

Abstract
The incidence and prevalence of non-alcoholic fatty liver disease (NAFLD) is constantly increasing. Despite this is apparently associated with the growing increase in obesity, insulin resistance and obesity-related metabolic disturbances their presence is not a necessary or sufficient condition to explain the accumulation of fat in the liver. Conversely, NAFLD is a predictor of other metabolic risks. NAFLD is currently the most frequent chronic liver disease but should not be considered benign or anecdotic because a considerable proportion of patients with NAFLD progress to cirrhosis and end-stage liver disease. Consequently, the search for alternative molecular mechanisms with therapeutic implications in NAFLD and associated disorders deserves a careful consideration. Mitochondria are possible targets as these organelles generate energy from nutrient oxidation. Some findings, generated in patients with extreme obesity and in murine models, support the notion that NAFLD could be a mitochondrial disease. This is plausible because mitochondrial dysfunction affects the accumulation of lipids in hepatocytes and promotes lipid peroxidation, the production of reactive oxygen species, the release of cytokines causing inflammation and cell death. Here we discuss basic research and mechanistic studies targeting the role of chemokine ligand 2 in liver inflammation and that of the paraoxonases in the oxidative stress. Their combination and association with mitochondrial dysfunction may uncover mechanisms underlying the progression of NAFLD and may help to identify novel therapeutic targets.
Keywords: Biomarkers, Cytokines, Inflammation, Metabolism, Mitochondrial dysfunction, Metabolic profiling, Obesity, Oxidation, Risk factors
Core tip: Recently acquired knowledge on the role of oxidation, inflammation and mitochondrial dysfunction in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) suggests a crucial role in the search for biological markers and therapeutic targets to alleviate the progression of the disease. Classically associated with obesity, the pathogenesis of NAFLD is extremely complex and mostly unknown. Consequently, the correct clinical management and prevention has not been established yet. Despite disparate results in the literature, mainly due to the different methods to measure the oxidative stress, there is currently no doubt that oxidative stress plays a crucial role. Similarly, the infiltration of monocytes, mainly due to the action of chemokines, is a relevant factor. However, to uncover the actual molecular mechanisms has proven to be more difficult than expected. The role of both oxidation and inflammation could be considered defensive and/or causative but the combination of these and other mechanisms may improve the understanding of NAFLD as a manifestation of mitochondrial disease. J. Camps and J. Joven. Chemokine ligand 2 and paraoxonase-1 in non-alcoholic fatty liver disease: the search for alternative causative factors.
WHAT WE TALK ABOUT WHEN WE TALK ABOUT NON-ALCOHOLIC FATTY LIVER DISEASE
Non-alcoholic fatty liver disease (NAFLD) is a complex disease characterized by intra-cytoplasmic fat in the liver higher than 5% of the total liver weight (steatosis) in patients without alcohol or drugs consumption, toxin exposure and/or viral disease. The fat droplets in hepatocytes may be large (macrovesicular), mixed large and small, or small (microvesicular). There is architectural integrity but inflammation, fibrosis, or apoptosis may be also found histologically[1]. Non-alcoholic steatohepatitis (NASH) is accepted as a distinct entity and the diagnosis requires steatosis accompanied by hepatocellular injury (ballooning) and lobular inflammation. Fibrosis is not necessarily required[2]. Liver biopsy is the only widely accepted method for the diagnosis of NAFLD, but some limitations may explain disparate results in the literature[3]. Further, its clinical indication is constrained to patients with the additional presence of risk factors for advanced disease. This is due to the fact that many physicians consider NAFLD as a benign condition and minimize its progressive course. Accordingly, searching for a reliable non-invasive marker is an important research goal. The value of imaging is promising but common laboratory tests (e.g., alanine aminotransferase) should be considered useless. Contributing factors are not reliable. A major role for gender and genetic predisposition is not likely[4] but increased age has an apparently high impact[5,6]. There is a clear association between obesity (and/or insulin resistance) and NAFLD but the importance of such association is still debatable because, even in extreme obesity the incidence of NAFLD is lower than 70%[7,8]. NASH is present in a high proportion of patients with NAFLD indicating the progression of the disease and a high potential to eventually lead to end-stage liver disease[5,9]. Of note, there are no formally established pharmacological treatment options[10] and research efforts in clinical trials are surprisingly low probably indicating the false notion that fighting against unhealthy lifestyle is sufficient for the management of NAFLD.
REGULAR EXERCISE, CALORIC LIMITATION AND ATTENTION TO THE MACRO AND MICRONUTRIENTS IN THE DIET ARE CONSISTENTLY BENEFICIAL IN NAFLD
NAFLD is an important cardiovascular risk factor. Consequently, regular aerobic exercise should be promoted for the management of NAFLD. It reduces hepatic lipids and this effect appears to be independent of improvements in insulin resistance, body weight reduction or decrease in adipose tissue volume[11]. Moreover, exercise is effective in ameliorating oxidative stress and inflammation. The mechanisms are complex but include a protective effect in the carbonylation of enzymes regulating lipid metabolism[12]. Other results suggest that exercise reduces hepatic inflammation, injury, and fibrosis by suppressing macrophage infiltration through mechanisms in which chemokine (C-C motif) ligand 2 (CCL2) plays a substantial role[13].
The consensus favours caloric restriction and sustained weight loss as the correct treatment for NAFLD[14]. Bariatric surgery appears to be a promising treatment. Patients experience a high, fast and sustained reduction in excess body weight. Further, it is likely to have potential benefit in ameliorating other factors that contribute to the pathogenesis of NAFLD (i.e., improvement of insulin sensitivity, dyslipemia and inflammation). Other benefits derived from changes in the secretion of adiponectin and gastrointestinal hormones have been also reported[8,15]. The combination of exercise with dietary restrictions is beneficial in NAFLD and the effect is probably mediated by a decrease in inflammatory factors and oxidative stress[16]. Additionally, qualitative and quantitative modifications of dietary composition with changes in either macro or micronutrients may be also important to modulate the clinical course of NAFLD[17]. Dietary advice, however, is complicated by the lack of reliable evidences in humans. For example, the assessment of the effects of dietary protein on NAFLD has been limited to animal models. Low fat diet is currently the preferred option but carbohydrate quantity and quality in diets clearly affect the management of NAFLD[18]. The differential role of saturated fat, monounsaturated fat, polyunsaturated fat, omega-3 fatty acids, and all-trans fat in the progression of NAFLD has been reported[19]. However, using metabolomics as an experimental tool, several studies have reported that the role of dietary cholesterol seems to be prominent in the initiation and progression of NAFLD in humans and in animal models[8,20-22]. Particularly, we detected changes in metabolite levels, using a non-targeted NMR-based metabolomics approach, that strongly support the idea that dietary cholesterol is a causative factor in the development of both liver steatosis and hepatic inflammation[21]. In this regard, we have also found in animal models that the continuous administration of polyphenols alleviate weight gain, liver steatosis, deleterious changes in the composition of liver tissue, and insulin resistance. Metabolites of polyphenols accumulate in immune cells and at the surface of hepatic lipid droplets, an effect associated with beneficial changes in the expression of miR-103, miR-107 and miR-122[23,24]. These findings may be also interpreted as an indication that the combination of anti-inflammatory and antioxidant effects provided by these bioactive compounds may be of value in the management of NAFLD. Besides, the underlying mechanisms are probably associated with mitochondrial dysfunction and indicated that CCL2 may have metabolic functions that may be used to modify the reversible components of the associated metabolic derangements[20-24]. Our results do not discard contributions by other cytokines but CCL2 was primarily investigated due to its recognized effect in macrophage infiltration of both liver and adipose tissue.
CCL2 IN LIVER INFLAMMATION, OBESITY, INSULIN RESISTANCE AND THE REGULATION OF METABOLISM: THE ROLE OF AUTOPHAGY
Available data indicate that inflammation, NAFLD, response to nutrients and autophagy are closely associated with the function of CCL2[24-31]. Several reports suggest that autophagy is likely to influence the quality of the immune responses that occur in response to nutritional stress in the context of a high fat diet-associated NAFLD[24-26]. CCL2 is a reliable marker of inflammation in hepatic derangements and correlated with the histological hepatic inflammation[27]. Moreover, some authors claim that CCL2 levels are significantly elevated in patients with NASH compared to NAFLD[28,29]. Particularly, the CCL2/CCR2 axis is important for perpetuating the hepatic inflammation and the pharmacological inhibition of CCL2 during chronic liver damage attenuates the development of steatosis[30,31].
The association between obesity and NAFLD should be seriously considered because obesity is a condition associated with a frightening disease burden. Although the search for a simple and acceptable nutritionally based therapeutic approach continues, other strategies should be explored[32]. The effect on liver morphology and function of obesity and the consequent development of NAFLD has received substantial attention in our laboratory and we failed to show a beneficial effect of peroxisome proliferator-activated receptors (PPAR) agonists (rosiglitazone and/or fenofibrate) on ob/ob and LDLR-double deficient mice. This illustrates the expected complexity in the regulation of lipid and glucose metabolism, the inflammatory response and the oxidative stress. Both drugs simultaneously activate pro-steatotic and anti steatotic metabolic pathways resulting in an aggravation of the hepatic lipid accumulation[33].
Inflammation, in this scenario, represents an indicator of the failure of adipose tissue in its major function of energy storage. We have recently developed an animal model overexpressing CCL2 that is designed to characterize the relationship between metabolism and cytokine gene expression. Our data suggest that dietary variations contribute to autophagy: animals overexpressing CCL2 displayed an increased number of autophagosomes and a differential rate of mitophagy as compared with CCL2 deficient animals[34]. The role of CCL2 varies in response to different metabolic conditions. Information about the regulation of autophagy by cytokines is based on relatively limited data obtained in mammalian cells and consequently requires animal models and further research in humans[35]. However, results in humans are difficult to replicate. In the context of NAFLD, the synergic role of both dietary energy ingestion and the tissue CCL2 response is readily observed[21-23]. Obese patients with steatosis display a higher number of autophagosomes in the liver and in obese patients without steatosis there are ill-defined changes in the morphology of mitochondria. Despite these effects are not related to inflammation in the liver, autophagy affects the inflammatory status of the adipocyte and the size of adipocytes (i.e., in patients with steatosis the size of adipocytes is higher than in those without liver alteration). Intriguingly, the degree of steatosis is heterogeneous among obese patients and a substantial amount of these patients does not develop neither NAFLD nor insulin resistance[8] (Figure 1). The correct interpretation of these findings requires more information. For example, the absence of autophagy in adipocytes, as identified in adipocyte-specific ATG7 knockout mice, or the mitigation of autophagy observed in CCL2 deficient mice, protect against high-fat diet-induced obesity and insulin resistance in mice[36,37]. However, other data report that autophagy may function to limit excessive inflammation in adipose tissue during obesity[38]. The function of autophagy in regulating intracellular lipid droplets (lipophagy) is also relevant in the liver of obese patients. Whether impaired lipophagy is a sufficient condition to cause NAFLD remains to be fully ascertained[39].
Figure 1.
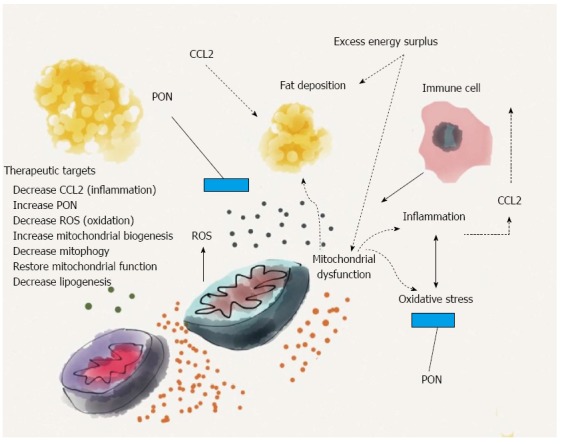
Several factors, including the excess energy surplus, inflammation and oxidation, are responsible for the deposition of fat in the liver. Chemokine (C-C motif) ligand 2 (CCL2) affects, either directly or through the migration of immune cells, the mitochondrial function. The net result is a higher production of reactive oxygen species (ROS), which is partially attenuated by the action of paraoxonases (PON). Experimental evidence suggests that both molecules are inversely interrelated in the regulation of hepatic inflammation, fat deposition and mitochondrial function. The underlying mechanisms may indicate an alternative approach in the search for therapeutic targets.
The regulatory network of autophagy in the development of NAFLD may also involve signalling through AMP-activated protein kinase (AMPK), which is a central regulator of cellular metabolism and affects glucose homeostasis, lipid metabolism, protein synthesis, and oxidative metabolism[25,33]. Moreover, the modulation of AMPK by metformin may have different effects according to the baseline metabolic context suggesting that the effect of metformin in NAFLD is not limited to the role of insulin sensitizer. The control of lipid metabolism by AMPK may be due to the decreased synthesis of malonyl CoA or a decrease in transcription of lipogenic genes that increases mitochondrial biogenesis[40]. Macrophages in adipose tissue are also important in lipolysis and lipid metabolism. Macrophages are cells that possess distinct non-inflammatory trophic functions, which are regulated by the metabolic context, and, as such, are closely associated with lipid accumulation and the development of metabolic diseases[41,42].
LINKAGE BETWEEN INFLAMMATION AND PARAOXONASE-1, A MULTI-FACETED ENZYME IN THE DEFENCE AGAINST OXIDATION
Paraoxonase-1 (PON1) and CCL2 are closely interrelated in the regulation of hepatic inflammation[42,43]. Moreover, in mice, CCL2 and PON1 are similarly distributed in tissues and co-localized under certain circumstances, suggesting a possibly coordinated role[44]. PON1 is synthesized by the liver and is bound to circulating high-density lipoproteins. The actual function of PON1 is still a matter of debate but, among other effects, protects lipoproteins and cells from lipid peroxidation by degrading specific oxidized cholesteryl esters and phospholipids. Oxidized lipids, in turn, inactivate PON1[45-48]. It is currently accepted that oxidative stress plays a pivotal role in the evolution of NAFLD to the more severe NASH. As mentioned earlier, there are morphological and functional disturbances in the mitochondria of patients with NAFLD, which promote an increased free radical production and lipid peroxidation[49]. In this context PON1 is related to inflammation, fibrosis and the regulation of PPARs[50]. It is therefore expected that low PON-1 activities may be associated with NAFLD. We have recently confirmed such association in a murine model of PON1 deficiency[51]. PON1 deficient mice fed a high fat high-cholesterol diet depicted oxidative stress and metabolic alterations resulting in NAFLD. Reduced and oxidized glutathione, 8-oxo-20-deoxyguanosine, malondialdehyde, 8-isoprostanes and protein carbonyl concentrations were all significantly increased. This was accompanied by a functional decrease in glycolysis, the urea cycle and the Krebs cycle and a significant increase in the pathways of triglyceride and phospholipid synthesis. The measurement of circulating PON1 may also bear diagnostic and prognostic importance when properly performed[52,53]. When this is added to algorithms using common liver laboratory tests the overall sensitivity and specificity increased to levels similar to those obtained with liver biopsy and histological examination. This is probably due to the fact that serum PON1, contrarily to the standard laboratory test values, decreases with the extent of liver derangement. Of note, the interrelations between CCL2 and PON1 may provide important clues to the understanding of non-communicable diseases. Particularly, the increase in the production of CCL2 by endothelial cells may be significantly attenuated by the action of PON1[54], a finding that further reinforce our hypothesis that both CCL2 and PON1 are important in the assessment of the pathogenesis of NAFLD.
CCL2 AND PON1 INTERPLAY ON MECHANISMS REGULATING MITOCHONDRIAL FUNCTION: THE IMPLICATIONS OF CONSIDERING NAFLD A MILD FORM OF MITOCHONDRIAL DISEASE
There are significant evidences in animal models indicating that CCL2 and PON1, as well as the combination of both, may be integrated in the correct functioning of mitochondria in the presence of high fat diets causing NAFLD[21,51]. Hepatocytes are rich in mitochondria and their structure and function are important for a healthy liver. In the mitochondria, oxidation of glucose and fat result in the production of energy but under normal conditions approximately 2% of mitochondrial oxygen consumption results in the production of reactive oxygen species (ROS). In conditions of increased fatty acid oxidation there is a higher production of ROS that may exceed the cell’s antioxidant capacity leading to oxidative stress and inflammation. As earlier reviewed, mitochondrial dysfunction contributes to the pathogenesis of NAFLD and this simple notion may provide alternative therapeutic opportunities because the perpetuation of ROS generation, oxidative stress and inflammation ultimately results in the development of NASH[55]. Whether changes in mitochondrial efficiency are a causative or adaptive response remains an open question. Interestingly, heterozygous mice deficient in mitochondrial trifunctional protein, characterized by defects in oxidation predispose mice to NAFLD and systemic insulin resistance. Consistent observations have also been made in an obese rodent model[56-58]. Cellular and mitochondrial metabolism are regulated by a number of transcription factors and co-regulators such as peroxisome proliferator-activated receptor-α coactivator 1 (PGC-1). Mice with liver-specific deletion of PGC-1 exhibit impaired mitochondrial oxidative capacity, mitochondrial dysfunction and NAFLD. PGC-1 is inhibited by oxidative stress and inflammation. Conversely, overexpression of hepatic PGC-1, which alleviates NAFLD, is one of the multiple effects of exercise and caloric restriction and may be mimicked by polyphenols and other drugs. One of the most popularly claimed mechanisms for PGC-1 activation depends on the role of sirtuins, which are interconnected with the action of AMPK and are currently considered attractive therapeutic targets in the management of NAFLD. Accordingly, sirtuins are associated with multiple and related functions acting in the pathogenesis of NAFLD such as mitochondrial function, hepatic fatty acid metabolism, insulin secretion and hepatic gluconeogenesis[59]. The number of mitochondria (i.e., the correct balance between mitochondrial biogenesis and mitophagy) is also important in the development of NAFLD[36,55]. Mitochondrial biogenesis is a multistep process requiring the coordinated action of both mitochondrial and nuclear-originated transcripts. Consequently, available activators of mitochondrial biogenesis may be useful adjuvants in the management of NAFLD and some evidence may support the use of metformin in doses and routes of administration that may differ from those used in the management of diabetes[60,61]. The mechanisms outlined have not been thoroughly investigated in humans and current therapeutic armamentarium is mainly based in changes in lifestyle (exercise and caloric restriction); the current pharmacological therapies being clearly insufficient. As a consequence, the treatment of NAFLD is an “intention” rather than a “goal” and this may be unacceptable in a context of rapid upsurge of obesity and associated metabolic derangements.
CONCLUSION
NAFLD should not be considered a benign condition. There is an unmet need to identify and modulate selective targets of treatment. We consider here, particularly, a requirement to direct therapeutic efforts towards the decrease in the deleterious effects of chemokines-related inflammation, the increase in the paraoxonase-induced ability to resist cellular oxidative stress and the early detection of mitochondrial dysfunction in order to improve the balance between mitochondrial biogenesis and mitophagy.
ACKNOWLEDGMENTS
Underlying research materials related to this manuscript are available on request from the corresponding authors.
Footnotes
Supported by Instituto de Salud Carlos III, No. PI08/1381, and No. PI11/00130; Carlos III Health Institute, Madrid, Spain; and the Fondo Europeo de Desarrollo Regional.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 15, 2014
First decision: August 15, 2014
Article in press: September 30, 2014
P- Reviewer: Akbulut S, Caboclo JLF, Jani K, Mutoh M, Protiva P S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 2.Bettermann K, Hohensee T, Haybaeck J. Steatosis and steatohepatitis: complex disorders. Int J Mol Sci. 2014;15:9924–9944. doi: 10.3390/ijms15069924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratziu V, Charlotte F, Heurtier A, Gombert S, Giral P, Bruckert E, Grimaldi A, Capron F, Poynard T. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128:1898–1906. doi: 10.1053/j.gastro.2005.03.084. [DOI] [PubMed] [Google Scholar]
- 4.Wilfred de Alwis NM, Day CP. Genes and nonalcoholic fatty liver disease. Curr Diab Rep. 2008;8:156–163. doi: 10.1007/s11892-008-0027-9. [DOI] [PubMed] [Google Scholar]
- 5.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Zain SM, Mohamed R, Cooper DN, Razali R, Rampal S, Mahadeva S, Chan WK, Anwar A, Rosli NS, Mahfudz AS, et al. Genome-wide analysis of copy number variation identifies candidate gene loci associated with the progression of non-alcoholic fatty liver disease. PLoS One. 2014;9:e95604. doi: 10.1371/journal.pone.0095604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rafiq N, Younossi ZM. Effects of weight loss on nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:427–433. doi: 10.1055/s-0028-1091986. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Gallego E, Guirro M, Riera-Borrull M, Hernández-Aguilera A, Mariné-Casadó R, Fernández-Arroyo S, Beltrán-Debón R, Sabench F, Hernández M, Del Castillo D, et al. Mapping of the circulating metabolome reveals α-ketoglutarate as a predictor of morbid obesity-associated non-alcoholic fatty liver disease. Int J Obes (Lond) 2015;39:279–287. doi: 10.1038/ijo.2014.53. [DOI] [PubMed] [Google Scholar]
- 9.Söderberg C, Stål P, Askling J, Glaumann H, Lindberg G, Marmur J, Hultcrantz R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology. 2010;51:595–602. doi: 10.1002/hep.23314. [DOI] [PubMed] [Google Scholar]
- 10.Rinella ME, Loomba R, Caldwell SH, Kowdley K, Charlton M, Tetri B, Harrison SA. Controversies in the Diagnosis and Management of NAFLD and NASH. Gastroenterol Hepatol (N Y) 2014;10:219–227. [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson NA, Sachinwalla T, Walton DW, Smith K, Armstrong A, Thompson MW, George J. Aerobic exercise training reduces hepatic and visceral lipids in obese individuals without weight loss. Hepatology. 2009;50:1105–1112. doi: 10.1002/hep.23129. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Duan Z, Hu H, Li G, Yan S, Wu J, Wang J, Yin D, Xie Q. Proteomic profile of carbonylated proteins in rat liver: exercise attenuated oxidative stress may be involved in fatty liver improvement. Proteomics. 2013;13:1755–1764. doi: 10.1002/pmic.201200522. [DOI] [PubMed] [Google Scholar]
- 13.Kawanishi N, Yano H, Mizokami T, Takahashi M, Oyanagi E, Suzuki K. Exercise training attenuates hepatic inflammation, fibrosis and macrophage infiltration during diet induced-obesity in mice. Brain Behav Immun. 2012;26:931–941. doi: 10.1016/j.bbi.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Loria P, Adinolfi LE, Bellentani S, Bugianesi E, Grieco A, Fargion S, Gasbarrini A, Loguercio C, Lonardo A, Marchesini G, et al. Practice guidelines for the diagnosis and management of nonalcoholic fatty liver disease. A decalogue from the Italian Association for the Study of the Liver (AISF) Expert Committee. Dig Liver Dis. 2010;42:272–282. doi: 10.1016/j.dld.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Vargas V, Allende H, Lecube A, Salcedo MT, Baena-Fustegueras JA, Fort JM, Rivero J, Ferrer R, Catalán R, Pardina E, et al. Surgically induced weight loss by gastric bypass improves non alcoholic fatty liver disease in morbid obese patients. World J Hepatol. 2012;4:382–388. doi: 10.4254/wjh.v4.i12.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh S, Tanaka K, Tsujimoto T, So R, Shida T, Shoda J. Regular exercise coupled to diet regimen accelerates reduction of hepatic steatosis and associated pathological conditions in nonalcoholic fatty liver disease. Metab Syndr Relat Disord. 2014;12:290–298. doi: 10.1089/met.2013.0143. [DOI] [PubMed] [Google Scholar]
- 17.Sofi F, Casini A. Mediterranean diet and non-alcoholic fatty liver disease: new therapeutic option around the corner? World J Gastroenterol. 2014;20:7339–7346. doi: 10.3748/wjg.v20.i23.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem. 2012;23:203–208. doi: 10.1016/j.jnutbio.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Blendis L, Halpern Z, Oren R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): a population based study. J Hepatol. 2007;47:711–717. doi: 10.1016/j.jhep.2007.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Rull A, Vinaixa M, Angel Rodríguez M, Beltrán R, Brezmes J, Cañellas N, Correig X, Joven J. Metabolic phenotyping of genetically modified mice: An NMR metabonomic approach. Biochimie. 2009;91:1053–1057. doi: 10.1016/j.biochi.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Vinaixa M, Rodríguez MA, Rull A, Beltrán R, Bladé C, Brezmes J, Cañellas N, Joven J, Correig X. Metabolomic assessment of the effect of dietary cholesterol in the progressive development of fatty liver disease. J Proteome Res. 2010;9:2527–2538. doi: 10.1021/pr901203w. [DOI] [PubMed] [Google Scholar]
- 22.Rull A, Rodríguez F, Aragonès G, Marsillach J, Beltrán R, Alonso-Villaverde C, Camps J, Joven J. Hepatic monocyte chemoattractant protein-1 is upregulated by dietary cholesterol and contributes to liver steatosis. Cytokine. 2009;48:273–279. doi: 10.1016/j.cyto.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Joven J, Espinel E, Rull A, Aragonès G, Rodríguez-Gallego E, Camps J, Micol V, Herranz-López M, Menéndez JA, Borrás I, et al. Plant-derived polyphenols regulate expression of miRNA paralogs miR-103/107 and miR-122 and prevent diet-induced fatty liver disease in hyperlipidemic mice. Biochim Biophys Acta. 2012;1820:894–899. doi: 10.1016/j.bbagen.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 24.Rull A, Camps J, Alonso-Villaverde C, Joven J. Insulin resistance, inflammation, and obesity: role of monocyte chemoattractant protein-1 (or CCL2) in the regulation of metabolism. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/326580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 26.Kwanten WJ, Martinet W, Michielsen PP, Francque SM. Role of autophagy in the pathophysiology of nonalcoholic fatty liver disease: a controversial issue. World J Gastroenterol. 2014;20:7325–7338. doi: 10.3748/wjg.v20.i23.7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsillach J, Bertran N, Camps J, Ferré N, Riu F, Tous M, Coll B, Alonso-Villaverde C, Joven J. The role of circulating monocyte chemoattractant protein-1 as a marker of hepatic inflammation in patients with chronic liver disease. Clin Biochem. 2005;38:1138–1140. doi: 10.1016/j.clinbiochem.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Haukeland JW, Damås JK, Konopski Z, Løberg EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K, Aukrust P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44:1167–1174. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Aragonès G, Ercilla A, Barreda M, Rull A, Beltrán-Debón R, Rodríguez-Gallego E, Alonso-Villaverde C, Camps J, Joven J. Human Duffy blood group alloantigen system influences the measurement of monocyte chemoattractant protein-1 (MCP-1) in serum but not in plasma. Clin Lab. 2012;58:185–188. [PubMed] [Google Scholar]
- 30.Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- 31.Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, López-Bravo M, Joven J, Ardavín C, Rodríguez-Fernández JL, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858–3867. doi: 10.4049/jimmunol.1302821. [DOI] [PubMed] [Google Scholar]
- 32.Joven J, Micol V, Segura-Carretero A, Alonso-Villaverde C, Menéndez JA. Polyphenols and the modulation of gene expression pathways: can we eat our way out of the danger of chronic disease? Crit Rev Food Sci Nutr. 2014;54:985–1001. doi: 10.1080/10408398.2011.621772. [DOI] [PubMed] [Google Scholar]
- 33.Rull A, Geeraert B, Aragonès G, Beltrán-Debón R, Rodríguez-Gallego E, García-Heredia A, Pedro-Botet J, Joven J, Holvoet P, Camps J. Rosiglitazone and fenofibrate exacerbate liver steatosis in a mouse model of obesity and hyperlipidemia. A transcriptomic and metabolomic study. J Proteome Res. 2014;13:1731–1743. doi: 10.1021/pr401230s. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez-Gallego E, Riera-Borrull M, Hernández-Aguilera A, Mariné-Casadó R, Rull A, Beltrán-Debón R, Luciano-Mateo F, Menendez JA, Vazquez-Martin A, Sirvent JJ, et al. Ubiquitous transgenic overexpression of C-C chemokine ligand 2: a model to assess the combined effect of high energy intake and continuous low-grade inflammation. Mediators Inflamm. 2013;2013:953841. doi: 10.1155/2013/953841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tous M, Ferré N, Rull A, Marsillach J, Coll B, Alonso-Villaverde C, Camps J, Joven J. Dietary cholesterol and differential monocyte chemoattractant protein-1 gene expression in aorta and liver of apo E-deficient mice. Biochem Biophys Res Commun. 2006;340:1078–1084. doi: 10.1016/j.bbrc.2005.12.109. [DOI] [PubMed] [Google Scholar]
- 36.Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rull A, Escolà-Gil JC, Julve J, Rotllan N, Calpe-Berdiel L, Coll B, Aragonès G, Marsillach J, Alonso-Villaverde C, Camps J, et al. Deficiency in monocyte chemoattractant protein-1 modifies lipid and glucose metabolism. Exp Mol Pathol. 2007;83:361–366. doi: 10.1016/j.yexmp.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology. 2012;153:5866–5874. doi: 10.1210/en.2012-1625. [DOI] [PubMed] [Google Scholar]
- 39.Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20:3–11. doi: 10.1038/cdd.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–830. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camps J, Rodríguez-Gallego E, García-Heredia A, Triguero I, Riera-Borrull M, Hernández-Aguilera A, Luciano-Mateo F, Fernández-Arroyo S, Joven J. Paraoxonases and chemokine (C-C motif) ligand-2 in noncommunicable diseases. Adv Clin Chem. 2014;63:247–308. doi: 10.1016/b978-0-12-800094-6.00007-8. [DOI] [PubMed] [Google Scholar]
- 43.Camps J, Marsillach J, Rull A, Alonso-Villaverde C, Joven J. Interrelationships between paraoxonase-1 and monocyte chemoattractant protein-1 in the regulation of hepatic inflammation. Adv Exp Med Biol. 2010;660:5–18. doi: 10.1007/978-1-60761-350-3_2. [DOI] [PubMed] [Google Scholar]
- 44.Rodríguez-Sanabria F, Rull A, Beltrán-Debón R, Aragonès G, Camps J, Mackness B, Mackness M, Joven J. Tissue distribution and expression of paraoxonases and chemokines in mouse: the ubiquitous and joint localisation suggest a systemic and coordinated role. J Mol Histol. 2010;41:379–386. doi: 10.1007/s10735-010-9299-x. [DOI] [PubMed] [Google Scholar]
- 45.Camps J, Marsillach J, Joven J. Measurement of serum paraoxonase-1 activity in the evaluation of liver function. World J Gastroenterol. 2009;15:1929–1933. doi: 10.3748/wjg.15.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackness MI, Arrol S, Abbott C, Durrington PN. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis. 1993;104:129–135. doi: 10.1016/0021-9150(93)90183-u. [DOI] [PubMed] [Google Scholar]
- 47.Aviram M, Rosenblat M, Billecke S, Erogul J, Sorenson R, Bisgaier CL, Newton RS, La Du B. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic Biol Med. 1999;26:892–904. doi: 10.1016/s0891-5849(98)00272-x. [DOI] [PubMed] [Google Scholar]
- 48.García-Heredia A, Marsillach J, Rull A, Triguero I, Fort I, Mackness B, Mackness M, Shih DM, Joven J, Camps J. Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: a nondirected metabolomic study. Mediators Inflamm. 2013;2013:156053. doi: 10.1155/2013/156053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solís Herruzo JA, García Ruiz I, Pérez Carreras M, Muñoz Yagüe MT. Non-alcoholic fatty liver disease. From insulin resistance to mitochondrial dysfunction. Rev Esp Enferm Dig. 2006;98:844–874. doi: 10.4321/s1130-01082006001100006. [DOI] [PubMed] [Google Scholar]
- 50.Marsillach J, Camps J, Ferré N, Beltran R, Rull A, Mackness B, Mackness M, Joven J. Paraoxonase-1 is related to inflammation, fibrosis and PPAR delta in experimental liver disease. BMC Gastroenterol. 2009;9:3. doi: 10.1186/1471-230X-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.García-Heredia A, Kensicki E, Mohney RP, Rull A, Triguero I, Marsillach J, Tormos C, Mackness B, Mackness M, Shih DM, et al. Paraoxonase-1 deficiency is associated with severe liver steatosis in mice fed a high-fat high-cholesterol diet: a metabolomic approach. J Proteome Res. 2013;12:1946–1955. doi: 10.1021/pr400050u. [DOI] [PubMed] [Google Scholar]
- 52.Camps J, Marsillach J, Joven J. The paraoxonases: role in human diseases and methodological difficulties in measurement. Crit Rev Clin Lab Sci. 2009;46:83–106. doi: 10.1080/10408360802610878. [DOI] [PubMed] [Google Scholar]
- 53.Ferré N, Camps J, Prats E, Vilella E, Paul A, Figuera L, Joven J. Serum paraoxonase activity: a new additional test for the improved evaluation of chronic liver damage. Clin Chem. 2002;48:261–268. [PubMed] [Google Scholar]
- 54.Mackness B, Hine D, Liu Y, Mastorikou M, Mackness M. Paraoxonase-1 inhibits oxidised LDL-induced MCP-1 production by endothelial cells. Biochem Biophys Res Commun. 2004;318:680–683. doi: 10.1016/j.bbrc.2004.04.056. [DOI] [PubMed] [Google Scholar]
- 55.Hernández-Aguilera A, Rull A, Rodríguez-Gallego E, Riera-Borrull M, Luciano-Mateo F, Camps J, Menéndez JA, Joven J. Mitochondrial dysfunction: a basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013;2013:135698. doi: 10.1155/2013/135698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rector RS, Morris EM, Ridenhour S, Meers GM, Hsu FF, Turk J, Ibdah JA. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology. 2013;57:2213–2223. doi: 10.1002/hep.26285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris EM, Meers GM, Booth FW, Fritsche KL, Hardin CD, Thyfault JP, Ibdah JA. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am J Physiol Gastrointest Liver Physiol. 2012;303:G979–G992. doi: 10.1152/ajpgi.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Menendez JA, Quirantes-Piné R, Rodríguez-Gallego E, Cufí S, Corominas-Faja B, Cuyàs E, Bosch-Barrera J, Martin-Castillo B, Segura-Carretero A, Joven J. Oncobiguanides: Paracelsus’ law and nonconventional routes for administering diabetobiguanides for cancer treatment. Oncotarget. 2014;5:2344–2348. doi: 10.18632/oncotarget.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menendez JA, Cufí S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressant metformin: less is more. Aging (Albany NY) 2011;3:348–362. doi: 10.18632/aging.100316. [DOI] [PMC free article] [PubMed] [Google Scholar]