

Abstract
Mycobacterium kansasii is the second most common mycobacterial cause of lung disease. Standard treatment consists of rifampin, isoniazid, and ethambutol for at least 12 months after negative sputum. Thus, shorter-duration therapies are needed. Moxifloxacin has good MICs for M. kansasii. However, good preclinical models to identify optimal doses currently are lacking. We developed a novel hollow fiber system model of intracellular M. kansasii infection. We indexed the efficacy of the standard combination regimen, which was a kill rate of −0.08 ± 0.05 log10 CFU/ml/day (r2 = 0.99). We next performed moxifloxacin dose-effect and dose-scheduling studies at a half-life of 11.1 ± 6.47 h. Some systems also were treated with the efflux pump inhibitor reserpine. The highest moxifloxacin exposure, as well as lower exposures plus reserpine, sterilized the cultures by day 7. This suggests that efflux pump-mediated tolerance at low ratios of the area under the concentration-time curve from 0 to 24 h (AUC0–24) to MICs is an early bacterial defense mechanism but is overcome by higher exposures. The highest rate of moxifloxacin monotherapy sterilization was −0.82 ± 0.15 log10 CFU/ml/day (r2 = 0.97). The moxifloxacin exposure associated with 80% of maximal kill (EC80) was an AUC0–24/MIC of 317 (the non-protein-bound moxifloxacin AUC0–24/MIC was 158.5). We performed Monte Carlo simulations of 10,000 patients in order to identify the moxifloxacin dose that would achieve or exceed the EC80. The simulations revealed an optimal moxifloxacin dose of 800 mg a day. The MIC susceptibility breakpoint at this dose was 0.25 mg/liter. Thus, moxifloxacin, at high enough doses, is suitable to study in patients for the potential to add rapid sterilization to the standard regimen.
INTRODUCTION
Mycobacterium kansasii is the third most common mycobacterial cause of chronic disease in the United States but the second most common after tuberculosis in other parts of the world (1–5). Although it has been associated with AIDS, worldwide there are many more cases of non-AIDS patients (1, 6). Data on treatments tested in randomized controlled clinical trials are scant. The recommended treatment in non-HIV-infected patients consists of isoniazid, rifampin, and ethambutol; this regimen was copied from that used to treat tuberculosis (4). It is recommended that patients receive therapy for more than 12 months after negative sputum, making the therapy duration even longer than that for tuberculosis (7). Therefore, it is important to identify a shorter-duration therapy. The quinolone moxifloxacin has been shown to have very good MICs in M. kansasii clinical isolates, with 90% of isolates having a MIC of ≤0.06 mg/liter (8). However, given the M. kansasii disease patient population sizes and social distribution and the lack of advocacy for this disease, it is unlikely that true randomized controlled clinical trials will be performed with this drug in the foreseeable future. One approach is to develop a good preclinical disease model whose results can be used in computer-aided clinical trial simulations (9).
The pathological lesions encountered in M. kansasii infection include a wide variety of lesions, such as necrotic and nonnecrotic granulomas, eosinophilic necrosis, neutrophilic abscesses, and characteristic folded bacilli within histiocytes (10). Bacilli in these lesions are encountered in both extracellular and intracellular locations. We were interested in designing a preclinical disease model that would reflect the bacilli within tissue macrophages. We created a novel hollow-fiber system model of M. kansasii that could enable the study of both disseminated and pulmonary disease. In the case of Mycobacterium tuberculosis, the hollow-fiber system model has been shown to have a quantitative forecasting accuracy of >94% in long-term clinical outcomes (11–17). We then utilized the M. kansasii hollow-fiber system in dose-effect studies and studied the possible presence of tolerant bacteria, based on findings with other mycobacteria, and then used the output in Monte Carlo simulations to identify the optimal dose (18–20).
MATERIALS AND METHODS
Organism.
M. kansasii (ATCC 12478) was purchased from the American Type Culture Collection (Manassas, VA). This commercially available isolate is the G133 Bostrom strain that is resistant to 100 mg/liter streptomycin. Prior to each experiment, the bacterial stock was thawed and incubated in Middlebrook 7H9 broth with 10% oleic acid-albumin-dextrose-catalase (OADC) and 100 mg/liter streptomycin at 37°C in a shaking incubator for 4 days to achieve exponential-phase growth.
Materials.
Hollow-fiber cartridges were purchased from FiberCell (Frederick, MD). RPMI 1640 and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA), as were rifampin, isoniazid, ethambutol, and resazurin (7-hydroxy-3H-phenoxazin-3-one 10-oxide). Fetal bovine serum (FBS) was procured from SAFC Biosciences (Sigma). FBS was heat inactivated prior to use. Moxifloxacin hydrochloride solution of 400 mg/250 ml in 0.8% saline was purchased from University of Texas Southwestern Medical Center Pharmacy and serially diluted using RPMI 1640 to the drug concentrations required for study.
Determination of MIC by broth dilution and resazurin assays.
M. kansasii cultures on day 4 of log-phase growth were adjusted to a McFarland standard of 0.5 and diluted to a bacterial density of 1.5 × 105 CFU/ml in Middlebrook 7H9 broth supplemented with 10% OADC. Nine ml of inoculum then was pipetted into test tubes. One ml of moxifloxacin was added to make final concentrations of 0, 0.0312, 0.0625, 0.125, 0.25, 0.5, 1.0, 2, 4, and 8 mg/liter in triplicate, after which tubes were incubated at 37°C under 5% CO2 for 7 days. On day 7, the cultures were washed to remove drug carryover, serially diluted, and plated onto Middlebrook 7H10 agar supplemented with 10% OADC. The cultures then were incubated at 37°C under 5% CO2 for 7 to 10 days, after which colonies were counted, and the minimum concentration associated with 99% inhibition was identified. The experiment was performed twice.
Moxifloxacin MICs also were identified using the resazurin assay based on a modification of the method of Palomino et al. (21). M. kansasii cultures were prepared and incubated with moxifloxacin at the same concentrations as those described above. On day 3, 100-μl cultures from each tube were placed on a microtiter plate, and 50 μl resazurin solution (final concentration of 0.001%, wt/vol) was added. Plates then were incubated at 37°C under 5% CO2 overnight, after which color change from blue to pink was recorded to identify the minimum concentration associated with 99% inhibition. The experiment was performed twice.
Hollow-fiber model of intracellular M. kansasii.
We have used the hollow fiber systems to develop a preclinical laboratory model that mimics human pharmacokinetics and the pathophysiological properties of M. tuberculosis and Mycobacterium avium complex (18–20, 22, 23). The pharmacokinetic system has been described in detail in the past (24). Here, we adapted this model for M. kansasii. Briefly, human-derived THP-1 macrophages (ATCC TIB-202) growing in RPMI 1640 medium, supplemented with 10% heat-inactivated FBS, were grown to a cell density of 1.5 × 106 cells/ml. The THP-1 macrophages next were infected with M. kansasii (cell density, 1.5 × 107 CFU/ml) and coincubated overnight at 37°C under 5% CO2, giving a bacillus-to-macrophage multiplicity of infection of 1:10. Infected macrophages were then washed twice with warm RPMI 1640 and 100 mg/liter streptomycin by centrifugation at 100 × g for 5 min to remove the extracellular bacteria and then examined in a hemocytometer for cell counts and viability after staining with trypan blue. The cell counts also were verified with an automated cell counter (Sceptor; EMD Millipore). We next inoculated each of the hollow-fiber systems with 20 ml of infected THP-1 macrophages into the peripheral compartment. The peripheral compartment of the hollow-fiber system is separated by semipermeable hollow fibers from a central compartment in which fresh RPMI 1640 with 10% FBS circulates. Each subsequent hollow-fiber study was performed twice, with 3 replicate hollow-fiber systems for each drug regimen/dose.
Validation of pharmacodynamics of the intracellular M. kansasii hollow-fiber model.
THP-1 macrophage were infected with M. kansasii, as described above, after which 20 ml of the infected cells was inoculated into the peripheral compartment of two hollow-fiber systems. Systems then were treated with the standard three-drug regimen, consisting of a human equivalent dose of rifampin at 600 mg a day, isoniazid at 300 mg a day, and ethambutol at 15 mg/kg/for 14 days, with pharmacokinetics as described before (12–14, 22). On days 3, 7, 10, and 14, the peripheral compartment of each system was sampled and washed twice to remove extracellular bacteria and drug carryover as described above, after which macrophages were ruptured and bacteria cultured on Middlebrook 7H10 agar supplemented with 10% OADC (23).
Dose-ranging studies.
The peripheral compartment of each hollow-fiber system was inoculated with 20 ml of M. kansasii-infected THP-1 macrophages prepared as described above. The central compartment was treated once daily with 1 of 8 human equivalent moxifloxacin doses, corresponding to 0 to 800 mg/day, administered daily to the central compartment of the hollow-fiber system under the control of a computerized syringe pump. The rates of dilution of the drug were set to achieve concentration-time profiles of the drug similar to those achieved in patients treated with the same doses, in this case a moxifloxacin half-life (± standard deviations) of 12 ± 1.3 h, which is encountered in patients based on moxifloxacin FDA licensing studies, was targeted. In addition, two hollow-fiber systems also were treated with reserpine (10 mg/liter) daily in addition to a moxifloxacin exposure of areas under the concentration-time curve from 0 to 24 h (AUC0–24) of 12.5 and 25 mg · h/liter daily. This concentration of reserpine did not kill M. kansasii in preliminary studies and was used as a broad-spectrum efflux pump inhibitor (i.e., it works against many families of drug efflux pumps). We sampled the central compartment of each hollow-fiber system at 1, 3, 5, 9, 12, 18, and 23 h after the first drug infusion to validate the drug concentrations achieved in each system. On days 3, 7, 10, and 14, the peripheral compartment of each system was sampled for macrophage count and M. kansasii quantitative culture. Macrophages were separated from media by centrifugation and washing in streptomycin-containing media followed by non-drug-containing media to remove extracellular bacteria and drug carryover. They then were ruptured using phosphate-buffered saline-Tween 20 (PBS-T; 0.05%, vol/vol) to release intracellular bacteria for culture on Middlebrook 7H10 agar supplemented with 10% OADC, as described previously (23). In addition, to enumerate the low-level and high-level moxifloxacin-resistant subpopulation, the Middlebrook 7H10 agar was supplemented with either 1.5× or 3× MIC of moxifloxacin.
Drug assay.
Central compartment samples were analyzed for moxifloxacin concentration using a liquid chromatographic technique with UV detection (292 nm) as described before (23). Levofloxacin was used as the internal standard. The assay was linear between 0.2 and 100 mg/liter (r2 = 0.999), and the relative standard deviations were within 5% (23). Assays for measuring rifampin, isoniazid, and ethambutol concentrations were as described before (12–14, 22).
Pharmacokinetic-pharmacodynamic (PK/PD) modeling.
Compartmental pharmacokinetic analysis of moxifloxacin drug concentrations from each hollow-fiber system was performed using a one-compartment model with first-order input and elimination in ADAPT II software (25). The output was utilized to calculate AUC0–24/MIC ratios. The inhibitory sigmoid maximum-effect (Emax) model was used to identify the relationship between the bacterial burden and moxifloxacin exposure.
Monte Carlo simulations to identify optimal dose and resistance breakpoints.
We utilized the population pharmacokinetic parameter estimates from 241 patients in South Africa, a country where M. kansasii infection is a problem (1, 26, 27). These parameters, including between-subject variability as a percentage of coefficient of variation, are clearance of 10.6 liters/h (18.7%), ka (absorption rate constant) of 1.59 h−1(69.9%), and volume of 114 liters. The volume has little between-subject variability in this study but has been shown to be up to 32% in other studies; the latter value was used (28). The penetration of moxifloxacin into lung epithelium lining fluid (ELF), bronchial secretions, and alveolar macrophages has been studied, generally with an AUC0–24 equivalent between serum and either ELF or bronchial secretions in pneumonia (28–30). The ratios of penetration in patients of these bronchial secretions and ELF bath-infected macrophages are similar to those of infected macrophages in the hollow-fiber system (23). These population pharmacokinetic parameter estimates and variances were utilized as the domain of input in subroutine PRIOR of ADAPT. A total of 10,000 patients, treated with doses of either 200 mg, 400 mg, 600 mg, or 800 mg of moxifloxacin, were examined for AUCs achieved with each dose. The moxifloxacin MIC distribution published by Guna et al. was used to calculate the AUC0–24/MIC ratios in order to identify the probability of attaining the AUC0–24/MIC associated with 80% of maximal effect (EC80), which was considered optimal (8, 14). In addition, the MIC above which ≥10% of patients failed to achieve the EC80 for the standard 400-mg dose and the optimal dose were identified as proposed susceptibility breakpoints (31).
RESULTS
The moxifloxacin MIC, based on broth dilution test as recommended by the CLSI, was 0.06 mg/liter on two occasions. This assay took 4 weeks from set up to reading the results. This MIC was similar to the MIC obtained by the modified resazurin colorimetric assay on two different occasions, as shown in Fig. 1. The total time from setting up the resazurin test to reading the MIC was 4 days.
FIG 1.

Resazurin assay for identification of Mycobacterium kansasii MIC. Resazurin is blue and nonfluorescent and is reduced to resorufin, which is pink and highly fluorescent when mycobacteria grow. Coincubation of M. kansasii with various concentrations of moxifloxacin in the assay revealed that the lowest (minimum) moxifloxacin concentration that inhibits growth (keeping resazurin blue) was 0.0625 mg/liter.
The kill-slope of the standard regimen combination therapy of isoniazid, rifampin, and ethambutol in the hollow-fiber system, based on serum pharmacokinetics of these drugs at standard doses, is shown in Fig. 2. The untreated controls grew at an overall rate of 0.0.2551 ± 0.0553 log10 CFU/ml/day. The standard therapy killed at a low rate of −0.080 ± 0.0514 log10 CFU/ml/day. The concentration-time profiles based on measurements of drug concentration in the hollow-fiber system were as described before (12–14, 22).
FIG 2.

Efficacy of standard combination treatment against intracellular M. kansasii. A standard three-drug regimen showed good kill rates compared to those of the untreated controls in the hollow-fiber model system.
The concentration-time profiles of moxifloxacin achieved in dose-effect studies in the hollow fiber systems were as shown in Fig. 3A. Modeling using a one-compartment model revealed the pharmacokinetic model predicted versus observed concentrations shown in Fig. 3B (r2 = 0.991). The systemic clearance was 15.4 ± 7.80 liters/h, a volume of 247.0 ± 70.2 liters, and an absorption constant of 4.15 ± 1.66 h−1, which translates to a half-life of 11.1 ± 6.47 h. The pharmacokinetic parameters achieved in each hollow-fiber system were utilized to calculate the AUC0–24 and AUC0–24/MIC ratios for PK/PD analysis.
FIG 3.

Pharmacokinetics of moxifloxacin in the hollow-fiber systems. (A) Concentration-time profiles of moxifloxacin achieved in the hollow-fiber system are shown for each system. (B) A one-compartment model with first-order input and elimination best described the model. Shown are the model predicted versus observed concentrations (r2 = 0.99), demonstrating that the model explained the data well.
Figure 4A shows the time-kill curves for each of the moxifloxacin monotherapy regimens. The steepest slope for moxifloxacin monotherapy shown in Fig. 4A was −0.82 ± 0.15 log10 CFU/ml/day at an AUC0–24 of 64.64 mg · h/liter (r2 = 0.97). Interestingly, the AUC0–24 for moxifloxacin plus reserpine of 12.90 mg · h/liter had the second steepest slope, −0.61 ± 0.19 log10 CFU/ml/day (r2 = 0.92) versus −0.19 ± 0.02 log10 CFU/ml/day (r2 = 0.98), for the higher AUC0–24 for moxifloxacin without reserpine of 16.05 mg · h/liter. This suggests that despite moxifloxacin's excellent efficacy, there is some efflux pump-induced tolerance, consistent with observations in other pathogenic mycobacteria and different pharmacophores (20, 32). The hollow-fiber system with higher AUC0–24 plus reserpine did worse in Fig. 4A, consistent with the level of efflux pump induction being inversely proportional to the concentration of the antibiotic inducing it, as noted with ethambutol and M. tuberculosis (19). The relationship between the AUC0–24/MIC ratio and M. kansasii burden is shown in Fig. 4B. At the end of the experiment, the inhibitory sigmoid Emax relationship was described using parameters shown in Table 1. Based on this relationship, the EC80 was calculated as an AUC0–24/MIC ratio of 317 and a nonprotein-bound ratio of 158.5, assuming 50% protein binding (29). There was no emergence of resistance during the 14 days of study.
FIG 4.
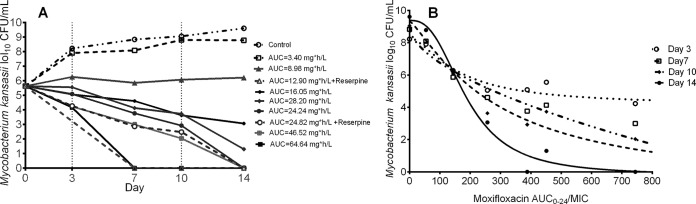
Moxifloxacin monotherapy dose-effect against intracellular M. kansasii. (A) Kill slopes of different moxifloxacin AUC0–24 exposures. An AUC of 8.98 (AUC0–24/MIC, ∼150; AUC0–24/MIC of free drug, 75), shown by the triangles, was associated with stasis effect or just holding the bacterial burden constant. (B) Inhibitory sigmoid Emax relationships between the AUC0–24/MIC ratio and M. kansasii burden for different days of sampling.
TABLE 1.
Day 14 inhibitory sigmoid Emax model parameters for moxifloxacin
| Parameter | Estimate | 95% CIa |
|---|---|---|
| Burden in non-treated control (log10 CFU/ml) | 9.39 | 7.91–10.87 |
| Emax (log10 CFU/ml) | 9.60 | 7.23–11.96 |
| Hill slope | 2.65 | 0.69–4.62 |
| AUC/MIC associated with 50% of Emax in mg · h/liter | 188.2 | 130.3–246.1 |
CI, confidence interval.
To put this into dosing context, Monte Carlo simulations revealed the results shown in Fig. 5. The cumulative fraction of response for 200 mg/day achieving or exceeding the EC80 was 13.81% (Fig. 5A), that of the standard dose of 400 mg was 67.33% (Fig. 5B), that of 600 mg was 88.27% (Fig. 5C), and that of 800 mg was 91.03% (Fig. 5D). Figure 5D also shows that the lowest MIC at which the target attainment falls below 90%, indicating that at least 10% of patients will not attain the EC80 (which defines the susceptibility breakpoint), was 0.125 mg/liter. This means that for resistance assays, the moxifloxacin critical concentration in Middlebrook media would be 0.25 mg/liter.
FIG 5.

Target attainment probability of different moxifloxacin doses. (A) Target attainment probability with 200 mg/day (lowest dose tested) at each MIC. The cumulative fraction of response is very low. (B) The standard dose of 400 mg/day failed to achieve a cumulative fraction of response of >30%. The susceptibility breakpoint, i.e., the MIC at which >90% achieve EC80, for the standard dose is 0.0625 mg/liter, which means the critical concentration would be 0.125 mg/liter at this dose. (C) A higher cumulative fraction of response was achieved with a dose of 600 mg/day but still was less than 90%. (D) Cumulative fraction of response with 800 mg/day moxifloxacin was >90%. The susceptibility breakpoint at this dose is a MIC of 0.125 mg/liter, which means the critical concentration for use in susceptibility assays at this dose is 0.25 mg/liter.
DISCUSSION
First, we report a novel preclinical model of M. kansasii that can be used to develop new treatment regimens. Given that it is unlikely that there will be clinical trials to test the efficacy of new drugs for the treatment of M. kansasii disease in the near future, the model offers a platform to examine drug efficacy. The model can be used for dose selection, to examine for resistance emergence, and to rank the kill rates of new regimens versus standard therapy.
Second, we demonstrate the emergence of antibiotic tolerance in replicating M. kansasii. This tolerance likely is induced by multiple different efflux pumps, similar to findings in M. avium and M. tuberculosis with several different pharmacophores (18–20). These efflux pumps can be overcome either with higher drug dose or the employment of efflux pump inhibitors. Early efflux pump induction also may be important in enabling the development of gene mutations associated with high-level drug resistance in the “antibiotic resistance arrow of time” (20, 32).
Third, we demonstrate that the efficacy of moxifloxacin against M. kansasii is high. Thus, moxifloxacin could be an important addition to the treatment regimen that could shorten therapy. Based on Monte Carlo simulations, we propose that this is best achieved by a clinical dose of 800 mg a day. Such doses have been administered in the clinic for tuberculosis with no increased toxicity compared to that of the standard dose (33–35). This higher dose would simultaneously achieve higher efficacy and overcome drug tolerance. Nevertheless, more safety studies will need to be performed for these nonapproved doses.
Fourth, we propose a new susceptibility breakpoint for moxifloxacin for M. kansasii. We propose a susceptibility breakpoint of 0.25 mg/liter to classify isolates from patients with a greater or lesser chance to respond to moxifloxacin therapy. In the case of M. tuberculosis, such susceptibility breakpoints derived from hollow-fiber studies and Monte Carlo simulations have redefined multidrug resistance and have been found to be accurate based on actual clinical studies that examined patient response (36–42). Thus, our proposed breakpoint likely will turn out to be accurate. The use of the resazurin assay would identify this much more rapidly than conventional methods. This relatively cost-effective assay does not need specialized equipment or highly skilled personnel to detect drug resistance.
There are some limitations to our study. We examined only one standard laboratory strain of M. kansasii in our experiments, whereas the AUC0–24/MIC ratios associated with optimal effect could be a distribution. Thus, it might change if a large number of clinical isolates with a range of MICs was tested. Second, there was no emergence of resistance to monotherapy. This is likely because our studies were for only 14 days, and longer-term therapy could lead to the emergence of resistance.
In summary, we present a new hollow-fiber model of intracellular M. kansasii for use to study potentially shorter therapy durations. We used it to identify moxifloxacin exposures associated with an optimal kill of intracellular M. kansasii.
ACKNOWLEDGMENTS
This work was funded by a grant from the National Institutes of Health/National Institute of General Medical Sciences Director New Innovator Award (1 DP2 OD001886) to T.G.
T. G. has worked as a consultant for Astellas Pharma US, Inc., on antifungal work. T.G. founded Jacaranda Biomed, Inc.
REFERENCES
- 1.Corbett EL, Churchyard GJ, Hay M, Herselman P, Clayton T, Williams B, Hayes R, Mulder D, De Cock KM. 1999. The impact of HIV infection on Mycobacterium kansasii disease in South African gold miners. Am J Respir Crit Care Med 160:10–14. doi: 10.1164/ajrccm.160.1.9808052. [DOI] [PubMed] [Google Scholar]
- 2.da Silva Telles MA, Chimara E, Ferrazoli L, Riley LW. 2005. Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J Med Microbiol 54:975–979. doi: 10.1099/jmm.0.45965-0. [DOI] [PubMed] [Google Scholar]
- 3.de Mello KG, Mello FC, Borga L, Rolla V, Duarte RS, Sampaio EP, Holland SM, Prevots DR, Dalcolmo MP. 2013. Clinical and therapeutic features of pulmonary nontuberculous mycobacterial disease, Brazil, 1993-2011. Emerg Infect Dis 19:393–399. doi: 10.3201/eid/1903.120735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 5.Horsburgh CR Jr, Selik RM. 1989. The epidemiology of disseminated nontuberculous mycobacterial infection in the acquired immunodeficiency syndrome (AIDS). Am Rev Respir Dis 139:4–7. doi: 10.1164/ajrccm/139.1.4. [DOI] [PubMed] [Google Scholar]
- 6.Shitrit D, Baum GL, Priess R, Lavy A, Shitrit AB, Raz M, Shlomi D, Daniele B, Kramer MR. 2006. Pulmonary Mycobacterium kansasii infection in Israel, 1999-2004: clinical features, drug susceptibility, and outcome. Chest 129:771–776. doi: 10.1378/chest.129.3.771. [DOI] [PubMed] [Google Scholar]
- 7.Griffith DE. 2007. Therapy of nontuberculous mycobacterial disease. Curr Opin Infect Dis 20:198–203. doi: 10.1097/QCO.0b013e328055d9a2. [DOI] [PubMed] [Google Scholar]
- 8.Guna R, Munoz C, Dominguez V, Garcia-Garcia A, Galvez J, de Julian-Ortiz JV, Borras R. 2005. In vitro activity of linezolid, clarithromycin and moxifloxacin against clinical isolates of Mycobacterium kansasii. J Antimicrob Chemother 55:950–953. doi: 10.1093/jac/dki111. [DOI] [PubMed] [Google Scholar]
- 9.Pasipanodya J, Gumbo T. 2011. An oracle: antituberculosis pharmacokinetics-pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother 55:24–34. doi: 10.1128/AAC.00749-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith MB, Molina CP, Schnadig VJ, Boyars MC, Aronson JF. 2003. Pathologic features of Mycobacterium kansasii infection in patients with acquired immunodeficiency syndrome. Arch Pathol Lab Med 127:554–560. doi: 10.1043/0003-9985(2003)127<0554:PFOMKI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 11.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis 190:1642–1651. doi: 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 12.Gumbo T, Louie A, Liu W, Brown D, Ambrose PG, Bhavnani SM, Drusano GL. 2007. Isoniazid bactericidal activity and resistance emergence: integrating pharmacodynamics and pharmacogenomics to predict efficacy in different ethnic populations. Antimicrob Agents Chemother 51:2329–2336. doi: 10.1128/AAC.00185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, Drusano GL. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother 51:3781–3788. doi: 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gumbo T, Siyambalapitiyage Dona CS, Meek C, Leff R. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother 53:3197–3204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. 2011. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 204:1951–1959. doi: 10.1093/infdis/jir658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. 2013. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis 208:1464–1473. doi: 10.1093/infdis/jit352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Critical Path Institute. 2013. Voluntary exploration data submission to the FDA. Hollow fiber system model of tuberculosis. Critical Path Institute, Tucson, AZ. [Google Scholar]
- 18.Gumbo T, Louie A, Liu W, Ambrose PG, Bhavnani SM, Brown D, Drusano GL. 2007. Isoniazid's bactericidal activity ceases because of the emergence of resistance, not depletion of Mycobacterium tuberculosis in the log phase of growth. J Infect Dis 195:194–201. doi: 10.1086/510247. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava S, Musuka S, Sherman C, Meek C, Leff R, Gumbo T. 2010. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis 201:1225–1231. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. 2012. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother 56:4806–4815. doi: 10.1128/AAC.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palomino JC, Martin A, Camacho M, Guerra H, Swings J, Portaels F. 2002. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 46:2720–2722. doi: 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. 2010. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic-pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother 54:1728–1733. doi: 10.1128/AAC.01355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. 2010. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother 54:2534–2539. doi: 10.1128/AAC.01761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srivastava S, Gumbo T. 2011. In vitro and in vivo modeling of anti-tuberculosis drugs and its impact on optimization of doses and regimens. Curr Pharm Des 17:2881–2888. doi: 10.2174/138161211797470192. [DOI] [PubMed] [Google Scholar]
- 25.D'Argenio DZ, Schumitzky A. 1997. ADAPT II. A program for simulation, identification, and optimal experimental design. User manual. Biomedical Simulations Resource. University of Southern California, Los Angeles, CA. [Google Scholar]
- 26.Corbett EL, Hay M, Churchyard GJ, Herselman P, Clayton T, Williams BG, Hayes R, Mulder D, De Cock KM. 1999. Mycobacterium kansasii and M. scrofulaceum isolates from HIV-negative South African gold miners: incidence, clinical significance and radiology. Int J Tuberc Lung Dis 3:501–507. [PubMed] [Google Scholar]
- 27.Zvada SP, Denti P, Sirgel FA, Chigutsa E, Hatherill M, Charalambous S, Mungofa S, Wiesner L, Simonsson US, Jindani A, Harrison T, McIlleron HM. 2014. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob Agents Chemother 58:503–510. doi: 10.1128/AAC.01478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon N, Sampol E, Albanese J, Martin C, Arvis P, Urien S, Lacarelle B, Bruguerolle B. 2003. Population pharmacokinetics of moxifloxacin in plasma and bronchial secretions in patients with severe bronchopneumonia. Clin Pharmacol Ther 74:353–363. doi: 10.1016/S0009-9236(03)00201-7. [DOI] [PubMed] [Google Scholar]
- 29.Stass H, Dalhoff A, Kubitza D, Schuhly U. 1998. Pharmacokinetics, safety, and tolerability of ascending single doses of moxifloxacin, a new 8-methoxy quinolone, administered to healthy subjects. Antimicrob Agents Chemother 42:2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Capitano B, Mattoes HM, Shore E, O'Brien A, Braman S, Sutherland C, Nicolau DP. 2004. Steady-state intrapulmonary concentrations of moxifloxacin, levofloxacin, and azithromycin in older adults. Chest 125:965–973. doi: 10.1378/chest.125.3.965. [DOI] [PubMed] [Google Scholar]
- 31.Gumbo T. 2010. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother 54:1484–1491. doi: 10.1128/AAC.01474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasipanodya JG, Gumbo T. 2011. A new evolutionary and pharmacokinetic-pharmacodynamic scenario for rapid emergence of resistance to single and multiple anti-tuberculosis drugs. Curr Opin Pharmacol 11:457–463. doi: 10.1016/j.coph.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruslami R, Ganiem AR, Dian S, Apriani L, Achmad TH, van der Ven AJ, Borm G, Aarnoutse RE, van Crevel R. 2013. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open-label, randomised controlled phase 2 trial. Lancet Infect Dis 13:27–35. doi: 10.1016/S1473-3099(12)70264-5. [DOI] [PubMed] [Google Scholar]
- 34.Pranger AD, van Altena R, Aarnoutse RE, van Soolingen D, Uges DR, Kosterink JG, van der Werf TS, Alffenaar JW. 2011. Evaluation of moxifloxacin for the treatment of tuberculosis: 3 years of experience. Eur Respir J 38:888–894. doi: 10.1183/09031936.00176610. [DOI] [PubMed] [Google Scholar]
- 35.Alffenaar JW, van Altena R, Bokkerink HJ, Luijckx GJ, van Soolingen D, Aarnoutse RE, van der Werf TS. 2009. Pharmacokinetics of moxifloxacin in cerebrospinal fluid and plasma in patients with tuberculous meningitis. Clin Infect Dis 49:1080–1082. doi: 10.1086/605576. [DOI] [PubMed] [Google Scholar]
- 36.Williamson DA, Roberts SA, Bower JE, Vaughan R, Newton S, Lowe O, Lewis CA, Freeman JT. 2012. Clinical failures associated with rpoB mutations in phenotypically occult multidrug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis 16:216–220. doi: 10.5588/ijtld.11.0178. [DOI] [PubMed] [Google Scholar]
- 37.van Ingen J, Aarnoutse R, de Vries G, Boeree MJ, van Soolingen D. 2011. Low-level rifampicin-resistant Mycobacterium tuberculosis strains raise a new therapeutic challenge. Int J Tuberc Lung Dis 15:990–992. doi: 10.5588/ijtld.10.0127. [DOI] [PubMed] [Google Scholar]
- 38.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. 2014. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother 69:2420–2425. doi: 10.1093/jac/dku136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. 2014. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother 58:6111–6115. doi: 10.1128/AAC.03549-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho J, Jelfs P, Sintchencko V. 2013. Phenotypically occult multidrug-resistant Mycobacterium tuberculosis: dilemmas in diagnosis and treatment. J Antimicrob Chemother 68:2915–2920. doi: 10.1093/jac/dkt284. [DOI] [PubMed] [Google Scholar]
- 41.Ocheretina O, Escuyer VE, Mabou MM, Royal-Mardi G, Collins S, Vilbrun SC, Pape JW, Fitzgerald DW. 2014. Correlation between genotypic and phenotypic testing for resistance to rifampin in Mycobacterium tuberculosis clinical isolates in Haiti: investigation of cases with discrepant susceptibility results. PLoS One 9:e90569. doi: 10.1371/journal.pone.0090569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christianson S, Voth D, Wolfe J, Sharma MK. 2014. Re-evaluation of the critical concentration for ethambutol antimicrobial sensitivity testing on the MGIT 960. PLoS One 9:e108911. doi: 10.1371/journal.pone.0108911. [DOI] [PMC free article] [PubMed] [Google Scholar]