in bacterial load and decreased abscess formation compared to the untreated control. We propose that DNAC-1, by exerting a bimodal effect on the cell membrane and cell wall, is a viable candidate in the development of combination therapy against many common bacterial pathogens.
INTRODUCTION
Staphylococcus aureus is a common bacterial pathogen that is associated with serious infections, including pneumonia, sepsis, osteomyelitis, and endocarditis. Ever since the 1960s, many of the S. aureus strains have become resistant to methicillin (designated MRSA) and related antibiotics, such as oxacillin and cephalosporins. Many of these MRSA infections have occurred in hospitals and long-term care facilities (hospital-acquired MRSA [HA-MRSA] strains). Recently, a more virulent and epidemiologically distinct form of MRSA called community-associated MRSA (CA-MRSA) has been problematic. More importantly, MRSA strains have now become resistant to the last line of antibiotics, including linezolid and vancomycin (1). Compounding the problem is a lack of new antimicrobials due to the continued egress of large pharmaceutical companies from the fields of antibiotic research and development (2).
An alternative approach to finding new antibiotic classes is to enhance the utility of already-available antibiotics using combinatorial screens. In order to do this, we and others have screened libraries of small molecules, including previously approved drugs and those facing patent expiry, in order to identify candidate compounds. Various synthetic oligo-acyl-lysyl (OAK) peptides (3) and molecules, like ticlopidine (4), have been shown to be effective in combination with β-lactams against a clinical MRSA strain. Here, we describe a novel candidate compound called DNAC-1 that acts both in monotherapy against MRSA and in combination therapy with β-lactams against MRSA and other Gram-positive as well as Gram-negative pathogens. DNAC-1 causes defects in membrane morphology, membrane depolarization, mislocalization of FtsZ, penicillin-binding protein 2 (PBP2), and PBP4, and the disruption of cell wall synthesis. We propose that DNAC-1 has a bimodal mechanism of action on the cell membrane and cell wall. In vivo studies with a murine subcutaneous model of infection treated with DNAC-1 and oxacillin revealed a significantly reduced bacterial load in the abscess model, thus supporting the utility of drug discovery based on combination therapy.
MATERIALS AND METHODS
Bacterial strains, strain construction, and media.
The strain used for compound screening was MRSA USA300 (5). We also tested clinical isolates of MRSA (CA-MRSA [20 strains] and HA-MRSA [9 strains]), S. aureus strain Mu50 (vancomycin-intermediate S. aureus [VISA]), Enterococcus faecalis (5 strains), Enterococcus faecium (5 strains), Escherichia coli (2 strains), Klebsiella pneumoniae (6), Klebsiella oxytoca (4), Acinetobacter baumannii, Pseudomonas aeruginosa (5 strains), and Enterobacter aerogenes obtained from the Dartmouth Hitchcock Medical Center, Lebanon, NH, and Achillion Pharmaceuticals, New Haven, CT, for their MICs against DNAC-1. The bacterial cells were grown in Mueller-Hinton medium (Difco) supplemented with calcium and magnesium sulfate for the MIC studies and in tryptone soy broth (TSB) (Difco) for all other assays.
MRSA strain COL was used to assess membrane integrity and permeability by fluorescence microscopy. MRSA strains BCBPM073 and BCBPM162 expressing superfolding green fluorescent protein (sfGFP)-PBP2 (9) and PBP4-yellow fluorescent protein (YFP) (8) fusions, respectively, were used to evaluate the defects in the localization of peptidoglycan synthesis enzymes. For FtsZ localization, a COL derivative expressing FtsZ-cyan fluorescent protein (CFP) ectopically from the spa locus under the control of the Pspac promoter was used (9), using 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for induction. In order to assess cell wall damage, we constructed a strain expressing a vraSR promoter fusion to sfgfp. Briefly, a 779-bp fragment encompassing the coding region of sgfp-p7 was amplified from pTRC99a-P7 (6) using the primers sgfp P3 EcoRV (GCGCGATATCATAAGGAGGATTCGTATGAGTAAAGGAGAAGAACTTTTC) and sgfp P2 NotI (GCTTAGCGGCCGCTTAATGGTGATGATGGTGATGGTCGACTTTGTATAG), digested with EcoRV and NotI (Fermentas), and used to replace the gfpmutP2 gene in pSG5082 (10), giving pFAST3. An 844-bp fragment containing the vraSR promoter region was amplified from COL using the primers PvraSRP1KpnI (GCTGCGGTACCCGGTGCTATTTCTGCGCC) and PvraSRP2XhoI (GCTGCCTCGAGACGTTCAACATAGTTCATAAC), digested with KpnI and XhoI, and cloned into the KpnI/XhoI restriction sites of pFAST3, upstream of sfgfp-p7, resulting in pPvraS, as confirmed by DNA sequencing. pPvraSR was electroporated into the S. aureus RN4220 strain to enable chromosomal integration at the vraSR promoter by homologous recombination, as confirmed by PCR and sequencing reactions; the resulting strain was named RNpPVra. Strain COLpPVra was obtained by transducing the integrated plasmid pPvraSR from RNpPVra to COL using phage 80α, as previously described (11).
Small-molecule screening assay.
We screened 45,000 compounds from a small-molecule collection at the ICCB-Longwood Screening Facility, a part of the New England Regional Centers of Excellence (NERCE), for inhibitory activity against MRSA USA300, using optical density at 620 nm (OD620) as the readout in a 384-well format with and without 16 μg/ml oxacillin (0.25× MIC of oxacillin against USA300). We used cefoxitin as a positive control (5), while cells grown with oxacillin or Mueller-Hinton broth (MHB) alone were used as the negative control. Per a CLSI protocol (29), bacteria in the 384-well plates were grown without shaking. Each well was scaled to the respective positive and negative control to normalize the percent survival using the following equation: % survival = [(OD of the sample − OD of the positive control)/(OD of the sample − OD of the negative control)] × 100. Compounds yielding <50% survival with USA300 were considered to be hits.
MICs by broth microdilution and macrodilution.
The MICs against different strains were determined according to CLSI guidelines and defined as the lowest concentration of antibiotic inhibiting visible growth. The MIC of DNAC-1 was between 2 μg/ml and 4 μg/ml for microdilution and macrodilution, respectively. We used 4 μg/ml as the MIC for DNAC-1 in this paper to avoid confusion between the two techniques.
Determining the bactericidal effect of DNAC-1 during growth.
USA300 cells were grown to the exponential phase (OD620 = 0.5 using an 18-mm borosilicate glass tube in a Spectronic 20 spectrophotometer) in MHB and back-diluted 100-fold. The samples were treated with either dimethyl sulfoxide (DMSO) as a control or 16 μg/ml DNAC-1 with and without 16 μg/ml oxacillin (diluted in DMSO). At specific time points (0, 2, 4, 7, and 24 h), dilutions were made and the cells plated on regular TSB agar with no antibiotic to enumerate the CFU/ml. Each data point represents the mean and standard deviation from three replicate experiments. The Kruskal-Wallis one-way analysis of variance test using GraphPad Prism was applied to test significance, with a P value of <0.05 considered significant.
Checkerboard analysis to test synergy of DNAC-1 with other antibiotics against MRSA.
In order to determine the lowest effective concentration of oxacillin and other antibiotics when combined with DNAC-1, a checkerboard method was used. Briefly, 2-fold dilutions of DNAC-1 from 32 μg/ml (8× MIC) were made in columns of 96-well plates, while 2-fold dilutions of oxacillin or other antibiotic compounds were made in rows to obtain different combinations of drug concentrations. MRSA USA300 was then added to microtiter wells at a concentration of 1 × 105 CFU/ml, incubated at 37°C for 24 h without shaking, and the MIC was defined according to the CLSI protocol. FICAB was defined as the fractional inhibitory concentration (FIC) of drug A in the presence of drug B, and FICBA was defined as the FIC of drug B in the presence of drug A. The FIC index was calculated by FICAB/FICBA. Synergy was defined as an FIC index of ≤0.5, an index between 0.5 and 4 suggests indifference, and an index of >4 suggests inhibition.
Assessing the toxicity of DNAC-1.
Three methods were used to assess cellular toxicity. First, yeast cells were exposed to 20× MIC (determined for USA300) of DNAC-1 to assess direct cellular toxicity based on a loss of viability. Second, 4% sheep red blood cells were exposed to 4× MIC of DNAC-1 and monitored for lysis at OD540 in an in vitro hemolysis assay to evaluate if DNAC-1 possesses any generalized membrane perturbation properties. Third, the release of lactate dehydrogenase from human bronchial epithelial cells (CFBE) exposed to DNAC-1 was monitored. In brief, the CFBE cells were maintained in RPMI 1640 medium (Sigma, St. Louis, MO) with 10% fetal bovine serum and grown with 5% CO2 in an incubator at 37°C. When confluent, the cells in the flask were released with trypsin, collected, and counted. DNAC-1 at predetermined concentrations (16×, 32×, and 64× MIC) in serum-free medium was added to the epithelial cells and incubated for 24 h at 37°C. Triton X-100 at a 2% final concentration was added to the positive-control wells. On the following day, the cytotoxicity reagents were prepared according to the manufacturer's protocol (CytoTox 96 nonradioactive cytotoxicity assay kit; Promega). One hundred microliters of the mixed-detection kit reagent was then added in rapid succession to each of the wells containing CFBE supernatant. The assay plates were then incubated at room temperature (RT) in the dark for 20 min and read at 490 nm using a plate reader.
Macromolecular analysis of cell wall synthesis.
To assay for cell wall synthesis, we measured the incorporation of [3H]lysine into peptidoglycan in the presence of chloramphenicol (25 μg/ml final concentration), which inhibits protein synthesis but allows peptidoglycan synthesis to continue, thus enabling us to adjudge pentapeptide synthesis of lipid II, a precursor of peptidoglycan, in the presence of DNAC-1 at 10× MIC. Ampicillin was used as a cell wall-active antibiotic control. The effects of DNAC-1 and ampicillin on peptidoglycan synthesis were measured using the radiolabeled precursor [3H]lysine (2.5 μCi/ml final concentration) added to mid-exponential-phase cultures (108 CFU/ml) of S. aureus strain ATCC 29213 in chemically defined medium (12), as previously described (13). The radiolabeled precursor was added to the bacterial culture immediately before the antibiotics were added. The negative controls for the macromolecular assays consisted of all reaction materials with no antibiotics added, with the resulting counts used as the 100% values. After an additional 20 min of incubation at 37°C in the presence or absence of antibiotics, the samples were removed for trichloroacetic acid precipitation and subsequent scintillation counter analyses to determine radioactive incorporation into the cell wall; the data were expressed as the percent inhibition of incorporation in comparison to a drug-free control.
Transmission electron microscopy.
USA300 cells grown in MHC at 37°C were treated with either compound DNAC-1 (2× MIC) or DMSO for 90 min, washed twice with phosphate-buffered saline (PBS), and processed for electron microscopy (EM) by fixing with a 10× volume of 2% glutaraldehyde-tannic acid (GTA)–1% paraformaldehyde in 0.1 M Na cacodylate buffer (pH 7.4), postfixed in 1% OsO4 in cacodylate buffer (pH 7.4), embedded, and serially dehydrated in ethanol. The samples were sectioned and stained with uranyl acetate and imaged using a JEOL transmission electron microscope (TEM) 1010 at 100 kV and 20,000× magnification. Thirty fields of each strain with nearly equatorial-cut surfaces were measured for cell wall thickness, and the results were expressed as the means ± standard deviations. Statistical significance was determined using the Kruskal-Wallis test, with a P value of <0.05 considered significant.
Fluorescence microscopy.
For the fluorescence microscopy experiments, strains were incubated overnight in TSB supplemented with either erythromycin (10 μg/ml) or kanamycin (200 μg/ml), when required, at 37°C and back-diluted to fresh TSB and allowed to grow until mid-exponential phase (OD600, ∼0.6). In each case, the cultures were divided among five flasks, and DNAC-1 (in DMSO) was added at either 0.5× (2 μg/ml), 2× (8 μg/ml), or 10× MIC (40 μg/ml); the two remaining flasks were kept as controls with DMSO or TSB alone. The cultures were incubated with DNAC-1 for 15 min or 1 h, after which the cells were pelleted, washed in PBS buffer, and mounted on microscope slides with pads of 1% agarose in PBS. To analyze the activation of the cell wall stress stimulon by DNAC-1, a control with vancomycin at 2× MIC (6 μg/ml) was also used. The displayed values were adjusted for each image for visualization purposes.
When staining was required to assess morphology, the cells were incubated with 2 μg/ml FM 4-64 [N-(3-triethylammoniumpropyl)-4-(p-diethylaminophenyl-hexatrienyl) pyridinium dibromide], Bodipy FL-vancomycin (2 μg/ml), and Hoechst 33342 (4 μg/ml) (all from Molecular Probes) for 5 min at room temperature with shaking and washed before being imaged. When assessing cell viability, the cells were stained with propidium iodide (0.5 μg/ml; Invitrogen) as described above. The cells were imaged using a Zeiss Axio Observer microscope equipped with a Photometrics CoolSNAP HQ2 camera (Roper Scientific) and Metamorph 7.5 software (Molecular Devices), or by structured illumination microscopy (SIM) or laser wide-field microscopy in an Elyra PS.1 microscope (Zeiss) with a scientific complementary metal oxide semiconductor (sCMOS) camera and 5 grating rotations for each channel. The SIM images were reconstructed and analyzed with the Zen software (Zeiss). For the quantification of the signal of fluorescent derivatives of PBPs at the septa, five images were analyzed per condition using ImageJ. Only cells with a complete septum were analyzed.
Assay for membrane potential.
The carbocyanine dye DiOC2 (3,3′-diethyloxacarbocyanine iodide; Life Technologies) was used as described previously (14) to assess membrane potential. S. aureus USA300 cultures were grown to the early exponential phase (OD620, 0.3) in MHB, incubated with 10 μM DiOC2 in PBS with 1% glucose at 24°C for 30 min in the dark, and then transferred to a 384-well plate. The cells were analyzed using a plate reader, Tecan M1000 (excitation [Ex], 485 nm; emission [Em], 680 nm). After establishing a baseline reading, either carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (positive control) or DNAC-1 was added to the wells, and the drop in red fluorescence was monitored over time.
Murine model of subcutaneous infection with MRSA USA300 to evaluate efficacy of DNAC-1.
Three groups of six female BALB/c mice each were used in the subcutaneous infection model. Early-exponential-phase USA300 cells grown in TSB (OD620, 0.35) were harvested, washed, and resuspended in saline to a final concentration of 109 CFU/ml. After shaving and decontaminating an area on the back of each animal with ethanol, each mouse in an assigned group was injected subcutaneously with 108 CFU of USA300 (corresponding to ∼90% infective dose [ID90] [15]) along with the following in a total volume of 100 μl: (i) a single dose of DNAC-1 (8 μg) in DMSO, (ii) DNAC-1 (8 μg) with oxacillin (16 μg) in DMSO, or (iii) a DMSO control. The mice were monitored daily for general appearance and the size of any abscesses and sacrificed at day 8. At sacrifice, an 8-mm biopsy punch was made from the lesions and divided in half, one half for fixation in 10% formalin for histological analysis with hematoxylin and eosin stain, according to standard histological procedure, and the remaining portion was homogenized to determine the number of CFU/g of tissue. The Kruskal-Wallis one-way analysis of variance test using GraphPad Prism was applied to test significance, with a P value of <0.05 considered significant.
RESULTS
Identification of DNAC-1 in a screen for compounds synergistic with β-lactams against MRSA USA300.
We previously demonstrated the effectiveness of using combination therapy consisting of a β-lactam and another cell wall-active antibiotic in killing MRSA (5). To identify small molecules that have an inhibitory activity either alone or in combination with a β-lactam, like oxacillin, we employed a mechanistically unbiased viability screen to assay 45,000 compounds at the New England Regional Centers of Excellence (NERCE) for activities that inhibited the growth of MRSA strain USA300 in the presence of a sub-MIC of oxacillin. Cefoxitin was used as a positive control (5), while DMSO alone served as the negative control. Growth inhibition at 37°C was assessed by a reduction in the OD620 at 24 h compared to that of the negative control. The Z′ value (the statistical effect size) of our assay was 0.9167. We initially identified 100 hits that showed >50% growth inhibition. Rescreening of the cherry-picked compounds using a threshold of 80% growth inhibition narrowed the list down to six candidate compounds. One of the more promising compounds, DNAC-1, which had the lowest MIC against USA300, was chosen for further studies and is shown in Fig. 1A.
FIG 1.

Structure of DNAC-1 (A) and loss of viability of MRSA USA300 in DNAC-1 with and without a sub-MIC of oxacillin (ox) (B). USA300 cells were grown to mid-log phase, back-diluted 100-fold, and treated with 16 μg/ml DNAC-1, 16 μg/ml DNAC-1 with 16 μg/ml oxacillin (0.25× MIC), or left untreated. The samples were taken at 0, 2, 4, 7, and 24 h, and a viable count was carried out. The asterisk denotes P values of <0.01, as determined by a Kruskal-Wallis one-way analysis of variance (ANOVA) test between DNAC-1 with oxacillin and the control.
DNAC-1 potentiates the effects of different classes of antibiotics but is synergistic only with cell wall-targeting antibiotics against MRSA.
DNAC-1 was identified in a screen designed to detect inhibitory activities in the presence of 0.25× MIC of oxacillin (16 μg/ml), which is submicrobicidal for MRSA. We wanted to verify whether DNAC-1 can also potentiate the anti-MRSA effects of other commonly used antibiotics, including cephalothin, cefoxitin, imipenem, norfloxacin, and vancomycin. As shown in Table 1, upon mixing the antibiotics from the list above at 0.25× MIC, the MIC of DNAC-1 dropped from 2 to 4 μg/ml to the range of 0.125 to 1 μg/ml. In particular, two antibiotics, oxacillin and cephalothin, at 0.25× MIC, dropped the MIC of DNAC-1 to 0.125 μg/ml, representing a 16- to 32-fold drop. We also evaluated the converse scenario by evaluating the decrease in the MICs of other classes of antibiotics with 0.5× MIC of DNAC-1, using a checkerboard analysis, as shown in Table 2. More specifically, the MIC of USA300 for oxacillin dropped from 64 to 0.25 μg/ml with DNAC-1 at 0.5× MIC, yielding a 256-fold potentiation in the activity of oxacillin. This translates to an FIC index of 0.5, indicating that the two drugs are synergistic. Although DNAC-1 potentiates the effect of some of the classes of drugs, including those that affect DNA synthesis, like norfloxacin, levofloxacin, and ciprofloxacin, as well as tetracycline, which disrupts protein synthesis, the FIC index is >0.5, thus indicating at best a partial synergism with norfloxacin (FIC index, 0.53), and indifference with levofloxacin, ciprofloxacin, and tetracycline, which have FIC indices of 1, 0.625, and 0.75, respectively. Thus, even though a sub-MIC of DNAC-1 potentiates the effects of other classes of antibiotics, it seems to be synergistic only with cell wall-active agents.
TABLE 1.
MICs of DNAC-1 with commonly used cell wall-active antibiotics at 0.25× MIC against MRSA USA300
| Drug A | MIC of drug A alone (μg/ml) | MIC of DNAC-1 in combination with 0.25× MIC of drug A (μg/ml) |
|---|---|---|
| DNAC-1 | 4 | NAa |
| Oxacillin | 64 | 0.125 |
| Cephalothin | 7.8 | 0.125 |
| Ceftriaxone | 62.5 | 1 |
| Cefoxitin | 15.6 | 1 |
| Vancomycin | 2 | 0.25 |
NA, not available.
TABLE 2.
MICs of different classes of commonly used antibiotics at 0.5× MIC of DNAC-1 against USA300
| Drug A (target) | DNAC-1 (2 μg/ml) | MIC of drug A (μg/ml) | Interactiona |
|---|---|---|---|
| Oxacillin (cell wall synthesis) | −/+ | 64/0.25b | Synergistic |
| Norfloxacin (DNA synthesis) | −/+ | 8/0.25 | Partially synergistic |
| Ciprofloxacin (DNA synthesis) | −/+ | 64/4 | Indifferent |
| Levofloxacin (DNA synthesis) | −/+ | 8/1 | Indifferent |
| Tetracycline (protein synthesis) | −/+ | 32/8 | Indifferent |
The relationship between the antibiotics was determined via the checkerboard analysis.
−, drug A alone; +, drug A plus 2 µg/ml DNAC-1.
DNAC-1 in combination with cell wall-active agents inhibits growth of clinically relevant Gram-positive and Gram-negative pathogens.
Using clinical isolates, we found that DNAC-1 inhibited the growth of HA-MRSA, CA-MRSA, VISA, Staphylococcus epidermidis, E. faecalis, and E. faecium (all Gram positive), with MIC values for DNAC-1 ranging from 0.03 to 4 μg/ml in the presence of a sub-MIC of oxacillin; these findings indicate the potentiating effect of DNAC-1 with oxacillin for Gram-positive bacteria (Table 3). We also tested the effect of DNAC-1 on Gram-negative bacteria with and without 0.25× MIC of ceftriaxone (16 μg/ml), which is a broad-spectrum cephalosporin commonly used against Gram-negative bacteria. In this scenario, DNAC-1 was also effective in combination with a sub-MIC of ceftriaxone against A. baumannii, P. aeruginosa, and E. aerogenes species (total of 7 strains) (Table 3). Importantly, all of the tested strains of E. coli, K. pneumoniae, and K. oxytoca containing an extended-spectrum β-lactamase (ESBL) (total of 8 strains) were also susceptible to this combination (Table 3).
TABLE 3.
MICs of DNAC-1 against clinically relevant bacteria in the presence of 0.25× MIC of oxacillin or 0.25× MIC of ceftriaxone
| Strain (no. of isolates) | Presence/absence of druga | No. of isolates with corresponding MIC (mg/ml) at MIC of DNAC-1 (μg/ml) of: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 32 | 16 | 8 | 4 | 2 | 1 | 0.5 | 0.25 | 0.12 | ||
| CA-MRSA (20) | − | 1 | 1 | 3 | 14 | 1 | ||||
| +OXA | 2 | 9 | 4 | 5 | ||||||
| HA-MRSA (10) | − | 1 | 1 | 3 | 5 | |||||
| +OXA | 4 | 5 | 1 | |||||||
| Mu50 (VISA) | − | 1 | ||||||||
| +OXA | 1 | |||||||||
| S. epidermidis (10) | − | 10 | ||||||||
| +OXA | 10 | |||||||||
| E. faecium (VRE) (5)b | − | 5 | ||||||||
| +OXA | 5 | |||||||||
| E. faecalis (5) | − | 1 | 4 | |||||||
| +OXA | 5 | |||||||||
| A. baumannii | − | 1 | ||||||||
| +CEFT | 1 | |||||||||
| P. aeruginosa (5) | − | 5 | ||||||||
| +CEFT | 5 | |||||||||
| E. aerogenes | − | 1 | ||||||||
| +CEFT | 1 | |||||||||
| E. coli (2) ESBL | − | 2 | ||||||||
| +CEFT | 2 | |||||||||
| K. pneumoniae (4) ESBL | − | 4 | ||||||||
| +CEFT | 4 | |||||||||
| K. oxytoca (2) ESBL | − | 2 | ||||||||
| +CEFT | 2 | |||||||||
OXA, oxacillin; CEFT, ceftriaxone.
VRE, vancomycin-resistant enterococcus.
DNAC-1 is bactericidal against MRSA.
To evaluate the effect of DNAC-1 on the viability of MRSA, we treated actively dividing cells of MRSA USA300 with 4× MIC of DNAC-1 with and without 0.25× MIC of oxacillin. As shown in Fig. 1B, the treatment of USA300 with DNAC-1 at 4× MIC for 2 h caused a dramatic 4-log reduction (99.95%) in the number of viable cells, and this reduction was maintained over a period of 24 h. A combination of 4× MIC of DNAC-1 with 0.25× MIC of oxacillin, however, revealed a 4-log reduction at 2 h, followed by complete elimination of all viable bacteria by 4 h, and this bactericidal activity was maintained at 24 h. These results indicate that DNAC-1 in combination with oxacillin appears to be more effective than is DNAC-1 alone in terms of bactericidal activity.
Assessing toxicity of DNAC-1.
We next assessed the toxicity of DNAC-1 toward eukaryotic cells in three different ways and made the following observations: (i) DNAC-1 showed no toxic effect on Candida albicans, even at 20× MIC (for USA300); (ii) using 2% Triton X-100 as a positive control, we found that DNAC-1 did not lead to any appreciable release of lactate dehydrogenase (LDH) from cultured human bronchial epithelial (CFBE) cells; and (iii) the incubation of 4% sheep erythrocytes with DNAC-1 at various concentrations (2× to 4× MIC against MRSA) also did not induce red blood cell (RBC) lysis, thus indicating that DNAC-1 at these concentrations does not perturb the eukaryotic cell membrane.
DNAC-1 affects the cell wall of MRSA USA300.
To assess the inhibitory effect of DNAC-1 on cell wall synthesis, we measured the incorporation of [3H]lysine into peptidoglycan in the presence of chloramphenicol, which inhibits protein synthesis, while allowing the incorporation of radiolabeled lysine into the pentapeptide of the peptidoglycan chain. Ampicillin was used as a positive control. As shown in Fig. 2, DNAC-1 led to a reduction in lysine incorporation into the nascent cell wall compared to the untreated control, at a level comparable to that with ampicillin.
FIG 2.
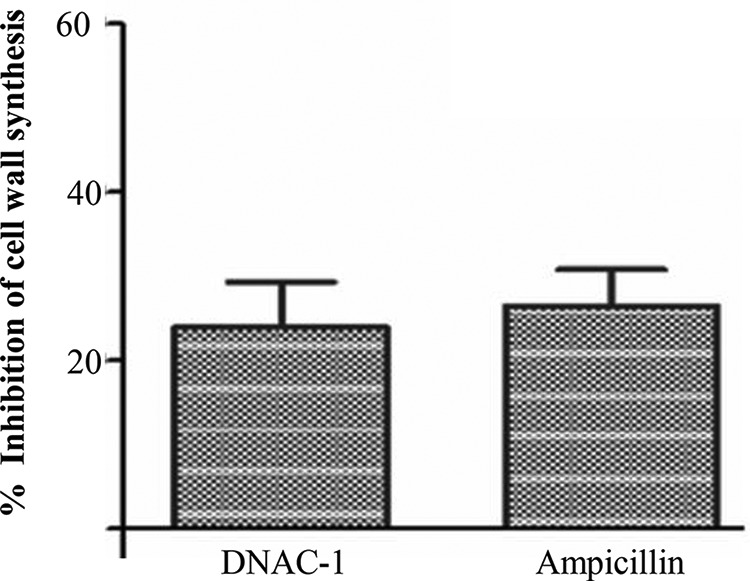
Percent inhibition of cell wall biosynthesis of S. aureus by DNAC-1, as measured by inhibition of radiolabeled lysine incorporation into nascent peptidoglycan. Bacteria were treated with either DNAC-1 or ampicillin (positive control) for 20 min in the presence of radioactive cell wall precursors. The data are presented as the percent inhibition compared to the untreated control.
To further assess the effect of DNAC-1 on cell wall morphology, USA300 cells were treated with the compound DNAC-1 (2× MIC) for 90 min, fixed, dehydrated, and observed under TEM. The cell wall thickness of the treated cells, evaluated in up to 30 different fields, was found to be increased compared to that of the untreated cells (Fig. 3A and B), with mesosome-like invaginations (11) of the cell membrane into the cytosol along the cell periphery (Fig. 3A, center) and along the septum (Fig. 3A, right).
FIG 3.

Effect of DNAC-1 on cell wall, as observed by TEM. USA300 cells were grown to mid-log phase and treated with DNAC-1 for 90 min or left untreated (control). The cells were then collected and processed for EM, as described in Materials and Methods. (A) Mesosome-like invaginations are seen both along the cell wall (arrow, center) and along the septum (arrow, right). (B) DNAC-1-treated cells have a significantly thicker cell wall. as demonstrated in the box-and-whisker plot showing the standard deviation. The asterisk represents a statistically significant difference, at a P value of <0.05, comparing the control to DNAC-1-treated cells.
DNAC-1 causes mislocalization of PBP2/4 and FtsZ and activation of the cell wall stress stimulon.
The results of the macromolecular synthesis assay and increased cell wall thickness in the TEM studies prompted us to question whether DNAC-1 affects the enzymes involved in cell wall synthesis. Accordingly, we examined the localization of PBP2 and PBP4, both of which are known to be involved in cell wall synthesis and antibiotic resistance (5, 7). A lack of PBP4 activity is also known to result in thickened cell walls (14, 16). Utilizing the previously created MRSA COL strains expressing functional GFP-PBP2 or PBP4-YFP (8, 9), the exposure of actively growing cells to DNAC-1 at either 0.5× or 2× MIC for 15 min showed a mislocalization of PBP2 and PBP4 away from the septum, where they normally reside in dividing cells (Fig. 4A, left and center). The quantification of PBP localization showed that only 3% (n = 696) of the DNAC-1-treated cells have PBP2 correctly localized at the septum versus 36% (n = 493) of the untreated cells, while 13% (n = 652) of the DNAC-1-treated cells have septal PBP4, compared to 27% (n = 591) of the untreated cells. Since PBP2 localization has been shown to depend on FtsZ (17), we thus determined if the delocalization of PBP2 upon DNAC-1 treatment is mediated via FtsZ. Accordingly, upon exposure of a strain containing CFP-tagged FtsZ (9) to 0.5× MIC of DNAC-1 for 15 min, we observed a mislocalization of FtsZ (Fig. 4A, right).
FIG 4.

Mislocalization of PBPs and FtsZ and activation of cell wall stress stimulon by DNAC-1. (A) Mislocalization of the PBP2, PBP4, and FtsZ was tracked using strains expressing either GFP-PBP2, PBP4-YFP, or FtsZ-CFP after treatment with 0.5× MIC of DNAC-1 for 15 min. Bars, 1 µm. (B) Activation of the vraSR operon was tested using strains containing a vraSR promoter driving sfGFP expression. In the presence of 2× MIC of DNAC-1, the mean ± standard deviation (SD) cell fluorescence was 3,594 ± 1,344 arbitrary units (a.u.) (n = 468), while in the presence of 2× MIC of vancomycin (positive control), the mean ± SD cell fluorescence was 23,911 ± 8,318 a.u. (n = 465). The left panel corresponds to cells in the absence of antibiotics, which showed a mean ± SD cell fluorescence of 2,196 ± 447 a.u. (n = 511). The P values are <0.0001 between the DMSO control and vancomycin-treated cells and <0.05 between the control and DNAC-1-treated cells. Bars, 10 µm.
Cell wall-active compounds have also been known to trigger the cell wall stress stimulon (CWSS) by upregulating the vraSR operon (18, 19). To assess if DNAC-1 would trigger a CWSS response, we constructed a COL strain with the gene encoding a fast-folding variant of GFP, sfGFP, fused to the promoter of the vraSR operon in its native chromosome locus. We found that upon treatment with 2× MIC of DNAC-1 for 30 min, the sfGFP fluorescence signal in the cells was increased approximately 1.6 times compared to that with the mock-treated cells, indicating that the CWSS was triggered (Fig. 4B). However, the vraSR response was weaker than that triggered by vancomycin at 2× MIC (Fig. 4B, center), a known activator of the cell wall stress stimulon (19).
DNAC-1 leads to mesosome formation, increased cellular permeabilization, and cell membrane depolarization in MRSA.
The membrane disruption seen in the EM study prompted us to further examine the effect of DNAC-1 on the membrane. To reduce any artifacts that may have been introduced into samples during TEM processing, we employed superresolution fluorescence microscopy of unfixed MRSA cells to investigate membrane defects. Within 15 min of exposure to DNAC-1 at 2× MIC, membrane staining of MRSA strain COL with FM 4-64 revealed numerous membrane spots and bulges into the cell cytosol compared to the untreated cells (Fig. 5A). These findings are in agreement with the mesosome-like structures visualized by TEM. No changes were observed in cell wall staining (Fig. 5B) using fluorescently labeled vancomycin. We also monitored changes in membrane permeability with propidium iodide staining upon treatment with DNAC-1 at 2× MIC (data not shown). The cells treated with DNAC-1 were permeabilized quickly, as revealed by propidium iodide staining after a 15-min exposure of DNAC-1. At 1 h of exposure, ∼55% of the bacterial cells showed propidium iodide staining, indicating a loss of cell membrane integrity.
FIG 5.

Disruption of cell membrane by DNAC-1. The cells were either untreated (control) or treated with 2× MIC of DNAC-1 for 15 min and stained with FM 4-64 to visualize the membrane (A) and Bodipy-FL-vancomycin (green) to visualize the cell wall and Hoechst 33342 dye (blue) to see the DNA (B).
To investigate if DNAC-1 disrupts the membrane potential, we used the dye DiOC2, which typically aggregates as a function of membrane potential. Upon treatment of USA300 with 2× to 8× MIC of DNAC-1 (8 to 32 μg/ml), there was gradual dissipation of the membrane potential, as monitored by the loss of red fluorescence, due to a loss of aggregation of DiOC2 (Fig. 6). However, this decrease in membrane potential was less than that of our positive control, CCCP, a known proton translocator supplied in the DiOC2 kit. Cell viability was also seen to parallel the decrease in membrane potential, as confirmed by the CFU counts at appropriate time points in the DNAC-1-treated cells compared with those of the untreated control (data not shown).
FIG 6.
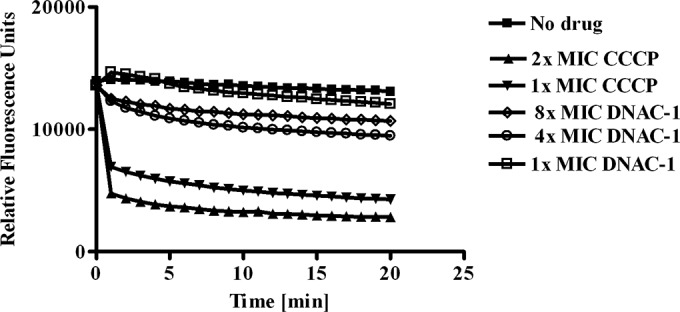
DNAC-1 causes membrane depolarization. Actively growing USA300 cells were back-diluted, resuspended in PBS with glucose, and treated with DiOC2 (14) for 30 min in the dark at 25°C. A loss of fluorescence was monitored over time upon the addition of increasing concentrations of DNAC-1 (4×, 8×, and 16× MIC). CCCP was used as a positive control.
In vivo efficacy of DNAC-1 in a murine subcutaneous model of infection.
To evaluate the efficacy of DNAC-1 in vivo, we utilized a murine subcutaneous infection model. As pharmacokinetic-pharmacodynamic (PK-PD) data were not available at the time of challenge, we treated mice with a single subcutaneous injection of USA300 containing either DNAC-1, DNAC-1 and oxacillin, or DMSO control in a ∼100-μl volume using PBS as a diluent. We inoculated three groups of six BALB/c mice each with 108 CFU of USA300 (representing the ∼ID90), with the first group (control) receiving bacteria alone with DMSO, the second group receiving bacteria and 8 μg of DNAC-1 (added right before injection) in a 100-μl volume, and the third group receiving bacteria along with DNAC-1 and 16 μg of oxacillin (also added right before injection). In the control group of mice treated with DMSO, subcutaneous abscesses developed in all mice (6/6) within 3 days of injection, while 2 of 6 animals developed smaller subcutaneous abscesses in the group treated with DNAC-1. In the group injected with a combination of DNAC-1 and oxacillin, none of the mice exhibited abscessed lesions (0/6). More importantly, in the mice treated with DNAC-1, the bacterial density was 3 × 103 CFU/g of tissue, whereas those treated with the DNAC-1–oxacillin combination harbored the lowest bacterial density, at 1.3 × 103 CFU/g of tissue (Fig. 7A). In contrast, the mean bacterial density recovered from the abscesses of the control group was the highest, at 9.3 × 109 CFU/g of tissue (Fig. 7A). Histological sections of abscessed tissues stained with hematoxylin and eosin from mice infected with the bacteria in DMSO alone revealed a disruption in skin architecture, clusters of bacteria (Fig. 7B, arrow), and massive polymorphonuclear leukocyte (PMN) infiltrates. In the mice treated with DNAC-1 alone, the number of bacteria was visibly reduced, and the alteration to the internal structures of the skin was also diminished compared to those of the mice inoculated with bacteria alone, while the number of inflammatory cells at the site of infection also appeared to be decreased. In the group treated with the DNAC-1–oxacillin combination, histological examination revealed a normal skin substructure, a lack of inflammatory cells, and no visible bacterial clusters below the epidermis (Fig. 7B).
FIG 7.
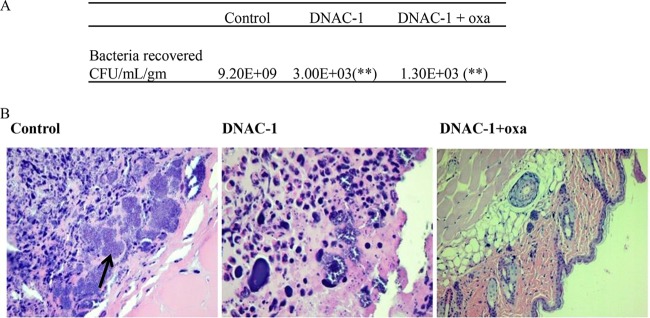
Efficacy of DNAC-1 in a murine subcutaneous infection model. Each group of mice (6 each) was injected with 108 CFU of USA300, along with DMSO (control), DNAC-1 alone, or DNAC-1 plus oxacillin (oxa). (A) Animals were examined for bacterial abscess for 8 days postinfection and then sacrificed. The lesions were biopsied and CFU recovered from the zone of infection. The asterisks denote P values of <0.05, as determined by a Kruskal-Wallis one-way ANOVA between DNAC-1 and the control and between DNAC-1 with oxacillin versus the control. (B) Histological changes in the punch biopsy sample. Note the presence of bacteria and loss of subcellular architecture in the control sample infected with bacteria alone. The original magnifications for the images are 40× (left and center) and 10× (right).
DISCUSSION
MRSA has become resistant to drugs, like vancomycin, daptomycin, and linezolid (2). Additionally, due to market pressure and diminished financial incentives, big pharmaceutical companies have reduced their research and development efforts for new antibiotics. This study was undertaken in an effort to identify new candidate compounds that improve the efficiencies of the currently available antimicrobials. Surprisingly, our candidate compound DNAC-1 demonstrates inhibitory activity not only in combination therapy with β-lactams but also on its own against many Gram-positive bacteria. In combination with ceftriaxone, DNAC-1 seems to be effective on a small collection of resistant Gram-negative clinical isolates. The benefit of identifying candidate compounds, such as DNAC-1, is 2-fold, as it adds to the arsenal of new broad-spectrum antibacterials and extends the usage of β-lactams to which many bacterial pathogens have already developed resistance.
In this study, a mechanistically unbiased screen was used to identify small molecules that work in conjunction with a β-lactam against MRSA. We hypothesized that if DNAC-1 has an effect on the cell wall, it should inhibit cell wall synthesis, similarly to ampicillin. This was confirmed by a macromolecular synthesis assay. Using the vraRS promoter driving a GFP fusion, we found that DNAC-1 induced the cell wall stress response operon, as has been found with vancomycin, albeit to a lower level. These data, in combination with the mislocalization of PBP2 and PBP4, which are cell wall synthesis enzymes, and FtsZ, which normally assembles into a ring-like structure at the septum during cell division to recruit PBP2 and other cell wall machinery to produce a new cell wall, highlight the cell wall as a plausible target for DNAC-1. However, we are unable to determine in our study if the effect of DNAC-1 is direct or secondary to primary damage elsewhere.
Bactericidal activity by way of cell wall synthesis inhibition is generally a slow process (20). However, our results with propidium iodide permeability demonstrate that the effect of DNAC-1 on cell viability is rapid (within 15 min). Coinciding with this early effect, we noticed membrane blebbing and mesosome-like structures in the TEM images of USA300 cells treated with DNAC-1, reminiscent of the membrane defect caused by cationic antimicrobial peptides (21). These mesosome-like structures are thought to be indicative of cell membrane damage. An analysis of the cell membrane potential revealed a rapid depolarization of the membrane, coinciding with a loss of cell viability. Given that the structural integrity of the cell membrane likely plays an important role in cell wall synthesis and turnover (22), we speculate that DNAC-1 may have a bimodal mechanism of action on the cell membrane and cell wall, similarly to what has been described for daptomycin (9). Based on this theory, the bactericidal action is a two-step process, which is initially a fast membrane depolarization and permeation step, followed by a slower disruption of cell wall integrity and/or de novo synthesis due to the mislocalization of PBPs.
Regarding the fast step, the constant proton gradient of the proton motive force (PMF) has to be established and maintained for viability of the bacterial cell (23). PMF is made up of the electric potential and the transmembrane proton gradient. The collapse of the transmembrane potential can have adverse effects on essential cellular functions, like ATP synthesis, even though dissipation of the PMF alone is not bactericidal (24). Therefore, the disruption in PMF must set the stage for perturbative events downstream.
In MRSA, the combined activities of PBP2, PBP2A, and PBP4 at the septum are essential for methicillin resistance. In the presence of cell wall-active agents, PBP2 is mislocalized (7). We previously showed that the simultaneous chemical inhibition of PBP2 and PBP4 transpeptidase activities results in not only decreased cross-linking of the cell wall but also leads to bactericidal activity in CA-MRSA (5). Thus, the mislocalization of PBP2/4 in an FtsZ-dependent manner and the disruption of cell wall synthesis by DNAC-1 might represent the second step leading to cell death. Taken together, the membrane depolarization and the PBP mislocalization point to a mechanism in which the disruption of the integrity of the membrane may interfere with active transport and localization of cell wall biosynthetic enzymes. This model suggests that the addition of a β-lactam to DNAC-1 would increase the killing efficiency due to a sequential impact on the cell membrane integrity and cell wall synthesis, similar to the mechanism proposed by Sewell and Brown to explain the synergy between wall teichoic acid inhibitors and β-lactams (25). Additionally, this might explain the broad-spectrum activity of DNAC-1 that we have demonstrated against both clinically relevant multidrug-resistant bacteria, including Gram-positive and Gram-negative species.
An alternative sequence of events can be envisioned in which the loss of PMF results in increased autolysis and, ultimately, cell death due to active degradation of the cell wall. It has been shown that in Bacillus subtilis, the acidification of the cell wall during growth regulates autolytic murein hydrolases (26). The current study suggests that the treatment of USA300 with DNAC-1 results in increased autolysis (data not shown). Taken together, these considerations are consistent with an alternative mechanism in which the activation of murein hydrolases results in the active breakdown of peptidoglycan.
Besides in vitro MIC studies, our in vivo murine model with MRSA USA300 showed that the combination of DNAC-1 with oxacillin is effective at reducing both the tissue bacterial density and abscess formation. Although we cannot rule out a role of increased host inflammatory response (27) causing enhanced bacterial killing by DNAC-1, as opposed to a direct effect of DNAC-1, tissue histology did not reveal an abundance of inflammatory cells at the site of injection in the presence of DNAC-1. It has been shown that oxacillin induces an altered virulence expression profile of CA-MRSA (28), which in turn can alter the immune response against the bacteria. In the mice treated with a combination of oxacillin and DNAC-1, there is a reduction in immune effector cells and abscess formation. As reported before, we believe that this might result from the downregulation of some of the immune effectors and the reduction in PMNs at the site of infection by oxacillin, resulting in reduced inflammatory damage to the tissue and hence preserving the morphological structure of the tissue. We are currently carrying out structure-activity relationship studies of DNAC-1 to enhance the efficacy of the drug, improve its solubility, and determine its direct target.
ACKNOWLEDGMENTS
We thank NERCE-BEID (grant U54AI057159) for facilitating this research and Matthew P. DeLisa (Cornell University) for plasmid pTRC99a-P7.
The work in the A.C. laboratory was partially funded by COBRE (NIH) grant P30GM106394 and the Pfeiffer Foundation. The M.G.P. laboratory was funded by grant ERC-2012-StG-310987 from the European Research Council, and J.M.M. was supported by fellowship SFRH/BD/71993/2010.
REFERENCES
- 1.Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. 2006. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 368:874–885. doi: 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- 2.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE Jr. 2004. Trends in antimicrobial drug development: implications for the future. Clin Infect Dis 38:1279–1286. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 3.Kaneti G, Sarig H, Marjieh I, Fadia Z, Mor A. 2013. Simultaneous breakdown of multiple antibiotic resistance mechanisms in S. aureus. FASEB J 27:4834–4843. doi: 10.1096/fj.13-237610. [DOI] [PubMed] [Google Scholar]
- 4.Farha MA, Leung A, Sewell EW, D'Elia MA, Allison SE, Ejim L, Pereira PM, Pinho MG, Wright GD, Brown ED. 2013. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem Biol 8:226–233. doi: 10.1021/cb300413m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Memmi G, Filipe SR, Pinho MG, Fu Z, Cheung AL. 2008. Staphylococcus aureus PBP4 is essential for β-lactam resistance in community-acquired MRSA. Antimicrob Agents Chemother 52:3955–3966. doi: 10.1128/AAC.00049-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher AC, DeLisa MP. 2008. Laboratory evolution of fast-folding green fluorescent protein using secretory pathway quality control. PLoS One 3:e2351. doi: 10.1371/journal.pone.0002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinho MG, Errington J. 2005. Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol Microbiol 55:799–807. doi: 10.1111/j.1365-2958.2004.04420.x. [DOI] [PubMed] [Google Scholar]
- 8.Loskill P, Pereira PM, Jung P, Bischoff M, Herrmann M, Pinho MG, Jacobs K. 2014. Reduction of the peptidoglycan crosslinking causes a decrease in stiffness of the Staphylococcus aureus cell envelope. Biophys J 107:1082–1089. doi: 10.1016/j.bpj.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, Roemer T. 2012. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med 4:126ra35. doi: 10.1126/scitranslmed.3003592. [DOI] [PubMed] [Google Scholar]
- 10.Pinho MG, Errington J. 2004. A divIVA null mutant of Staphylococcus aureus undergoes normal cell division. FEMS Microbiol Lett 240:145–149. doi: 10.1016/j.femsle.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Oshida T, Tomasz A. 1992. Isolation and characterization of a Tn551-autolysis mutant of Staphylococcus aureus. J Bacteriol 174:4952–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terleckyj B, Willett NP, Shockman GD. 1975. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect Immun 11:649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattingly SJ, Daneo-Moore L, Shockman GD. 1977. Factors regulating cell wall thickening and intracellular iodophilic polysaccharide storage in Streptococcus mutans. Infect Immun 16:967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finan JE, Archer GL, Pucci MJ, Climo MW. 2001. Role of penicillin-binding protein 4 in expression of vancomycin resistance among clinical isolates of oxacillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 45:3070–3075. doi: 10.1128/AAC.45.11.3070-3075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamber S, Schwartzman J, Cheung AL. 2010. Role of PknB kinase in antibiotic resistance and virulence in community-acquired methicillin-resistant Staphylococcus aureus strain USA300. Infect Immun 78:3637–3646. doi: 10.1128/IAI.00296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyke AW, Ward JB, Hayes MV, Curtis NA. 1981. A role in vivo for penicillin-binding protein-4 of Staphylococcus aureus. Eur J Biochem 119:389–393. doi: 10.1111/j.1432-1033.1981.tb05620.x. [DOI] [PubMed] [Google Scholar]
- 17.Pinho MG, Errington J. 2003. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol Microbiol 50:871–881. doi: 10.1046/j.1365-2958.2003.03719.x. [DOI] [PubMed] [Google Scholar]
- 18.Kuroda M, Kuwahara-Arai K, Hiramatsus K. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strain Mu3 and Mu50 by CDNA differential hybridization method. Biochem Biophys Res Commun 269:485–490. doi: 10.1006/bbrc.2000.2277. [DOI] [PubMed] [Google Scholar]
- 19.Utaida S, Dunman PM, Macapagal D, Murphy E, Projan SJ, Singh VK, Jayaswal RK, Wilkinson BJ. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719–2732. doi: 10.1099/mic.0.26426-0. [DOI] [PubMed] [Google Scholar]
- 20.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedrich CL, Moyles D, Beveridge TJ, Hancock RE. 2000. Antibacterial action of structurally diverse cationic peptides on Gram-positive bacteria. Antimicrob Agents Chemother 44:2086–2092. doi: 10.1128/AAC.44.8.2086-2092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemper MA, Urrutia MM, Beveridge TJ, Koch AL, Doyle RJ. 1993. Proton motive force may regulate cell wall-associated enzymes of Bacillus subtilis. J Bacteriol 175:5690–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurdle JG, O'Neill AJ, Chopra I, Lee RE. 2011. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol 9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Y-Q, Mukhipadhyay K, Yeaman MR, Adler-Moore JP, Bayer AS. 2005. Functional interrelationships between cell membrane and cell wall in antimicrobial peptide-mediated killing of Staphylococcus aureus. Antimicrob Agents Chemother 49:3114–3121. doi: 10.1128/AAC.49.8.3114-3121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sewell EW, Brown ED. 2014. Taking aim at wall teichoic acid synthesis: new biology and new leads for antibiotics. J Antibiot (Tokyo) 67:43–51. doi: 10.1038/ja.2013.100. [DOI] [PubMed] [Google Scholar]
- 26.Calamita HG, Ehringer WD, Koch AL, Doyle RJ. 2001. Evidence that the cell wall of Bacillus subtilis is protonated during respiration. Proc Natl Acad Sci U S A 98:15260–15263. doi: 10.1073/pnas.261483798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverstein R, Wood JG, Xue Q, Norimatsu M, Horn DL, Morrison DC. 2000. Differential host inflammatory responses to viable versus antibiotic-killed bacteria in experimental microbial sepsis. Infect Immun 68:2301–2308. doi: 10.1128/IAI.68.4.2301-2308.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudkin JK, Laabei M, Edwards AM, Joo HS, Otto M, Lennon KL, O'Gara JP, Waterfield NR, Massey RC. 2014. Oxacillin alters the toxin expression profile of community-associated methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 58:1100–1107. doi: 10.1128/AAC.01618-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.CLSI. 2006. Performance standards for antimicrobial susceptibility testing. Approved standard M100-S16 CLSI, Wayne, Pa. [Google Scholar]