

Abstract
One way to speed up the TB drug discovery process is to search for antitubercular activity among compound series that already possess some of the key properties needed in anti-infective drug discovery, such as whole-cell activity and oral absorption. Here, we present MGIs, a new series of Mycobacterium tuberculosis gyrase inhibitors, which stem from the long-term efforts GSK has dedicated to the discovery and development of novel bacterial topoisomerase inhibitors (NBTIs). The compounds identified were found to be devoid of fluoroquinolone (FQ) cross-resistance and seem to operate through a mechanism similar to that of the previously described NBTI GSK antibacterial drug candidate. The remarkable in vitro and in vivo antitubercular profiles showed by the hits has prompted us to further advance the MGI project to full lead optimization.
INTRODUCTION
Tuberculosis (TB), one of the oldest known infections, is still the second leading cause of mortality worldwide (1). The World Health Organization (WHO) estimated that there were 8.6 million new TB cases in 2012 and 1.3 million TB deaths, with a particularly high incidence among HIV-coinfected individuals (2).
The current standard directly observed treatment short course (DOTS) (3) consists of 2 months of treatment with isoniazid (INH), rifampin (Rf), pyrazinamide (PZA), and ethambutol (EMB) followed by 4 additional months of INH and Rf (4). Nevertheless, resistance to isoniazid, as well as to the combination of isoniazid and rifampin, a situation defining multidrug resistance (MDR), is common. Additionally, the emergence of novel strains which are resistant to these two drugs and to one of the three most commonly employed injectables has given rise to a novel category, the extensively drug-resistant (XDR) (5) strain. One step further, total drug resistance (TDR), is defined by resistance to all second-line drug classes. TDR cases have been increasingly reported in the clinic (6, 7).
Over the past decade, drug discovery and development efforts have increased, fueled by the upcoming threat of drug resistance in combination with the expansion of the HIV pandemic. These realities highlight an urgent need for more effective and tolerable treatments for drug-susceptible and drug-resistant disease in addition to latent TB infection.
To take advantage of the broad expertise of GSK in antibacterial drug discovery, a large subset of compounds representative of the wide chemical diversity generated in the GSK novel bacterial topoisomerase inhibitor (NBTI) initiative (8, 9, 10, 11) were evaluated in vitro against Mycobacterium tuberculosis. This exercise resulted in the identification of novel M. tuberculosis DNA gyrase inhibitors (MGIs), a new family of promising compounds with potential for the treatment of TB disease already reported in the literature as antimycobacterial and antibacterial agents (12, 13, 14, 15). Recently, AstraZeneca has reported anti-TB activity for NBTIs (16).
Here, we introduce MGIs as new advanced leads against TB as evidenced by their attractive in vivo and in vitro antitubercular profiles in addition to their lack of cross-resistance with fluoroquinolones (FQs).
MATERIALS AND METHODS
General aspects and ethics statement.
All of the experiments were approved by the Diseases of the Developing World, GSK Ethical Committee. All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EU and the GSK policy on the care, welfare, and treatment of animals. Specific-pathogen-free 6- to 8-week-old female C57BL/6j mice (18 to 20 g) were obtained from Harlan Interfauna (Iberica, Spain). The experiments were performed at the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited GSK laboratory animal science animal facilities in Tres Cantos (Madrid, Spain). The mice were kept in air-conditioned facilities with 15 air changes per hour. Room temperature and relative humidity were 22 ± 3°C and 40 to 70%, respectively. The mice were accommodated in groups of up to five individuals in TecniplastH type IV cages with autoclaved dust-free corncob bedding (Panlab, Barcelona, Spain). The mice were maintained under a 12-h light/dark period. Autoclaved tap water and an irradiated pelleted diet were provided ad libitum. The compounds used in these in vivo studies were prepared as suspensions in 1% methyl cellulose. The antitubercular standards used in the efficacy study were moxifloxacin (Sequoia Research Products Ltd.) and isoniazid (Sigma Aldrich), prepared as a solution in 20% Captisol-water and in water, respectively.
Compound synthesis.
Compounds 1 (17), 2, and 3 (12) were prepared following a previously described synthetic pathway (see the supplemental material).
In vitro assays. (i) Bacterial strains and culture.
M. tuberculosis strains (H37Rv, Beijing 1237, CDC1551, Erdman, and 8 clinical strains), Mycobacterium canetti, and Mycobacterium bovis BCG were grown at 37°C in Middlebrook 7H9 broth supplemented with 0.025% Tween 80 and 10% albumin-dextrose-catalase (ADC) or on Middlebrook 7H10 plates supplemented with 10% oleic acid-ADC (OADC). Escherichia coli DH5α was grown in LB broth. Ampicillin, hygromycin (Hyg), and sucrose (Suc) reagents were used for plasmid selection or recombination experiments.
(ii) DNA manipulation, plasmids, and transformation.
General molecular biology procedures were used as described previously (18) or following the manufacturer's instructions. Escherichia coli DH5α and M. tuberculosis H37Rv competent cells were prepared for electroporation as described previously (19).
(iii) MIC determination.
M. tuberculosis H37Rv ATCC 27294 was used for all the studies and was grown and tested as described earlier (20). The MIC was determined as described previously (20).
(iv) Killing assays.
Bacteria were grown at 37°C in 7H9 Middlebrook-ADC-Tween 80 to mid-exponential–exponential phase and then diluted in 10 ml fresh Middlebrook 7H9-ADC-Tween 80 to 5 × 105 CFU/ml. Incubation was continued after the addition of compounds at 20× MIC. At specified time points, aliquots of cultures were withdrawn, serially diluted in 7H9 Middlebrook-ADC-Tween 80, and plated on solid culture medium (7H10-OADC). Plates were then incubated at 37°C, and CFU were counted after 3 to 4 weeks.
(v) Generation rate of spontaneous resistant mutants.
The MICs in solid medium (Middlebrook 7H10-OADC) were determined in 24-well plates with serial dilutions of the compound of interest. Five microliters of bacterial culture containing 105 CFU/ml was added per well. Plates were incubated at 37°C for 20 days for M. tuberculosis H37Rv. MIC values are the minimum concentration of the compound which inhibits 90% bacterial growth.
In order to determine the generation rate of spontaneous resistant mutants to the compounds, bacteria were grown at 37°C in fresh Middlebrook 7H9-ADC-Tween 80 to the midexponential phase and then diluted in fresh Middlebrook 7H9-ADC-Tween 80 to 5 × 108 CFU/ml. Middlebrook 7H10-OADC plates with 20× MIC of each compound were inoculated with 108, 107, 106, and 105 CFU/plate, and the plates were incubated at 37°C during 3 to 4 weeks. The frequency of appearance of resistant mutants was calculated, and isolated colonies were restreaked onto new plates filled with Middlebrook 7H10-OADC agar containing the drugs and on plates without the drugs as a growth control in order to isolate the colonies.
(vi) Characterization of M. tuberculosis H37Rv MGI-resistant mutants.
Single colonies were used to amplify and sequence the quinolone resistance-determining region (QRDR). DNAs of the 21 resistant mutants were extracted from liquid cultures grown in Middlebrook 7H9-ADC-Tween 80. One milliliter of culture was centrifuged, and the pellet was resuspended in 500 μl of distilled water, incubated at 90°C for 2 h, and then filtered by using 0.22-μm filters. Five microliters of the supernatant was used as a source of genomic DNA for amplification of the QRDR. PCRs were performed by using a PuReTaq Ready-To-Go PCR beads kit (GE Healthcare Life Sciences). The QRDR was amplified with the primers described previously (21): gyrA1 (5′-CAGCTACATCGACTATGCGA-3′) and gyrA2 (5′-GGGCTICGGTGTACCTCAT-3′) for gyrA and gyrB1 (5′-CCACCGACATCGGTGGATT-3′) and gyrB2 (5′-CTGCCAClTGAGTTTlGTACA-3′) for gyrB. Amplification was performed for 40 cycles (94°C for 1 min, 55°C for 1 min, and 72°C for 1 min) to generate a fragment of 320 bp for gyrA and a fragment of 427 bp for gyrB.
PCR products were purified and sequenced using the dRhodamine Terminator cycle sequencing ready reaction kit (Applied Biosystems) in an ABI PRISM 7700 sequence detection system (Applied Biosystems). The sequences obtained were analyzed using Lasergene software (DNA and protein sequence analysis software; DNAStar).
(vii) Reversion from mutant to wild-type gyrA.
The system pNIL/pGOAL (22) was used. Briefly, the gyrA wild type from H37Rv was amplified with the primers gyrA19 (5′-GATCACTCCTAACACTCGTACCCGGC-3′) and gyrA20 (5′-GATCGTGGGAGACCACCATGGATCCC-3′) and cloned in pGEM-T Easy (Promega). A 3-kb NotI/ScaI fragment from pGEM-T gyrA was cloned into the NotI site of p1NIL. The PacI cassette from pGOAL19 (Hyg PAg85-lacZ Phsp60-sacB) was then cloned into the single PacI site to generate the suicide delivery vector pEGSK3. Vector DNA was used to electroporate M. tuberculosis H37Rv. Hygromycin- and kanamycin-resistant transformants (single crossovers) were inoculated in Middlebrook 7H9-ADC-Tween 80 and incubated for 2 weeks at 37°C. Serial dilutions were plated onto 2% sucrose plates. Plates were incubated for 3 to 4 weeks. Sucr colonies were then streaked onto plates with and without hygromycin or with and without an MGI compound to identify the different phenotypes.
(viii) Inhibition of M. tuberculosis DNA gyrase by compounds.
M. tuberculosis DNA gyrase activity was measured by supercoiling of pBR322 DNA (Inspiralis) similarly to previously described methods (23); 0.3 U of M. tuberculosis DNA gyrase holoenzyme (Inspiralis) was used per reaction mixture (25 μl), as this was the amount required to almost fully relax 500 ng of pBR322 DNA over 60 min at 37°C. In a total reaction mixture volume of 25 μl, 0.3 U of M. tuberculosis DNA gyrase in 50 mM Tris (pH 7.9) containing 5 mM dithiothreitol (DTT) and 30% (vol/vol) glycerol was added to the reaction buffer containing 50 mM HEPES (pH 7.9), 6 mM magnesium acetate, 4 mM DTT, 1 mM ATP, 100 mM potassium glutamate, 2 mM spermidine, 0.05 mg/ml bovine serum albumin (BSA), and 500 ng of relaxed pBR322 DNA. For inhibition studies, compounds were used at the appropriate concentrations by diluting down from 10 mM master stocks prepared in Me2SO (the final assay concentration was 1% [vol/vol] Me2SO for each compound dilution). For negative controls, reaction mixtures were set up in the absence of M. tuberculosis DNA gyrase. Each experiment was done at least twice with similar results.
Several fluoroquinolones were tested as pharmacology tools. The fitted 50% inhibitory concentrations (IC50s) for gatifloxacin (0.36 ± 0.1 μM), ciprofloxacin (4.90 ± 0.8 μM), moxifloxacin (2.50 ± 0.8 μM), and levofloxacin (5.15 ± 0.8 μM) were in general consensus with literature data (24).
In vivo efficacy assessment.
Specific-pathogen-free, 8- to 10-week-old female C57BL/6 mice were purchased from Harlan Laboratories and were allowed to acclimate for 1 week. The experimental design has been previously described (25). In brief, mice were intratracheally infected with 105 CFU/mouse (M. tuberculosis H37Rv strain). Products were administered for 8 consecutive days starting 1 day after infection. Lungs were harvested 24 h after the last administration. All lung lobes were aseptically removed, homogenized, and frozen. Homogenates were plated in 10% OADC-7H11 medium for 14 days at 37°C.
Molecular modeling and docking calculations.
The M. tuberculosis gyrase A homology model was built using the Staphylococcus aureus X-ray structure (Protein Data Bank [PDB] code 2XCS) as the template. Molecular modeling and energy minimization experiments were performed using MOE (26). For ligands, three-dimensional (3D) structures were constructed using the Builder module of MOE and optimized by energy minimization using the MMFF94X force field. All of the compounds were subjected to conformational searches for their lowest energy conformations as the docking input. Molecular docking experiments were carried out with the GOLD program (v4.01) (27). For each compound, the best pose was selected based on the evaluation of their best scores and similar interactions within the active site.
RESULTS
Screen of NBTI chemical diversity.
In order to determine the potential of NBTIs as novel antitubercular drugs, a large subset of 3,000 compounds representative of the wide chemical diversity generated in the project (>10,000 compounds) was selected for evaluation in vitro against M. tuberculosis H37Rv.
The M. tuberculosis MIC results obtained showed a high hit rate of active compounds (68% of them with MIC values lower than 10 μM). The most potent derivatives matched or improved the MIC values for currently used TB drugs, including last-generation FQs. Overall, 29% of the compounds had MICs of <1 μM, and 18% had MICs of <0.1 μM.
Similarly to those of NBTIs, the general structures of TB-active compounds were divided into three different parts according to the three topologically important areas in terms of interaction with the target gyrase (8). First, a left-hand side (LHS) is responsible for important contacts with the gyrase DNA substrate. Second, a right-hand side (RHS) is embedded into the gyrase enzyme, in addition to potentially providing some bacterial target selectivity. Last, a central linker unit (CU) establishes key interactions with the gyrase (8) and offers the possibility of modulating important physicochemical properties (solubility [CHIlogD]) that can impact human ether-à-go-go-related gene (hERG) inhibition (for hERG inhibition reviews, see reference 28).
These initial structure-activity observations, inherited from the predecessor antibacterial program, helped to schematize synthesis (details to be reported elsewhere) and to rationalize how to balance antimycobacterial potency with oral exposure, safety, and synthetic complexity. These efforts resulted in the identification of the 7-substituted-1,5-naphthyridin-2-one core as a privileged LHS, the N-ethyl-4-aminopiperidines as a linker, and monocyclic aromatic rings with different substitution patterns as the best RHS option, in contrast to NBTIs bearing bicyclic rings as the RHS (15). These three favorable features were combined in the synthesis of analogues 2 and 3 (Fig. 1).
FIG 1.

Structures of the best balanced NBTI/MGI hits.
Antitubercular profile.
Compound 1, as the best NBTI direct screening representative, and novel synthetic entities 2 and 3 were selected to be progressed to a panel of in vitro and in vivo studies to validate their potential as antitubercular drugs. The extracellular MICs were found to be 0.5 μM for compound 1, 0.08 μM for compound 2, and <0.01 μM for compound 3 (moxifloxacin MIC, 0.15 μM). In terms of intracellular antitubercular activity against M. tuberculosis THP1-infected macrophages, these compounds have shown very good activity (MIC of <0.02 μM for compounds 2 and 3).
MICs of the three key MGI compounds against a panel of M. tuberculosis clinical and laboratory strains were then established (Tables 1 and 2) in order to have an early indication of the potential of these compounds to address MDR strains. MICs were found to be higher than those for the reference M. tuberculosis H37Rv strain, while still being within a range acceptable for further MDR consideration.
TABLE 1.
MICs of compounds 1, 2, and 3 against 8 clinical strains of M. tuberculosis
| Compound | MIC (μg/ml) against strain: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| H37Rv | Clin1 | Clin2 | Clin3 | Clin4 | Clin5 | Clin6 | Clin7 | Clin8 | |
| 1 | 0.29 | 2.44 | 3.66 | 3.66 | 1.22 | 1.22 | NDa | 4.88 | 0.30 |
| 2 | 0.04 | 0.13 | 0.56 | 0.26 | 0.26 | 0.22 | 0.22 | 0.09 | 0.56 |
| 3 | ≤0.01 | 0.02 | 0.11 | 0.04 | 0.04 | 0.04 | 0.02 | 0.02 | 0.09 |
| Ciprofloxacin | 0.08 | 0.31 | 0.08 | 0.31 | 0.12 | 0.16 | 0.08 | 0.31 | 0.23 |
| Ofloxacin | 0.6 | 0.3 | 0.3 | 0.5 | 0.3 | 0.5 | 0.3 | 0.6 | 0.5 |
| Moxifloxacin | 0.1 | 0.08 | 0.04 | 0.06 | 0.04 | 0.06 | 0.04 | 0.08 | 0.04 |
ND, not determined.
TABLE 2.
MICs of key MGI compounds against wild-type M. tuberculosis laboratory strains and M. bovis BCG
| Compound | MIC (μg/ml) against strain: |
|||||
|---|---|---|---|---|---|---|
| H37Rv | Beijing 1237 | Canettii | CDC1551 | Erdman | BCG | |
| 2 | 0.04 | 0.15 | 0.15 | 0.15 | 0.15 | 0.12 |
| 3 | ≤0.01 | 0.03 | 0.04 | 0.04 | 0.04 | ≤0.01 |
| Ciprofloxacin | 0.2 | 0.08 | 0.16 | 0.12 | 0.12 | 0.04 |
| Ofloxacin | 0.6 | 0.3 | 0.5 | 0.3 | 0.5 | 0.16 |
| Moxifloxacin | 0.08 | 0.04 | 0.08 | 0.08 | 0.08 | ≤0.02 |
As a next step in compound evaluation, the bactericidal potentials of the new MGI compounds were studied by determining the time-kill kinetic curves of compounds 2 and 3 and comparing them with that of linezolid as a bacteriostatic model drug, while isoniazid and a GSK InhA direct inhibitor were used as bactericidal controls (Fig. 2). Results show that these new MGIs are bactericidal as defined by the ability of a compound to reduce 99.9% of the bacterial population in 7 days (for in vitro activities of fluoroquinolones, see reference 29).
FIG 2.

Time-kill curves of M. tuberculosis H37Rv in 7H9ADC medium at 20× MIC of compounds 2 and 3. Linezolid was used as a bacteriostatic model drug while isoniazid and a GSK InhA direct inhibitor were used as bactericidal controls.
In terms of the potential for spontaneously resistant mutant generation using compound 1 as a model molecule, a key consideration in any target-based program, the frequency of spontaneously resistant mutant generation was found to be 7.4 × 10−8 mutants/CFU, a number clearly lower than that of isoniazid and similar to that of rifampin. As a direct consequence of these assays, 21 independent mutant colonies spontaneously resistant to our compounds were isolated and characterized. Sixteen of them had a single point mutation in the quinolone resistance-determining regions (QRDR) (30). Thirteen were found in gyrA, where 8 different mutations were identified (H87Q, P50T, E79G, D89N, R98H, S95G, D89G, and H52R), and 3 in gyrB, where two different mutations were observed (D495N and D495A). Only one of the mutations found in the QRDR of gyrA (D89N) has been previously reported (31) as being related to fluoroquinolone resistance.
More in-depth sequencing identified point mutations in gyrA but outside the QRDR in the remaining 5 strains. The mutations were H368R, I348S, and S178L (Fig. 3).
FIG 3.

Mapping of resistance mutations in the GyrA-GyrB model from M. tuberculosis. The M. tuberculosis gyrase A homology model was built using the S. aureus X-ray structure (PDB code 2XCS).
MICs in the spontaneous mutants were found to be between 10 and >200 times higher than in the wild type, with cross-resistance between the three described MGIs being a common finding not associated to FQs (ciprofloxacin, ofloxacin, and moxifloxacin). The only exception was found in the D89 mutants, which show a slight but noticeable cross-resistance (2- to 8-fold higher MICs for ciprofloxacin and moxifloxacin, compared to the 20- to 300-fold increase in the MICs for our compounds) (Table 3).
TABLE 3.
MICs of the GyrA mutants are represented as the ratios of the MICs obtained for each compound in relation to the wild type
| Compound | MICmut/MICH37Rv ratio for GyrA mutants |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MGIr |
FQr |
||||||||||||
| H87Q | P50T | R98H | S95G | D89G | H52R | E79G | H368R | I348S | S178L | S91P | D94G | A90V | |
| 1 | 31 | 10.7 | 31 | 11 | 83 | 83 | 128 | 21 | 11 | 11 | NTa | NT | NT |
| 2 | 17 | 8.3 | 25 | 8 | >266 | 67 | 32 | 8 | 8 | 8 | 1 | 0.1 | 0.1 |
| 3 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | >2 | 1 | 1 |
| Ofloxacin | 2 | 1 | 1 | 1 | 3 | 2 | 2 | 0.8 | 1.5 | 1 | 9 | 13.5 | 9 |
| Moxifloxacin | 1 | 1 | 1 | 2 | 3 | 1 | 4 | 0.8 | 1 | 1 | NT | N T | 25 |
| Ciprofloxacin | >1.5 | ≥1 | ≥1 | >1.5 | >3.8 | >1.5 | 2 | ≥1 | >1.5 | ≥1 | >25 | 74 | 25 |
| INH | 1 | 1 | 1 | 1 | 1 | 1 | NT | 1 | 0.8 | 1 | 1 | 1 | 1 |
NT, not tested.
As a further check on the potential for cross-resistance in the clinic, five mutants resistant to FQs were also isolated and characterized. In these mutants, the most commonly encountered FQr mutations described in clinical isolates in gyrA (S91P, A90V, and D94G) (30, 31, 32, 33, 34, 35) were found. Of the laboratory mutants with resistance against FQs that were selected, only S91P mutants showed a slight cross-resistance with some of the lead MGIs, while D94G and A90V mutants gave rise to a hypersensitive phenotype to most of the MGIs tested.
In order to univocally correlate gene mutations with resistance, a system with a suicide plasmid vector for mutant reversal (E79G, H87Q, S95G, H368R, and I348S) was used. We used the system described by Parish and Stoker (22). Briefly, a plasmid with the wild-type M. tuberculosis H37Rv gyrA was transformed into the mutants, and a first homologous recombination took place. Hygr recombinants were selected. A second homologous recombination occurred, and the double recombinants were Hygs-Sucr. Afterwards, gyrA was amplified and sequenced, and MGI resistance was determined. As a example, in the case of the E79G mutant, 15% of the Hygs-Sucr transformants were sensitive for our compounds, and all of them have the wild-type phenotype (Table 4; see also Fig. S1 in the supplemental material). The same process was repeated with the other mutants (H87Q, S95G, H368R, and I348S), and there was a perfect correlation between loss of mutation and reversion to sensitivity (data not shown).
TABLE 4.
Ratios of the MICs obtained in the different strains isolated in the reversion study of the E79G mutant for compounds 1 and 2 in relation to those for the wild-type and polymorphism in codon 79 of the gyrA gene
| Compound | MICmut/MICH37Rv ratio for strain: |
|||||
|---|---|---|---|---|---|---|
| H37Rv | E79G mutant | Single crossover | Double crossover |
|||
| Clon 6 | Clon 8 | Clon 31 | ||||
| 1 | 1 | 128 | 32 | 1 | 32 | 1 |
| 2 | 1 | 32 | 16 | 2 | 16 | 1 |
| Ofloxacin | 1 | 2 | 1 | 1 | 1 | 1 |
| gyrA codon 79 polymorphism | GAG | GGG | NDa | GAG | GGG | GAG |
ND, not determined.
Additionally, the mode of action of a representative set of NBTI and MGI compounds was evaluated in the M. tuberculosis DNA gyrase supercoiling assay. Determination of the concentration-dependent inhibitory effect of compounds 1, 2, and 3 yielded the following IC50s: 2.80 ± 0.65 μM, 10.98 ± 3.41 μM, and 5.4 ± 1.2 nM, respectively. Compound 2 exhibited a poorer correlation between the M. tuberculosis DNA gyrase IC50 and the H37Rv MIC. Further studies are necessary to address this behavior. Most of the compounds with MICs against M. tuberculosis also inhibited the supercoiling activity of M. tuberculosis DNA gyrase, thus confirming their mode of inhibition (Fig. 4). Given the potential for toxicity due to cross-inhibition with human gyrase, a representation of this set was tested in the human topoisomerase II alpha (36). Results showed >30-fold more potency in M. tuberculosis DNA gyrase than against the human enzyme.
FIG 4.
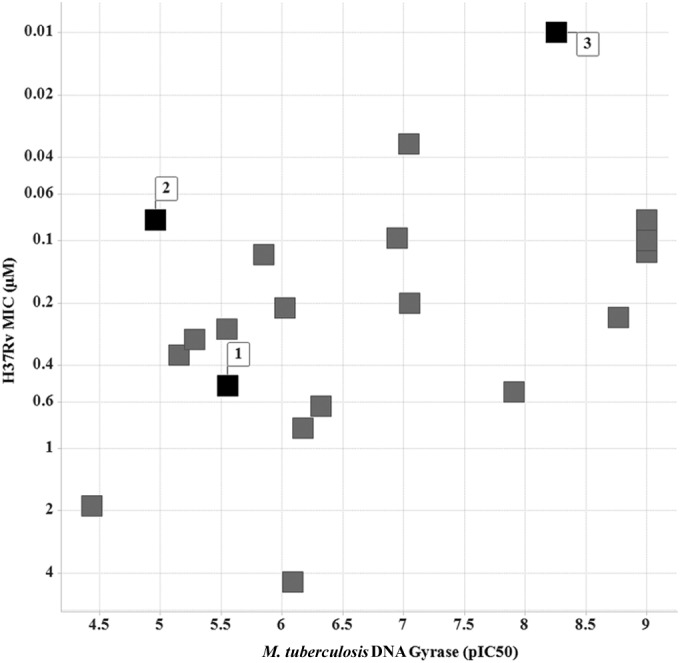
Plot of the concentration-dependent inhibitory effect of a representation of MGIs and NBTIs against M. tuberculosis DNA gyrase expressed as pIC50 versus H37Rv MIC (μM). Compounds 1, 2, and 3 are indicated as black squares.
Once the potential of the new MGI leads for MDR treatment had been clarified, compounds 1, 2, and 3 were further assessed in an acute infection mouse model of tuberculosis (25), measuring the fold reduction in CFU in the lung (Fig. 5) versus the CFU count for untreated controls. A preliminary profiling of selected compounds showed how the compounds were endowed with desirable pharmacokinetic and toxicological profiles (Table 5). The fold reductions in CFU were compared to those achieved with gold standard drugs, such as isoniazid (25 mg/kg body weight orally [p.o.], once a day [u.i.d.]) and moxifloxacin (100 mg/kg p.o., u.i.d.). Both MGIs 2 (50 mg/kg p.o., twice a day [b.i.d.]) and 3 (50 mg/kg subcutaneously [s.c.], b.i.d.) showed a significant CFU reduction in the lungs of infected mice (>2 log inhibition), showing potencies approaching those of the reference compounds. However, 1 (100 mg/kg p.o., b.i.d.), a compound in theory representing a good balance between in vitro anti-TB potency and pharmacokinetic profile (Table 5), failed to show activity at the maximum dose tolerated. This lack of activity might due to the poorer intracellular antitubercular profile exhibited by compound 1 than by MGIs 2 and 3.
FIG 5.
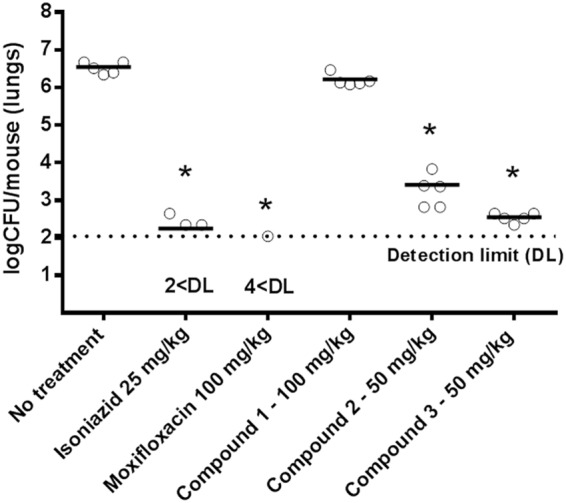
Antitubercular activity of isoniazid, moxifloxacin, and compounds 1, 2, and 3 in an acute infection murine model. Each circle represents data from an individual mouse. *, a P value of >0.05 was considered significant. Data were analyzed by a one-factor analysis of variance (ANOVA) and a Games-Howell post hoc test, since the P value for the Levene test was <0.05.
TABLE 5.
Profiles for representative NBTI/MGI compounds
| Characteristica | Results for compound: |
||
|---|---|---|---|
| 1 | 2 | 3 | |
| H37Rv MIC (μM) | 0.5 | 0.08 | <0.01 |
| Intracellular H37Rv MIC90 (μM) | 1.85 | <0.02 | <0.02 |
| M. tuberculosis DNA gyrase IC50 (μM) | 2.80 | 10.98 | 5.40 |
| HepG2 Tox50 (μM) | >100 | >100 | 67.6 |
| Chrom logD pH7.4 (37) | 1.51 | 3.73 | 4.36 |
| Solubility CLND (μM)b | 354 | ≥386 | ≥335 |
| In vitro CLi (ml/min · g) | |||
| Mousec | 1.09 | 3.8 | 20.84 |
| Humanc | <0.53 | 0.6 | 0.75 |
| In vivo CL (ml/min/kg)c | NT | 88.3 | 132.5 |
| Tmax (h)c | 0.75 | 0.25 | 0.25 |
| Cmax (μg/ml)c | 2.7 | 2.0 | 0.08 |
| DNAUC oral (μg · h/ml)/mg/kgc | 0.1 | 0.1 | 0.01 |
| % Fc | NT | 47.8 | 10.0 |
| hERG PXpress IC50 (μM) (38, 39, 40) | >50 | 2.85 | 1.32 |
Tox50, 50% cytotoxic concentration; CLND, chemiluminescent nitrogen detection; CLi, intrinsic clearance; CL, clearance; Tmax, time to maximum concentration of drug in serum; Cmax, maximum concentration of drug in serum; DNAUC, dose-normalized area under the blood-concentration time curve; NT, not tested.
CLND solubility values that are within 85% of maximum possible concentration (as determined from dimethyl sulfoxide [DMSO] stock concentration).
See supplemental material.
DISCUSSION
As a consequence of the long-term efforts GSK has dedicated to the novel bacterial topoisomerase inhibitor (NBTI) field, new antibacterials with novel modes of action, no cross-resistance with FQs, oral drug-likeness, and good safety profiles have been identified (8, 9, 10, 11). With this exercise as a starting point for TB drug discovery, a representative set of this collection (3,000 compounds) was screened against M. tuberculosis, resulting in the selection of compound leads 1 to 3 for further antitubercular profiling. Compound 1 was identified as the best balanced NBTI both in terms of in vitro anti-TB potency and pharmacokinetic profile, while compounds 2 and 3 were designed as novel entities incorporating key lessons learned from the already known NBTI chemical diversity.
The compounds tested were bactericidal in vitro (10× MIC) and showed a low rate of generation of spontaneous resistant mutants (similar to that of rifampin). As no evidence supporting the fact that MGI compounds killed M. tuberculosis through inhibition of the M. tuberculosis DNA gyrase was available to date and no assays for the TB enzyme were available at that time, as an alternative strategy, we sought to isolate laboratory resistant mutants to these compounds both to determine if there were single-point mutations in the M. tuberculosis DNA gyrase-resistant mutants and to test for potential cross-resistance between MGIs, NBTIs, and FQs. All 21 MGI-resistant strains isolated were found to harbor mutations in either GyrA or GyrB. Selected gyrA mutant reversion to the gyrA wild type resulted in resistant phenotype reversal to sensitive, a situation affecting the 5 mutants tested. These results strongly suggest that MGIs bind to DNA gyrase proximal to the FQ binding site and that inhibition of this enzyme is the lethal event leading to the bactericidal action of MGIs.
Interestingly, mutants with mutations outside the QRDR of gyrA have also been characterized. These mutations might be responsible for changes in the conformation of the protein due to steric impediments or interactions.
Recently, an M. tuberculosis DNA gyrase supercoiling assay was set up in-house. This biochemical assay demonstrated that this class exerts its action via inhibition of M. tuberculosis DNA gyrase.
During the preparation of the manuscript, a closely related class of N-linked aminopiperidinyl-based gyrase inhibitors was described by AstraZeneca (16). M. tuberculosis DNA gyrase supercoiling and the cleavage complex assays confirmed their mode of action.
Following the identification of compounds 1, 2, and 3 as potent antimycobacterials (Table 5), the compounds were evaluated for their ability to treat TB infection in a murine animal model of infection. The in vivo antitubercular profiles of compounds 2 and 3 showed a significant reduction of the CFU in the lungs of acutely infected mice in addition to exhibiting a level of efficacy comparable to that of the gold standard drugs (isoniazid and moxifloxacin) (Fig. 5).
The studies reported here resulted in the establishment of a TB-specific lead optimization effort. The current work is focused on the design of novel TB-tailored molecules that can combine the numerous factors governing successful drug design, i.e., in vitro and in vivo potency, oral exposure, an absence of cardiovascular liabilities related to hERG inhibition (10, 11), and a lack of clastogenic potential deriving from cross-activity with host topoisomerases. Further progress will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
The research leading to these results has received funding from the Global Alliance for TB Drug Development and from the European Commission Seventh Framework Programme ORCHID project no. 261378.
We thank T. Kaneko, K. Mdluli, Z. Ma, and C. Cooper (TB Alliance) for their helpful discussions and expertise in the field. We thank the pharmacology department for providing the data presented. We also thank all the scientists from the NBTI team (Antibacterial DPU and Infectious Disease Therapeutic Area Unit, GlaxoSmithKline) for their collaboration and help.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03913-14.
REFERENCES
- 1.World Health Organization. 2012. Global tuberculosis report 2012. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.World Health Organization. 2013. Global tuberculosis report 2013. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.World Health Organization. 2006. The stop TB strategy. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/strategy/en/. [Google Scholar]
- 4.World Health Organization. 2009. Treatment of tuberculosis: guidelines for national programs, 4th ed WHO/HTM/TB/2009.420 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 5.World Health Organization. 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response. WHO/HTM/TB/2010.3 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 6.Migliori GB, Centis R, D'Ambrosio L, Spanevello A, Borroni E, Cirillo DM, Sotgiu G. 2012. Totally drug-resistant and extremely drug-resistant tuberculosis: the same disease? Clin Infect Dis 54:1379–1380. doi: 10.1093/cid/cis128. [DOI] [PubMed] [Google Scholar]
- 7.Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, Ziazarifi AH, Hoffner SE. 2009. Emergence of new forms of totally drug-resistant tuberculosis bacilli: super extensively drug resistant tuberculosis or totally drug-resistant strains in Iran. Chest 136:420–425. doi: 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- 8.Bax B, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewi CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–943. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 9.Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, Bax BD. 2010. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol 17:1152–1153. doi: 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- 10.Miles TJ, Axten JM, Barfoot C, Brooks G, Brown P, Chen D, Dabbs S, Davies DT, Downie DL, Eyrisch S, Gallagher T, Giordano I, Gwynn MN, Hennessy A, Hoover J, Huang J, Jones G, Markwell R, Miller WH, Minthorn EA, Rittenhouse S, Seefeld M, Pearson N. 2011. Novel amino-piperidines as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg Med Chem Lett 21:7489–7495. doi: 10.1016/j.bmcl.2011.09.117. [DOI] [PubMed] [Google Scholar]
- 11.Miles TJ, Barfoot C, Brooks G, Brown P, Chen D, Dabbs S, Davies DT, Downie DL, Eyrisch S, Giordano I, Gwynn MN, Hennessy A, Hoover J, Huang J, Jones G, Markwell R, Rittenhouse S, Xiang H, Pearson N. 2011. Novel cyclohexyl-amides as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg Med Chem Lett 21:7483–7488. doi: 10.1016/j.bmcl.2011.09.114. [DOI] [PubMed] [Google Scholar]
- 12.Alemparte-Gallardo C, Ballell-Pages L, Barros-Aguirre D, Cacho-Izquierdo M, Castro-Pichel J, Fiandor-Roman JM, Hennessy AJ, Pearson ND, Remuinan-Blanco MJ. July 2009. Naphthyridin-2(1H)-one compounds useful as antibacterials. International patent application WO 2009/090222 A1 20090723. [Google Scholar]
- 13.Alemparte-Gallardo C, Barros-Aguirre D, Cacho-Izquierdo M, Fiandor-Roman JM, Remuinan Blanco MJ. November 2009. Preparation of tricyclic nitrogen containing compounds as antibacterials. International patent application WO 2009/141399 A1 20091126.
- 14.Alemparte-Gallardo C, Barros-Aguirre D, Cacho-Izquierdo M, Fiandor-Roman JM, Lavandera Diaz JL, Remuinan-Blanco MJ. July 2010. Naphthyridin-2(1H)-one compounds as antibacterials and their preparation and use for the treatment of bacterial infections. International patent application WO 2010/081874 A1 20100722.
- 15.Barros D. December 2009. Recent advances in TB drug development, novel Mtb DNA gyrase inhibitors: MGIs. In Proceedings of the 40th Union World Conference on Lung Health, Cancun, Mexico. [Google Scholar]
- 16.Hameed P, Patil V, Solapure S, Sharma U, Madhavapeddi M, Raichurkar A, Chinnapattu M, Manjrekar P, Shanbhag G, Puttur J, Shinde V, Menasinakai S, Rudrapatana S, Achar V, Awasthy D, Nandishaiah R, Humnabadkar V, Ghosh A, Narayan C, Kaur P, Sharma S, Wirngren J, Hoffner S, Panduga V, Kumar CNN, Reddy J, Ganguly S, Bharath Bheemarao SU, Mukherjee K, Arora U, Gaonkar S, Coulson M, Waterson D, Sambandamurthy VK, Desousa SM. 2014. Novel N-linked aminopiperidine based gyrase inhibitors with improved hERG and in vivo efficacy against Mycobacterium tuberculosis. J Med Chem 57:4889–4905. doi: 10.1021/jm500432n. [DOI] [PubMed] [Google Scholar]
- 17.Ballell L, Barros D, Brooks G, Castro J, Dabbs S, Daines RA, Davies DT, Fiandor Roman JM, Giordano I, Hennessy AJ, Hoffman JB, Jones GE, Miles TJ, Pearson ND, Pendrak I, Remuinan Blanco MJ, Rossi JS, Zhang L. January 2008. Derivatives and analogues of N-ethylquinolones and N-ethylazaquinolones as antibacterial agents and their reparation. International patent application WO 2008/009700 A1 20080124.
- 18.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Parish T, Stoker NG. 2001. Mycobacterial protocols. Springer Science+Business Media, New York, NY. [Google Scholar]
- 20.Martin A, Camacho M, Portaels F, Palomino JC. 2003. Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob Agents Chemother 47:3616–3619. doi: 10.1128/AAC.47.11.3616-3619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takiff HE, Salazar L, Guerrero C, Philipp W, Huang WM, Kreiswirth B, Cole ST, Jacobs WR, Telenti A. 1994. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob Agents Chemother 38:773–780. doi: 10.1128/AAC.38.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parish T, Stoker NG. 2000. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146(Pt 8):1969–1975. [DOI] [PubMed] [Google Scholar]
- 23.Aubry A, Pan XS, Fisher LM, Jarlier V, Cambau E. 2004. Mycobacterium tuberculosis DNA gyrase: interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob Agents Chemother 48:1281–1288. doi: 10.1128/AAC.48.4.1281-1288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginsburg A, Grosset JH, Bishai WR. 2003. Fluoroquinolones, tuberculosis, and resistance. Lancet Infect Dis 3:432–442. doi: 10.1016/S1473-3099(03)00671-6. [DOI] [PubMed] [Google Scholar]
- 25.Rullas J, García JI, Beltrán M, Cardona P-J, Cáceres N, García-Bustos JF, Angulo-Barturen I. 2010. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob Agents Chemother 54:2262–2264. doi: 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chemical Computing Group. 2010. Molecular operating environment molecular modeling program (MOE), version 2010.04. Chemical Computing Group, Montreal, QC, Canada. [Google Scholar]
- 27.Cambridge Crystallographic Data Centre. GOLD, version 4.01. CCDC, Cambridge, United Kingdom. [Google Scholar]
- 28.Jamieson C, Moir EM, Rankovic Z, Wishert GJ. 2006. Medicinal chemistry of hERG optimizations: highlights and hang-ups. J Med Chem 49:5029–5046. doi: 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- 29.Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharath S, Balasubramanian V. 2007. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother 51:576−582. doi: 10.1128/AAC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maruri F, Sterling TR, Kaiga AW, Blackman A, van der Heijden YF, Mayer C, Cambau E, Aubry A. 2012. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J Antimicrob Chemother 67:819–831. doi: 10.1093/jac/dkr566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Groll A, Martin A, Jureen P, Hoffner S, Vandamme P, Portaels F, Palomino JC, Da Silva PA. 2009. Fluoroquinolone resistance in Mycobacterium tuberculosis and mutations in gyrA and gyrB. Antimicrob Agents Chemother 53:4498–4500. doi: 10.1128/AAC.00287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mokrousov I, Otten T, Manicheva O, Potapova Y, Vishnevsky B, Navskaya O, Rastogi N. 2008. Molecular characterization of ofloxacin-resistant Mycobacterium tuberculosis strains Russia. Antimicrob Agents Chemother 52:2937–2939. doi: 10.1128/AAC.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubry A, Veziris N, Cambau E, Truffot-Pernot C, Jarlier V, Fisher M. 2006. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicrob Agents Chemother 50:104–112. doi: 10.1128/AAC.50.1.104-112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin X, Yu Z. 2010. Mutation characterization of gyrA and gyrB genes in levofloxacin-resistant Mycobacterium tuberculosis clinical isolates from Guangdong Province in China. J Infect 61:150–154. doi: 10.1016/j.jinf.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Sun Z, Zhang J, Zhang X, Wang S, Zhang Y, Li C. 2008. Comparison of gyrA gene mutations between laboratory-selected ofloxacin-resistant Mycobacterium tuberculosis strains and clinical isolates. Int J Antimicrob Agents 31:115–121. doi: 10.1016/j.ijantimicag.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Singh PK, Chan P, Hibbs M, Segura D, Vazquez MJ, Thomas DA, Theobald AJ, Gallagher KT, Hassan NJ. 2011. High-yield production and characterization of biologically active GST tagged human topoisomerase IIa protein in insect cells for the development of a high-throughput assay. Protein Expr Purif 76:165–172. doi: 10.1016/j.pep.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Young RJ, Green DVS, Luscombe CN, Hill AP. 2011. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 16:822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 38.De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. 2005. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J 26:590−597. doi: 10.1093/eurheartj/ehi092. [DOI] [PubMed] [Google Scholar]
- 39.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. 2003. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58:32−45. doi: 10.1016/S0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 40.Pollard CE, Valentin JP, Hammond TG. 2008. Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective. Br J Pharmacol 154:1538–1543. doi: 10.1038/bjp.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.