

Abstract
Helicobacter pylori (H. pylori) have long been associated with a spectrum of disease outcomes in the gastro-duodenal system. Heterogeneity in bacterial virulence factors or strains is not enough to explain the divergent disease phenotypes manifested by the infection. This review focuses on host genetic factors that are involved during infection and eventually are thought to influence the disease phenotype. We have summarized the different host genes that have been investigated for association studies in H. pylori mediated duodenal ulcer or gastric cancer. We discuss that as the bacteria co-evolved with the host; these host gene also show much variation across different ethnic population. We illustrate the allelic distribution of interleukin-1B, across different population which is one of the most popular candidate gene studied with respect to H. pylori infections. Further, we highlight that several polymorphisms in the pathway gene can by itself or collectively affect the acid secretion pathway axis (gastrin: somatostatin) thereby resulting in a spectrum of disease phenotype
Keywords: Helicobacter pylori, Gastric cancer, Duodenal ulcer, Cytokine, Acid secretion
Core tip: Helicobacter pylori infection results in diverse clinical outcomes. While duodenal ulcer is characterized by hyperacidity, gastric cancer results in hypoacidity. Virulence factors of the bacteria and its own genetic heterogeneity variability does not explain the divergent spectrum of disease manifestation. In this review, we highlight the host genetic factors that are involved and elicited by the bacteria. We discuss the different association studies performed with respect to gastric cancer and duodenal ulcer and further delineate the signaling cue of these inflammatory response pathway gene products to the gastrin: somatostatin axis that is known to regulate acid secretion.
INTRODUCTION
Helicobacter pylori (H. pylori), originally considered to be a native microbiota of the gut, colonize the human gut without much adverse consequences. However, colonization of H. pylori is strongly associated with increased risk of several diseases like duodenal ulcer, noncardia gastric adenocarcinoma and gastric mucosa associated lymphoid tissue lymphoma[1,2]. Interestingly, while patients with duodenal ulcer have antral- predominant gastritis with little mucosal atrophy and hyperacidity; patients with gastric ulcer almost invariably have corpus predominant gastritis and hypoacidity with various degree of mucosal atrophy[3]. These facts translate into the notion that duodenal ulcer is negatively associated with gastric cancer, while the reverse is the case for gastric ulcer[4,5]. Thus, it is clear that the progression of H. pylori infection leads to different disease outcomes, with completely opposite phenotypes. To add to this disease heterogeneity, a large group of individuals’ remains infected but are asymptomatic. Such diverse clinical representation seems not only to raise the question as “TO BE OR NOT TO BE” diseased when exposed to H. pylori but also speculate about the nature of the disease manifested.
PROCESS OF PERSISTENCE IN THE GUT: MIMICKING THE HOST
The answer to the above question is complicated and depends on how the bacteria and its host in this case the humans, interact. To cause pathogenesis, H. pylori have to persistently colonize the human stomach and fight the hostility of acidity, peristalsis, compete with other microbes and evade innate and adaptive immunity. H. pylori resist acid by hydrolyzing urea to yield ammonia and by regulating gene expression to respond to changes in pH. H. pylori expresses multiple paralogous outer membrane proteins (OMPs), many of which are phase variable; several of which appear to bind to receptors on the surface of gastric epithelial cells and could diminish the rate of bacterial wash- out as a result of peristalsis[6,7]. H. pylori produce antibacterial peptides that reduce competition from the other microbes[8]. To persist, H. pylori must evade the immune response; most H. pylori adhere superficially to the epithelial cell layer, where immune effectors are not easily accessible. Adherence of H. pylori to gastric epithelial cells stimulates numerous signaling pathways[9], and many H. pylori strains secrete toxins or other effector molecules[10,11]. H. pylori elicit a humoral immune response[12], and tissue infiltration by mononuclear and polymorphonuclear leukocytes, occurs in all humans who are persistently colonized[13]. Approaches such as signature-tagged mutagenesis and microarray tracking of transposon mutants have led to the identification of > 100 bacterial genes required for gastric colonization[14-16]. The expression of several of these genes is up-regulated during growth of H. pylori in the gastric environment[17]. To establish pathogenesis, H. pylori requires colonizing efficiently; which it had achieved through the years by co evolving with the humans. Co-evolution studies indicated that H. pylori have evolved and migrated along with human out of Africa 58000 years ago and has been accommodating itself with the humans since, thus giving rise to a heterogeneous bacterium whose virulence also varies geographically[18]. The heterogeneous bacterial virulence genes are thus an important aspect to understand the heterogeneity of the disease.
H. PYLORI BACTERIAL FACTORS IN DISEASE DEVELOPMENT: THE HALF TRUTH
H. pylori strains isolated from unrelated individuals exhibit a high level of genetic diversity[2,19]. Nucleotide sequences of conserved genes are 92%-99% identical among different H. pylori strains, but several H. pylori genes are more highly diverse in sequence. In addition to variation in the sequences of individual genes among H. pylori strains, there is considerable variation in gene content[1,20,21]. H. pylori strains can be broadly categorized into 2 groups: strains that express multiple factors that interact with host tissue (including proteins encoded by the cag PAI, active forms of VacA, and OMPs such as BabA) and strains that lack these factors[22,23]. Strains with intermediate properties have been identified, although less frequently than expected.
H. pylori strains that express multiple “interaction factors” (CagA+, s1-VacA+ BabA+, OipA + strains) are predicted to be highly interactive with the host, whereas strains that lack these factors would be relatively non-interactive. Concordant with these predictions, CagA+, s1-VacA+, OipA + and BabA+ strains are associated with increased gastric mucosal inflammatory cell infiltration and increased gastric epithelial injury, compared with strains that do not express these factors[1,22]. In addition, the colonization density of CagA+, s1-VacA+, and BabA+ strains is typically higher than that of strains that do not express these factors[24]. H. pylori strains expressing multiple interaction factors and strains that lack these factors might occupy different niches in the gastric environment, or each could have selective advantages at different times during prolonged colonization. Recently, a novel virulence factor that is a VirB4-homologue called dupA (duodenal ulcer-promoting gene) was identified and associated with an increased risk for duodenal ulcer and a reduced risk for gastric atrophy and cancer in both Asian and Western countries[25].
The presence or absence of the above mentioned virulence factors alone or in combination does not explain its association to the diverse H. pylori mediated disease globally as the contributions of these pathogenic markers with disease susceptibility have differing geographical patterns. For example, in Western countries multiple segments of CAG EPIYA-C is associated gastric cancer while a single segment of EPIYA-D is so prevalent in East Asian countries that it is difficult to associate it to disease manifestation and differentiate between simple gastritis and gastric cancer[18].
The bacterial virulence factors, though explains the pathogenesis of the bacteria over other strains, but cannot account for the wide disease heterogeneity. As mentioned earlier, H. pylori exert diverse effects on the host gastric acid secretion in the gut. It can result in increased, decreased or unchanged acid secretion[26]. The specific effect of H. pylori on acid secretion depends on the pattern of gastritis induced by the infection. Studies have shown that inhibition of gastric acid pharmacologically can lead to a shift from an antrum-predominant pattern (duodenal ulcer phenotype) to a corpus-predominant one with onset of gastric atrophy (gastric cancer phenotype).
Host genetic factors: Heterogeneity of gene
H. pylori cause its damage by initiating chronic inflammation in the gastric mucosa. This inflammation is mediated by an array of pro- and anti-inflammatory cytokines. Genetic polymorphisms directly influence inter-individual variation in the magnitude of cytokine response, and this clearly contributes to an individual’s ultimate clinical outcome. In case of H. pylori infection, it is reasonable to speculate that the most relevant candidate genes would be the ones whose products were involved in handling the H. pylori exposure/attack (innate and adaptive immune responses) and ones that mediated the resulting inflammation. As previously mentioned, H. pylori induced gastritis is associated with 3 main phenotypes that correlate closely with clinical outcome: duodenal ulcer phenotype, benign phenotype, and gastric cancer phenotype. Thus, it was clear that an endogenous agent that was up-regulated in the presence of H. pylori, has a profound proinflammatory effect, and was also an acid inhibitor that would be the most relevant host genetic factor to be studied. Interleukin 1 beta (IL1B) fit this profile perfectly because, not only is it one of the earliest and most important proinflammatory cytokines in the context of H. pylori infection, it is also the most powerful acid inhibitor known[27].
Being the most eligible candidate gene IL1B received much attention from all the researchers studying H. pylori mediated duodenal ulcer and gastric cancer. Three bi-allelic polymorphisms have been reported in IL1B, all representing C>T base transition. These are -511C>T, -31C>T and +3954C>T from the transcriptional initiation site[28,29]. The association studies with IL1B in different disease population infected with H. pylori are summarized in the Table 1. Interestingly, these polymorphisms have been shown to significantly affect gastric mucosal IL1B production in response to H. pylori infection[30]. In context of functional polymorphisms in IL1B, that might influence H. pylori mediated disease outcome; novel functional polymorphisms were recently reported at upstream of the IL1B gene at -1464 and -3737 nt region[31,32]. It has been suggested that a G>C change at this locus, together with the previously reported single nucleotide polymorphism (SNP)’s at -511 and -31 region, modulates the transcriptional activity of IL1B. However, no further association studies have been conducted with these polymorphisms in either duodenal ulcer or gastric cancer patients. Because IL1B is both pro inflammatory as well as a potent inhibitor of gastric acid secretion, the promoter polymorphisms may be a possible answer to the question as to why only a small proportion of individuals develop a body predominant gastritis and chronic acid hyposecretion[33]. Association of polymorphisms that increases the level of IL1B with either gastric cancer or its precancerous state have been established in American[34], Portugese[35], Japanese[36] and Mexican[37] patients. This demonstration of the association in a number of geographically and ethnically diverse populations suggest that it may be of fundamental importance in the pathway leading to H. pylori mediated gastroduodenal diseases. Chakravorty et al[38], depicted that H. pylori infected individuals with duodenal ulcers in eastern Indian had a significantly higher frequency of the -511T/T homozygotes with an age and sex adjusted odds ratio of 4.22 (95%CI: 1.8-9.5) and -31 C/C homozygotes with an odds ratio of 2.16 (95%CI: 1.12-4.16), when compared with individuals with normal mucosa. In contrast to these observations, reports from Spanish Caucasian and Japanese populations suggest the protective role of IL1B-511 T/T genotype towards H. pylori mediated duodenal ulcer[36,39]. However, these associations were statistically non-significant. On the other hand, reports from Korean and Japanese populations failed to show such association[40,41]. In the case of gastric cancer, reports from China and Japan showed the association of IL1B-511C/C and -31T/T genotypes with the risk of gastric cancer[42,43], which represented the opposite homozygous risk genotypes that Chakravorty et al[38] observed in our duodenal ulcer patients. On the contrary, among Caucasians the IL1B-511T/T and -31 C/C genotypes were reported to be strongly associated with the risk of gastric cancer[44]. Thus, the results of epidemiological studies on the association of IL1B polymorphisms with duodenal ulcer and gastric cancer from different populations are conflicting[45], and also the study populations used were small and sometimes not clearly defined with respect to ethnicity[39-41]. A recent meta analysis with IL1B -31C/T polymorphism in duodenal ulcer subjects points out that there is no overall association of IL1B with duodenal ulcer but significant protection of -31C/C against duodenal ulcer patient was evident when the results were analyzed by ethnicity[46]. This meta analysis also emphasizes the influence of the ethnic background while conducting the association studies in different populations.
Table 1.
Summary of the gene and their locus studied with respect to Helicobacter pylori mediated gastro-duodenal disease
| Gene | Locus/polymorphism | Susceptible/protection | Population/ethnicity | Disease | Ref. |
| IL1B | C>T -511, C>T -31, C>T +3954 | Susceptible | Scottish, Polish, American, Portugese, Japanese and Mexican | Gastric cancer | di Giovine et al, 1992; Stokkers et al, 1998; El omar et al, 2000; El omar et al, 2003; Machado et al, 2001; Furuta et al, 2002; Garza-Gonzalez et al, 2003 |
| C>T -511/-31T>C | Susceptible | Indian, American, Japanese, Chinese | Duodenal ulcer | Charkarvorty et al, 2006; Kato et al, 2001; Lee et al, 2003; Furuta et al, 2002 | |
| ILRN | 86bp VNTR/ intron 2 | Susceptible | Indian, American, Japanese, Chinese | Gastric cancer | Tarlow et al, 1993 |
| TNFα | G/G-308 | Susceptible | Indian, American, Japanese, Chinese | Duodenal ulcer | Kuntsmann et al, 1999 |
| A/A-308 | Susceptible | American, Japanese, Chinese | Gastric cancer | El Omar et al, 2003 | |
| T/T-857 | Susceptible | American, Japanese, Chinese | Duodenal ulcer/gastric ulcer | Zambon et al, 2005 | |
| T/T-1031 | Susceptible | American, Japanese, Chinese | Antral inflamation | Zambon et al, 2005 | |
| A/A -238 | Protective | American, Japanese, Chinese | Gastric cancer | Jang et al, 2001 | |
| IL6 | G/G-174 | Susceptible | American, Japanese, Chinese | Gastritis | Lobbo gatti, 2005 |
| IL8 | A/A-251 | Susceptible | Chinese, Venezuelan | Gastric cardia carcinogenesis, Dysplasia | Savage et al, 2004; Kato et al, 2006 |
| G/G + 396 | Susceptible | Chinese | Gastric cardia carcinogenesis | Savage et al, 2005 | |
| IL-10 | ATA-592/-819/-1082 | Susceptible | Noncardia gastric cancer | Rad et al, 2010 | |
| XPD | rs1318 (lys751Gln) | Susceptible | Guangxi population, Chinese | GAA | Long et al, 2010 |
| XRCC4 | rs1805377 (ser298Asn) | Susceptible | Chinese | GAA | Long et al, 2010 |
| HLA class II | DQB1*0301 | Susceptible | GAA | Lee et al | |
| HLA | DQA1*0102 | Protective | Gastric atrophy and Intestinal GAA | Azuma et al, magunson et al | |
| class II | |||||
| HLA class II | DRB1*1601 | Susceptible | GAA | Magnusson et al | |
| cox-2 | G>A, -899 GC+CC | Susceptible | Hexi area of Gansu province, Chinese | GAA | Ke-Xiang et al |
| TLR-1, TLR-2, TLR-4 | Suceptibilty | Cohorts in Germany and Netherlands | H. pylori infection | Mayerle et al, 2013 | |
| TLR-4 | A>G-896 | Susceptible | Caucasian | Increased inflamatory response, Hypochlorhydria | Arbor et al, 2000; Hold et al, 2009; El omar, 2003 |
| Bmp6 | Susceptible | Caucasian | Gastric cancer | Katoh et al, 2006 | |
| Gdf15 | Susceptible | Caucasian | Gastric cancer | Katoh et al, 2006 | |
| Runx3 | Susceptible | Caucasian | Gastric cancer | Kaoh et al, 2006 | |
| Cdh1 | Susceptible | Caucasian | Gastric cancer | Guilford et al, 1998 |
IL: Interleukin 1 beta; TNF: Tumor necrosis factor; H. pylori: Helicobacter pylori.
Other functional polymorphisms in different cytokine genes have also been investigated. Kunstmann et al[47] reported for the first time the association of Tumor necrotic factor alpha (TNFα) -308 G/G genotype with H. pylori mediated duodenal ulcer. More recently, it has been shown that carriers of the TNFα-308 A allele are at high risk for gastric cancer[34], whereas the TNFα-238 A allele seems to have a protective function against the risk of cancers[48]. Zambon et al[49] reported the association of TNFα -857 T/T genotype with H. pylori mediated duodenal ulcer as well as gastric ulcer and TNFα-1031 T/T genotype with antral inflammation. Individuals with the homozygous G allele at position IL6-174 have been shown to produce higher levels of IL6 than those with the C/C genotype and this genotype is associated with the high mucosal levels of IL6 in H. pylori-associated gastritis[50]. Another important cytokine that plays a central role in the pathogenesis of H. pylori-induced diseases is IL8. Savage et al[51] suggest that the homozygous polymorphic variants of IL8 (-251A/A and +396G/G) increase the risk for gastric cardia carcinogenesis in the high-risk Chinese population. These variants could confer an altered IL8 expression pattern or interact with environmental factors to increase the risk for inflammation and hence gastric cancer. Recently, Kato et al[52] reported that in Venezuelan subjects, there was a significant effect of IL8 -251A allele on the prevalence of H. pylori mediated dysplasia (P = 0.021). The OR associated with the A-allele was 1.34 (95%CI: 0.82-2.18) for heterozygotes and 2.00 (95%CI: 1.13-3.56) for homozygotes, compared with the TT genotype. Furthermore, there was a statistically significant interaction between the number of A-alleles and H. pylori Cag A genotype (P = 0.009), suggesting that the A-allele increased the risk of dysplasia only when Cag A was present. IL10 is an anti-inflammatory cytokine that down-regulates IL1B, TNF-α, Interferon-γ, and other proinflammatory cytokines. Relative deficiency of IL10 may result in a Th-1-driven hyperinflammatory response to H. pylori with greater damage to the gastric mucosa. Homozygosity for the low-IL10 ATA haplotype (based on 3 promoter polymorphisms at positions -592, -819, and -1082) increased the risk of noncardia gastric cancer with an odds ratio of 2.5 (95%CI: 1.1-5.7) El-Omar et al[34] studied the effect of having an increasing number of proinflammatory genotypes (IL-1B-511*T, IL-1RN*2*2, TNFα-308*A, and IL10 ATA/ATA) on the risk of nongastric cancer. The risk increased progressively so that presence of 3 or 4 of these polymorphisms increased the odds ratio for gastric cancer 27-fold. The fact that H. pylori is a prerequisite for the association of these polymorphisms with malignancy demonstrates that, in this situation, inflammation is indeed driving carcinogenesis.
Though these studies form important pieces of the puzzle, but unfortunately do not reflect the true story. This is because that no association studies from the same geographical region have been conducted for both diseases. Either, studies have been reported with duodenal ulcer or cancer patients. Moreover, the allelic diversity in the population itself is a critical question as it seems to vary depending on ethnicity as reflected from the several exonic SNP in IL1B studied in the HAPMAP project(data not shown for Indian population[53,54] (Figure 1). A disease marker for the Asian population might not be a disease marker for the Caucasian population. Hence, more studies should be conducted keeping this bias in mind to get clearer picture.
Figure 1.
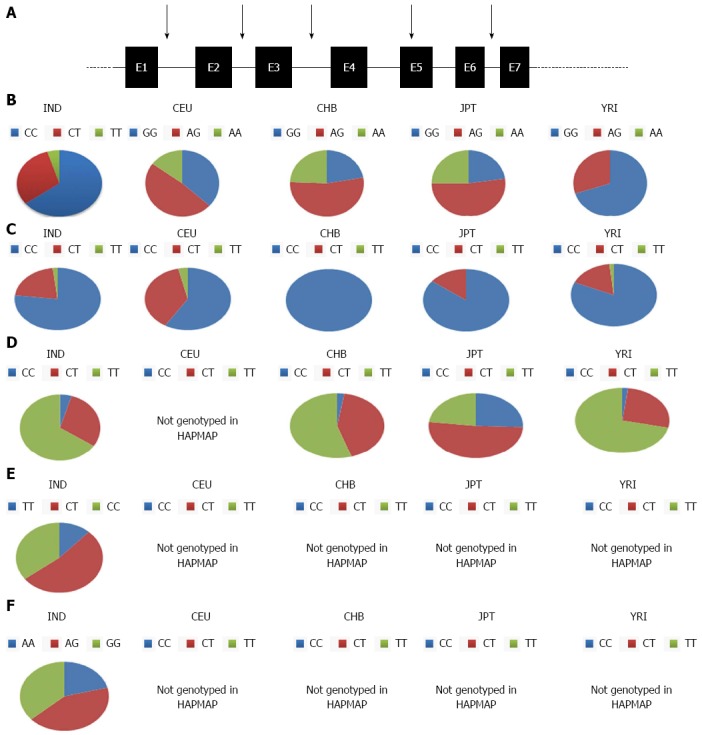
Genotypic allelic distribution of interleukin 1 beta. IL-1B exonic polymorphisms investigated in Indian as well as the HAPMAP project have been illustrated (A), allelic distribution of (B) rs000025 (C) rs 1143629 (D) rs 3136558 (E) rs 1143634 (F) rs1143643 in Indian (IND), Caucasian (CEU), CEPH (Utah residents with ancestry from northern and western Europe), Yoruba in Ibadan, Nigeria (YRI) Japanese in Tokyo, Japan (JPT), Han Chinese in Beijing, China (CHB) are plotted as pie charts. This figure depicts the variation in allelic frequencies in different population. IL-1B: Interleukin 1 beta.
ORCHESTRA OF SIGNALING MOLECULES AND H. PYLORI: A GENETIC DISSECTION OF THE DISEASE
H. pylori, being non invasive establishes the pathogenesis through its virulence factors by initiating an orchestra of signaling molecules. Using the type IV secretion system[10], H. pylori injects CagA protein and peptidoglycan (PGN) into human gastric epithelial cells. CagA is phosphorylated at the Glu-Pro-Ile-Tyr-Ala (EPIYA) motif by Src-family protein kinases to interact with PTPN11 (SHP2) protein tyrosine phosphatase. CagA-SHP2 complex interacts with GRB2 adaptor molecule to activate the SOS-RAS-RAF-MEK-ERK signaling cascade for the regulation of cell growth and differentiation (Figure 2A). PGN derived from H. pylori is recognized by the cytoplasmic pathogen-recognition molecule NOD1 to activate the NFκB signaling cascade for the transcriptional activation of cytokines in epithelial cells[55] (Figure 2C). H. pylori activate the CagA-SHP2-ERK and peptidoglycan-NOD1-NFκB signaling cascades in gastric epithelial cells using type IV secretion system. The pathogenesis of H. pylori can also be established through the OipA signaling cascade that involves p38/STAT1 to activate IRF1 or CREB or NFκB that activates various cytokine genes including IL8 (Figure 2B).
Figure 2.
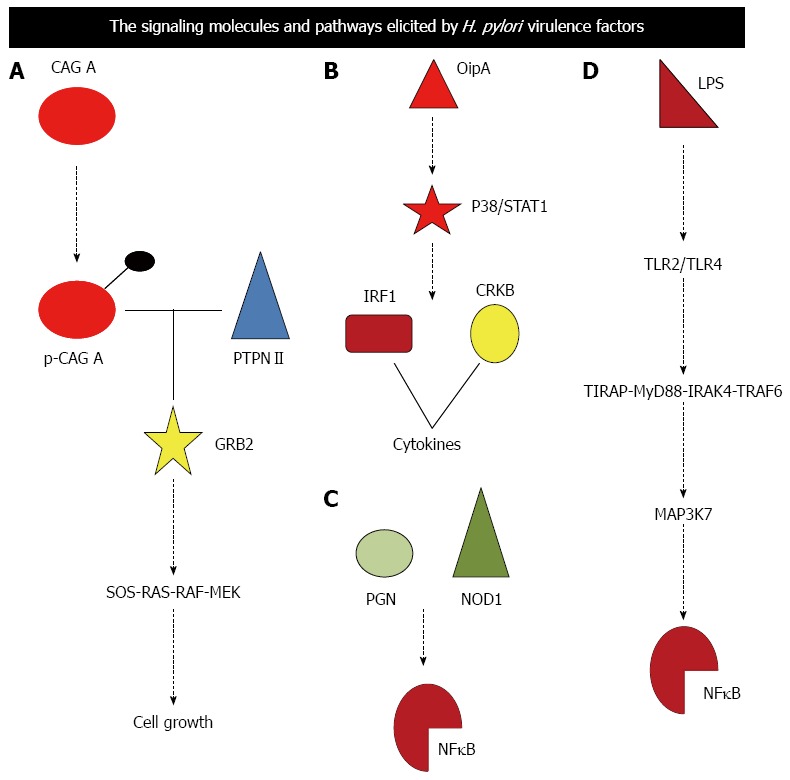
Signaling molecules and pathway elicited by Helicobacter pylori virulence factors are illustrated. A-D: Depicts signaling pathway elicited by Cag A, OipA, PGN and LPS respectively. Cag A: Cytotoxin; PGN: Peptidoglycan; LPS: Lipopolysaccheride; PTPN11: Protein tyrosine phosphatase non-receptor type 11; TLR: Toll like receptor; GRB2: Growth factor receptor-bound protein 2; SOS: Son of sevenless homolog; NFkB: Nuclear factor kappa beta; TIRAP: Toll-interleukin 1 receptor domain containing adaptor protein; MYD88: Myeloid differentiation primary response gene 88; IRAK4: Interleukin-1 receptor-associated kinase 4; TRAF6: TNF receptor associated factor; MAP3K7: Mitogen-activated protein kinase kinase kinase 7; NOD1: Nucleotide-binding oligomerization domain-containing protein 1.
Lipopolysaccharide (LPS) derived from the cell wall of H. pylori is also recognized by the cell-surface pathogen recognition receptor TLR2 and TLR4 on gastric epithelial cells and immune cells, respectively[56]. LPS-TLR signals are transduced through the TIRAP-MYD88-IRAK4-TRAF6 complex to MAP3K7 serine/threonine kinase for the NFkB and AP-1 signaling activation[57,58]. H. pylori activate the TRAF6-MAP3K7-NFkB and TRAF6-MAP3K7-AP-1 signaling cascades in epithelial and immune cells[57,58] (Figure 2D).
H. pylori induce the elevation of IL6 and TNFα in the gastric mucosa as mentioned above. IL6 and TNFα then induce the up regulation of WNT5A and WNT10B, respectively, to activate the WNT signaling pathway (Figure 3A). FGF7 is implicated in mucosal repair during chronic H. pylori infection. SHP2 function is dysregulated due to CagA injection by H. pylori. FGF signaling is activated in the gastric mucosa with H. pylori infection at the multiple levels due to FGF7 up-regulation and SHP2 dysregulation (Figure 3B).
Figure 3.
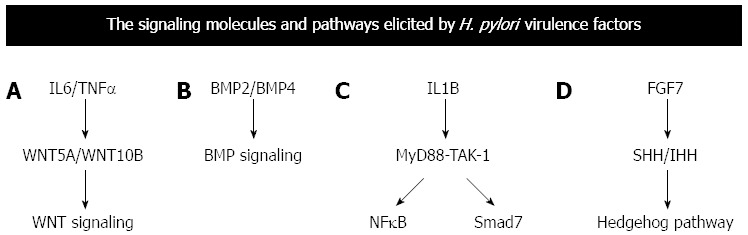
Signaling pathway elicited by the virulence factors elicits various transcription factors or cytokines that in turn influences other molecular cascades. A-D: Depicts common molecular cascades elicited by cytokines or messengers. The cytokines or other molecular messengers are expressed due to signaling pathways described in Figure 2. IL-1B: Interleukin 1 beta; TNFα: Tumor necrosis factor alpha; BMP: Bone morphogenetic protein; WNT: Wingless-type; BMP: Bone morphogenetic protein; IHH: Indian hedgehog; MYD88: Myeloid differentiation primary response gene 88; NFκB: Nuclear factor kappa beta.
SHH and PTCH1 are expressed in the parietal cells[59]. SHH-dependent proliferation of parietal cells plays a key role for the maintenance of homeostasis in the gastric mucosa with chronic H. pylori infection (Figure 3C). BMP2 and BMP4 are secreted from infiltrating inflammatory cells to activate the BMP-IHH signaling loop in the surface epithelial cells[60]. Up regulation of SHH and IHH explains the mechanism of Hedgehog signaling activation in the gastric mucosa with H. pylori infection (Figure 3D).
SNPs of genes implicated in H. pylori related signaling cascades have been searched for with the candidate gene approach to identify risk factors of gastric cancer[35,44]. SNPs of PTPN11 gene encoding SHP2 phosphatase mediating the CagA induced ERK signaling activation, TLR4 gene encoding Toll like receptor for H. pylori recognition have been associated with gastric cancer. Recently, a GWAS study conducted by Mayerle et al[61] in two large population based cohorts in Germany and Netherlands identified the toll like receptor locus on 4p14 to be significantly associated with H. pylori sero-positivity. This locus codes for TLR1, TLR6 and TLR 10, with TLR1 being of importance with respect to H. pylori infection. It has been shown that high level of TLR1 expression corroborated with higher colonization of H. pylori. However, such studies have been restricted to the Caucasian population and have to be conducted in other populations (like the Asian and African) where the density of Helicobacter pylori infection is higher. Moreover, the association of TLR1 in H. pylori associated disease background was not looked into.
Arbour et al[62] described a functional polymorphism at position +896 in exon 4 of the TLR4 gene (dbSNP ID: rs4986790). This A>G transition results in replacement of a conserved aspartic acid residue with glycine at amino acid 299 (Asp299Gly) resulting in alteration at the extracellular domain of the TLR4 receptor. This renders carriers hyporesponsive to LPS challenge by either disrupting transport of TLR4 to the cell membrane or by impairing ligand binding or protein interactions. Recent work demonstrates that defective signaling through the TLR4 receptor ultimately leads to an exaggerated inflammatory response with severe tissue destruction, even though the initial immune response may be blunted. This is due to inadequate production of IL10-secreting type 1 regulatory cells. Hold et al[56] recently hypothesized that the TLR4+896A>G polymorphism would be associated with an exaggerated and destructive chronic inflammatory phenotype in H. pylori-infected subjects. This phenotype would be characterized by gastric atrophy and hypochlorhydria, the hallmarks of subsequent increased risk of gastric cancer. In a recent study, EL-Omar et al[63] tested the effect of this polymorphism on the H. pylori-induced gastric phenotype and the risk of developing premalignant and malignant outcomes. The authors assessed associations with premalignant gastric changes in relatives of gastric cancer patients, including those with hypochlorhydria and gastric atrophy. Two independent white population-based case-control studies of upper GI tract cancer were also genotyped. TLR4+896G carriers had a 7.7-fold (95%CI: 1.6-37.6) increased odds ratio for hypochlorhydria; the polymorphism was not associated with gastric acid output in the absence of H. pylori infection. Carriers also had significantly more severe gastric atrophy and inflammation. Sixteen percent of gastric cancer patients in the initial study and 15% of the noncardia gastric cancer patients in the replication study had 1 or 2 TLR4 variant alleles vs 8% of both control populations (combined OR = 2.4; 95%CI: 1.6 -3.4). In contrast, prevalence of TLR4+896G was not significantly increased in esophageal squamous cell (2%; OR = 0.4) or adenocarcinoma (9%; OR = 0.8) or gastric cardia cancer (11%; OR = 1.2). The association of TLR+896A>G polymorphism with both gastric cancer and its precursor lesions implies that it is relevant to the entire multistage process of gastric carcinogenesis, which starts with H. pylori colonization of the gastric mucosa. Subjects with this polymorphism have an increased risk of severe inflammation and, subsequently, development of hypochlorhydria and gastric atrophy, which are regarded as the most important precancerous abnormalities. This severe inflammation is initiated by H. pylori infection, but it is entirely feasible that subsequent co-colonization of an achlorhydric stomach by a variety of other bacteria may sustain and enhance the microbial inflammatory stimulus and continue to drive the carcinogenic process.
Katoh et al[64] selected 18 genes involved in signaling based on bioinformatics (WNT4, MARK3, CELSR1, ROR2, DAAM1, MAML2, MAML3, FGF6, FGF19, GLI3, BMP6, BMP7, GDF15, RUNX1, RUNX3, POU2F3, POU5F1 and GATA4 genes) and investigated their expression in gastric cancer. They concluded that the SNPs of BMP6, GDF15 and RUNX3 genes were associated with gastric cancer. BMP6 are GDF15 are BMP family ligands[64], while RUNX3 is a transcription factor mediating BMP-induced upregulation of Hedgehog family ligand IHH[64]. No such studies have been conducted in case of duodenal ulcer. Germline mutations of CDH1 gene[65] as well as SNPs of genes encoding components of the cytokine signaling network and stem cell signaling network are associated with human gastric cancer. Transcriptional up-regulation of ligands for the stem cell signaling network as well as epigenetic silencing of negative regulators of the stem cell signaling network are known to induce carcinogenesis during chronic H. pylori infection.
H. PYLORI AND ACID SECRETIONS: THE END POINT
The end effect of the differential diseases caused by H. pylori infection involves differential acid induced in these gastric diseases. Acute infection results in hypochlorhydria, whereas chronic infection results in either hypo- or hyperchlorhydria. The mechanism by which H. pylori infection stimulates the molecular orchestra in host resulting in hypochlorhydria or hyperchlorhydria[66] may thus help us to understand the disease development. The process by which H. pylori attenuates acid secretion is known to be multifactorial and includes: (1) direct inhibition of the parietal cell (and perhaps ECL cell) by a constituent of the bug (e.g., vacuolating cytotoxin, lipopolysaccharide, or acid-inhibitory factor); and (2) indirect inhibition of parietal cell function as a result of changes in cytokines as well as hormonal, paracrine, and neural regulatory mechanisms[67,68]. In preliminary studies, it has been shown that H. pylori activate CGRP sensory neurons coupled to stimulation of somatostatin and thus inhibition of gastrin, histamine, and acid secretion[69]. H. pylori itself inhibits human H+K+ ATPaseα -subunit gene expression. It also elicits secretion of at least 2 cytokines, IL1B and TNFα that directly inhibit parietal cell secretion[70]. As discussed in the earlier part of the review, there is evidence that TLR and IL1B polymorphisms are associated with H. pylori mediated duodenal ulcer or gastric cancer. With this perspective in mind, it becomes extremely important to investigate the influence of these genetic polymorphisms associated with H. pylori mediated gastric diseases to the acid secretion pathway. In order to understand how these molecules are perturbed by the bug in individuals with different genetic background, it’s critical to evaluate whether small changes at each point on the molecular pathway add up to effect the disease phenotype or a major individual change in the pathway dictates the course of the disease.
Chronic infection with H. pylori may be associated with either decreased or increased acid secretion[71], depending on the severity and distribution of gastritis. Reduced acid secretion, at the onset, is thought to be due to functional inhibition of parietal cells by either products of H. pylori itself or, more likely, products of the inflammatory process, as discussed above for acute infection[72,73]. Acid hypersecretion lasts at least 8 wk and is due to hypergastrinemia- induced increases in parietal and ECL cell masses[74]. With time, atrophy of oxyntic glands with loss of parietal cells may occur, resulting in irreversible achlorhydria. Approximately 10% to 15% of patients chronically infected with H. pylori have antral predominant inflammation. These patients, who are predisposed to duodenal ulcer, produce increased amounts of acid as a result of reduced antral somatostatin content and elevated basal and stimulated gastrin secretion[27]. The mechanism by which somatostatin secretion is decreased is not known but may involve cytokines induced by the inflammation and/or the production of N-methyl histamine, a selective H3-receptor agonist, by H. pylori[75,76]. One may speculate that the H3-receptor agonist could diffuse across the antral mucosa to interact with H3 receptors on antral somatostatin cells, causing inhibition of somatostatin secretion, and, thus, stimulation of gastrin secretion. Gastrin, in turn, stimulates histamine secretion from ECL cells leading to enhanced acid secretion. Both interleukin- 8 and platelet-activating factor are up-regulated in HP-infected mucosa and are capable of stimulating gastrin release from isolated rabbit and canine G cells[70,77].
Several studies also regard IL1 gene polymorphism as a bridge to link it to gastric cancer as overexpression of IL1B induces hypochlorhydria[71]. The hypotheses for IL1 gene polymorphisms are based on the assumption that carriers of these genotypes are associated with increased levels of IL1B in the gastric mucosa in response to H. pylori infection[30]. Increased IL1B levels in the gastric corpus would also theoretically result in enhanced suppression of gastric acid secretion, allowing more rapid development of gastric atrophy and an increase in the risk of developing gastric cancer[78]. This is because low gastric acid resulted in a change of the colonized place of H. pylori from gastric antrum to the corpus; unfortunately, corpus-predominant gastritis with bacterial overgrowth was at increased risk of atrophic gastritis and even gastric cancer. Similarly, a difference in expression pattern of IL1B is also observed in antrum and corpus region, with the level of IL1B being higher in corpus atrophy which is normally associated with gastric ulcer and cancer.
It has also been suggested that altered regulation of gastric acid secretion and associated hormonal factors may play an important role in the pathogenesis of H. pylori related duodenal ulcer[27]. However, the mechanism of this inhibitory process is not completely understood. In order for IL1B to modulate gastric acid expression, it would be logical to expect that it functions by modulating the several intermediates that are involved in modulating gastric acid secretion like gastrin, somatostatin, histamine, H+K+ATpase etc.
It is also known that IL1B regulates somatostatin levels in ulcer patients, thus implying the role of IL1B mediated modulation of somatostatin as a pathway for regulation of gastin. That somatostatin plays an important role in duodenal ulcer, is supported by observations which illustrate that the number of D cells and the level of somatostatin in the tissue of the ulcer patients were remarkably reduced in comparison with those in non-ulcer patients (P < 0.01 and P < 0.05, respectively). The G: D cells and gastrin: somatostatin ratios in ulcer patients were much higher than those in the non-ulcer control group. It is considered that the reduction of D cells and the relative lack of somatostatin in duodenal ulcer patients might have a role in the mechanism of the duodenal ulceration[79]. In concordance with these results it has been demonstrated in few studies, that following eradication of H. pylori, there was an increase in somatostatin mRNA and a concomitant decrease in gastrin mRNA in patients with duodenal ulcers[80]. Many studies have reported the effect of the IL1B on gastrin indirectly. Chakravorty et al[81], and Datta De et al[82,83] have illustrated the mechanism of action of IL1B on gastrin directly in AGS cells. The limitation of these study was use of the cell line rather than the whole organism, as a lot of other environmental and genetic factors would influence these pathways and hence have its effect on gastrin or other acid regulating factors like histamine, acetylcholine etc. However, the expression level of these intermediate signaling molecules were checked in duodenal ulcer patients and the hypothesis that high expression of gastrin would mean low levels of NFkB and Smad7 seemed to correlate. There have also been reports where the roles of TLR in ECL cells have been investigated[84].
The lacunae in these studies are that they concentrate on one pathway ignoring the other possible pathways where these polymorphisms might influence. Like, the role of neurotransmitter acetylcholine in the perspective of H. pylori mediated disease has never been looked into. In the whole organism there would be compensatory pathways present that would try bringing the system back to its normal mode. Thus, merely linking the polymorphism to acid secretion or elucidating the path of action does not end the study. However, it marks the beginning of a more systemic approach to understand the disease. Researchers have evaluated an individual molecules or pathway and tried to understand acid regulation in H. pylori mediated disease, which makes these studies incomplete. Moreover, most of these studies are not performed in any disease models. The major setback being that there are no disease models for H. pylori mediated gastric disease.
TO BE OR NOT TO BE
The wide spectrum of disease outcome in H. pylori mediated infection thus has a very complicated explanation and is still not clearly understood. The heterogeneity in the bacteria, due its co-evolution with humans has made it to be geographically variable in different parts of the world. Thus there is variation in the virulence factors itself and also the dose of the virulence factors that effects its association to the disease. Changes in TLR due to polymorphism would result in difference in eliciting the inflammatory responses. Further, polymorphisms in the inflammatory gene like IL1B, TNFα, can partially explain the susceptibility of the host type to gastroduodenal diseases. However, polymorphisms in these inflammatory genes show a lot of variation within different population and hence cannot be considered as stable disease markers. The link between the genotypes found to be susceptible in gastric cancer or duodenal ulcer to the respective phenotypes is an interesting method to understand the opposite phenotypes developed in H. pylori mediated gastroduodenal diseases. For example perturbation of the gastrin: somatostatin ratio would change the acid output in the gut. Thus changes in any gene that would influence the expression of this axis would influence the disease outcome. This is summarized in Figure 4, which shows that small perturbations can magnify the effect and result in a certain phenotype that is characteristics of the disease. Moreover, the role of miRNA controlling this axis should be keenly looked into.
Figure 4.
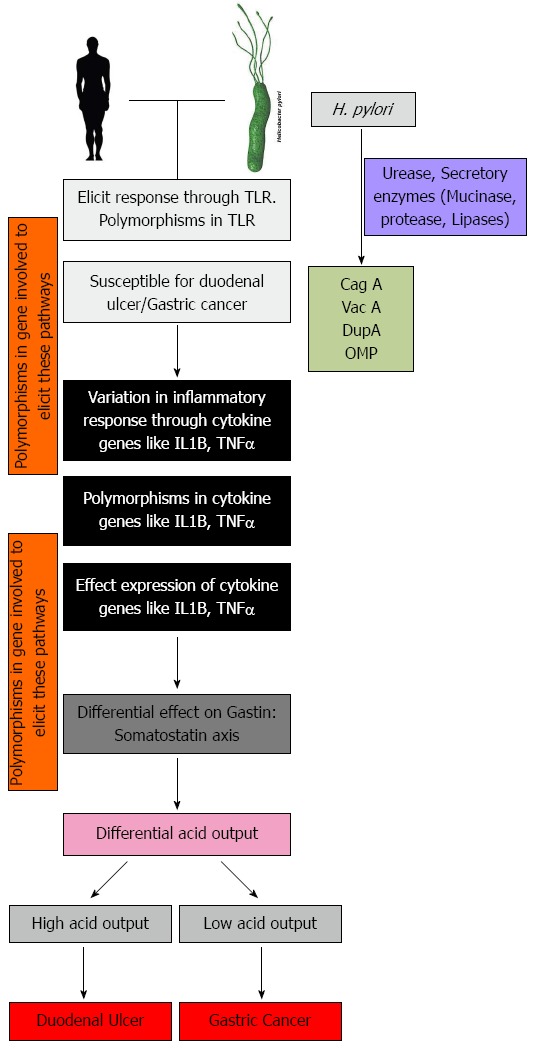
These model illustrates the different important host genes that are involved when Helicobacter pylori infection is established. Variation in any one or several of these gene add to heterogeneity in disease manifestation. H. pylori: Helicobacter pylori; TLR: Toll like receptor; IL-1B: Interleukin 1 beta; TNFα: Tumor necrosis factor alpha.
From these facts, it can be speculated that the proneness of a person to develop a disease would depend on the variability of the gene at multiple level.
Footnotes
Conflict-of-interest: The authors have no conflict of interest related to the manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 4, 2014
First decision: August 6, 2014
Article in press: January 8, 2015
P- Reviewer: Braden B, Kim H, Pang XH, Rabelo-Goncalves EMA, Youn HS S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Atherton JC, Cao P, Peek RM, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 2.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 4.Dixon M. Pathology of Gastritis and Peptic Ulceration. In: Mobley HLT, Mendz GL, and Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington, DC: ASM press; 2001. pp. 459–469. [Google Scholar]
- 5.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 6.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 7.Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pütsep K, Brändén CI, Boman HG, Normark S. Antibacterial peptide from H. pylori. Nature. 1999;398:671–672. doi: 10.1038/19439. [DOI] [PubMed] [Google Scholar]
- 9.Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci USA. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 11.Cover TL, Cao P, Lind CD, Tham KT, Blaser MJ. Correlation between vacuolating cytotoxin production by Helicobacter pylori isolates in vitro and in vivo. Infect Immun. 1993;61:5008–5012. doi: 10.1128/iai.61.12.5008-5012.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Perez GI, Dworkin BM, Chodos JE, Blaser MJ. Campylobacter pylori antibodies in humans. Ann Intern Med. 1988;109:11–17. doi: 10.7326/0003-4819-109-1-11. [DOI] [PubMed] [Google Scholar]
- 13.Dooley CP, Cohen H, Fitzgibbons PL, Bauer M, Appleman MD, Perez-Perez GI, Blaser MJ. Prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic persons. N Engl J Med. 1989;321:1562–1566. doi: 10.1056/NEJM198912073212302. [DOI] [PubMed] [Google Scholar]
- 14.Baldwin DN, Shepherd B, Kraemer P, Hall MK, Sycuro LK, Pinto-Santini DM, Salama NR. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect Immun. 2007;75:1005–1016. doi: 10.1128/IAI.01176-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo BP, Mekalanos JJ. Rapid genetic analysis of Helicobacter pylori gastric mucosal colonization in suckling mice. Proc Natl Acad Sci USA. 2002;99:8354–8359. doi: 10.1073/pnas.122244899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, Haas R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813–822. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castillo AR, Woodruff AJ, Connolly LE, Sause WE, Ottemann KM. Recombination-based in vivo expression technology identifies Helicobacter pylori genes important for host colonization. Infect Immun. 2008;76:5632–5644. doi: 10.1128/IAI.00627-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010;7:629–641. doi: 10.1038/nrgastro.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- 20.Cao P, Cover TL. Two different families of hopQ alleles in Helicobacter pylori. J Clin Microbiol. 2002;40:4504–4511. doi: 10.1128/JCM.40.12.4504-4511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogura M, Perez JC, Mittl PR, Lee HK, Dailide G, Tan S, Ito Y, Secka O, Dailidiene D, Putty K, et al. Helicobacter pylori evolution: lineage- specific adaptations in homologs of eukaryotic Sel1-like genes. PLoS Comput Biol. 2007;3:e151. doi: 10.1371/journal.pcbi.0030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Doorn LJ, Figueiredo C, Mégraud F, Pena S, Midolo P, Queiroz DM, Carneiro F, Vanderborght B, Pegado MD, Sanna R, et al. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology. 1999;116:823–830. doi: 10.1016/s0016-5085(99)70065-x. [DOI] [PubMed] [Google Scholar]
- 24.Atherton JC, Tham KT, Peek RM, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis. 1996;174:552–556. doi: 10.1093/infdis/174.3.552. [DOI] [PubMed] [Google Scholar]
- 25.Lu H, Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology. 2005;128:833–848. doi: 10.1053/j.gastro.2005.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Go MF. Review article: natural history and epidemiology of Helicobacter pylori infection. Aliment Pharmacol Ther. 2002;16 Suppl 1:3–15. doi: 10.1046/j.1365-2036.2002.0160s1003.x. [DOI] [PubMed] [Google Scholar]
- 27.El-Omar EM, Oien K, El-Nujumi A, Gillen D, Wirz A, Dahill S, Williams C, Ardill JE, McColl KE. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- 28.di Giovine FS, Takhsh E, Blakemore AI, Duff GW. Single base polymorphism at -511 in the human interleukin-1 beta gene (IL1 beta) Hum Mol Genet. 1992;1:450. doi: 10.1093/hmg/1.6.450. [DOI] [PubMed] [Google Scholar]
- 29.Stokkers PC, van Aken BE, Basoski N, Reitsma PH, Tytgat GN, van Deventer SJ. Five genetic markers in the interleukin 1 family in relation to inflammatory bowel disease. Gut. 1998;43:33–39. doi: 10.1136/gut.43.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, Yamaoka Y. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123:1793–1803. doi: 10.1053/gast.2002.37043. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Wilkins LM, Aziz N, Cannings C, Wyllie DH, Bingle C, Rogus J, Beck JD, Offenbacher S, Cork MJ, et al. Single nucleotide polymorphisms in the human interleukin-1B gene affect transcription according to haplotype context. Hum Mol Genet. 2006;15:519–529. doi: 10.1093/hmg/ddi469. [DOI] [PubMed] [Google Scholar]
- 32.Duff GW. Genetic variation in cytokines and relevance to inflammation and disease. In: Balkwill F, editor. The Cytokine Network Frontiers in Molecular Biology. Oxford: Oxford University Press; 2000. p. 152. [Google Scholar]
- 33.Robert A, Olafsson AS, Lancaster C, Zhang WR. Interleukin-1 is cytoprotective, antisecretory, stimulates PGE2 synthesis by the stomach, and retards gastric emptying. Life Sci. 1991;48:123–134. doi: 10.1016/0024-3205(91)90405-z. [DOI] [PubMed] [Google Scholar]
- 34.El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–1201. doi: 10.1016/s0016-5085(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 35.Machado JC, Pharoah P, Sousa S, Carvalho R, Oliveira C, Figueiredo C, Amorim A, Seruca R, Caldas C, Carneiro F, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology. 2001;121:823–829. doi: 10.1053/gast.2001.28000. [DOI] [PubMed] [Google Scholar]
- 36.Furuta T, Shirai N, Takashima M, Xiao F, Sugimura H. Effect of genotypic differences in interleukin-1 beta on gastric acid secretion in Japanese patients infected with Helicobacter pylori. Am J Med. 2002;112:141–143. doi: 10.1016/s0002-9343(01)01036-1. [DOI] [PubMed] [Google Scholar]
- 37.Garza-González E, Hold G, Pérez-Pérez GI, Bosques-Padilla FJ, Tijerina-Menchaca R, Maldonado-Garza HJ, el-Omar E. [Role of polymorphism of certain cytokines in gastric cancer in Mexico. Preliminary results] Rev Gastroenterol Mex. 2003;68:107–112. [PubMed] [Google Scholar]
- 38.Chakravorty M, Ghosh A, Choudhury A, Santra A, Hembrum J, Roychoudhury S. Interaction between IL1B gene promoter polymorphisms in determining susceptibility to Helicobacter pylori associated duodenal ulcer. Hum Mutat. 2006;27:411–419. doi: 10.1002/humu.20299. [DOI] [PubMed] [Google Scholar]
- 39.García-González MA, Lanas A, Savelkoul PH, Santolaria S, Benito R, Crusius JB, Peña AS. Association of interleukin 1 gene family polymorphisms with duodenal ulcer disease. Clin Exp Immunol. 2003;134:525–531. doi: 10.1111/j.1365-2249.2003.02325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SG, Kim B, Choi W, Lee I, Choi J, Song K. Lack of association between pro-inflammatory genotypes of the interleukin-1 (IL-1B -31 C/+ and IL-1RN *2/*2) and gastric cancer/duodenal ulcer in Korean population. Cytokine. 2003;21:167–171. doi: 10.1016/s1043-4666(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 41.Kato S, Onda M, Yamada S, Matsuda N, Tokunaga A, Matsukura N. Association of the interleukin-1 beta genetic polymorphism and gastric cancer risk in Japanese. J Gastroenterol. 2001;36:696–699. doi: 10.1007/s005350170033. [DOI] [PubMed] [Google Scholar]
- 42.He X, Jiang L, Fu B, Zhang X. [Relationship between interleukin-1B and interleukin-1 receptor antagonist gene polymorphisms and susceptibility to gastric cancer] Zhonghua Yi Xue Zazhi. 2002;82:685–688. [PubMed] [Google Scholar]
- 43.Takagi A, Deguchi R, Kobayashi K, Miwa T. Cytokine expressions and H. pylori-associated gastric mucosal lesion. Keio J Med. 2002;51 Suppl 2:51–52. doi: 10.2302/kjm.51.supplement2_51. [DOI] [PubMed] [Google Scholar]
- 44.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 45.El-Omar EM, Oien K, Murray LS, El-Nujumi A, Wirz A, Gillen D, Williams C, Fullarton G, McColl KE. Increased prevalence of precancerous changes in relatives of gastric cancer patients: critical role of H. pylori. Gastroenterology. 2000;118:22–30. doi: 10.1016/s0016-5085(00)70410-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhang BB, Wang J, Bian DL, Chen XY. No association between IL-1β -31 C/T polymorphism and the risk of duodenal ulcer: a meta-analysis of 3793 subjects. Hum Immunol. 2012;73:1200–1206. doi: 10.1016/j.humimm.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 47.Kunstmann E, Epplen C, Elitok E, Harder M, Suerbaum S, Peitz U, Schmiegel W, Epplen JT. Helicobacter pylori infection and polymorphisms in the tumor necrosis factor region. Electrophoresis. 1999;20:1756–1761. doi: 10.1002/(SICI)1522-2683(19990101)20:8<1756::AID-ELPS1756>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 48.Jang WH, Yang YI, Yea SS, Lee YJ, Chun JH, Kim HI, Kim MS, Paik KH. The -238 tumor necrosis factor-alpha promoter polymorphism is associated with decreased susceptibility to cancers. Cancer Lett. 2001;166:41–46. doi: 10.1016/s0304-3835(01)00438-4. [DOI] [PubMed] [Google Scholar]
- 49.Zambon CF, Basso D, Navaglia F, Belluco C, Falda A, Fogar P, Greco E, Gallo N, Rugge M, Di Mario F, et al. Pro- and anti-inflammatory cytokines gene polymorphisms and Helicobacter pylori infection: interactions influence outcome. Cytokine. 2005;29:141–152. doi: 10.1016/j.cyto.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Lobo Gatti L, Zambaldi Tunes M, de Lábio RW, Silva LC, de Arruda Cardoso Smith M, Marques Payão SL. Interleukin-6 polymorphism and Helicobacter pylori infection in Brazilian adult patients with chronic gastritis. Clin Exp Med. 2005;5:112–116. doi: 10.1007/s10238-005-0074-3. [DOI] [PubMed] [Google Scholar]
- 51.Savage SA, Abnet CC, Mark SD, Qiao YL, Dong ZW, Dawsey SM, Taylor PR, Chanock SJ. Variants of the IL8 and IL8RB genes and risk for gastric cardia adenocarcinoma and esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2004;13:2251–2257. [PubMed] [Google Scholar]
- 52.Kato I, van Doorn LJ, Canzian F, Plummer M, Franceschi S, Vivas J, Lopez G, Lu Y, Gioia-Patricola L, Severson RK, et al. Host-bacterial interaction in the development of gastric precancerous lesions in a high risk population for gastric cancer in Venezuela. Int J Cancer. 2006;119:1666–1671. doi: 10.1002/ijc.21979. [DOI] [PubMed] [Google Scholar]
- 53.International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 54.Indian Genome Variation Consortium. The Indian Genome Variation database (IGVdb): a project overview. Hum Genet. 2005;118:1–11. doi: 10.1007/s00439-005-0009-9. [DOI] [PubMed] [Google Scholar]
- 55.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 56.Hold GL, Rabkin CS, Gammon MD, Berry SH, Smith MG, Lissowska J, Risch HA, Chow WH, Mowat NA, Vaughan TL, et al. CD14-159C/T and TLR9-1237T/C polymorphisms are not associated with gastric cancer risk in Caucasian populations. Eur J Cancer Prev. 2009;18:117–119. doi: 10.1097/CEJ.0b013e3283101292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, Peek RM, Blanke SR, Chen LF. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, Akanuma M, Shiratori Y, Omata M. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119:97–108. doi: 10.1053/gast.2000.8540. [DOI] [PubMed] [Google Scholar]
- 59.Saitoh T, Kirikoshi H, Mine T, Katoh M. Proto-oncogene WNT10B is up-regulated by tumor necrosis factor alpha in human gastric cancer cell line MKN45. Int J Oncol. 2001;19:1187–1192. doi: 10.3892/ijo.19.6.1187. [DOI] [PubMed] [Google Scholar]
- 60.Bleuming SA, Kodach LL, Garcia Leon MJ, Richel DJ, Peppelenbosch MP, Reitsma PH, Hardwick JC, van den Brink GR. Altered bone morphogenetic protein signalling in the Helicobacter pylori-infected stomach. J Pathol. 2006;209:190–197. doi: 10.1002/path.1976. [DOI] [PubMed] [Google Scholar]
- 61.Mayerle J, den Hoed CM, Schurmann C, Stolk L, Homuth G, Peters MJ, Capelle LG, Zimmermann K, Rivadeneira F, Gruska S, et al. Identification of genetic loci associated with Helicobacter pylori serologic status. JAMA. 2013;309:1912–1920. doi: 10.1001/jama.2013.4350. [DOI] [PubMed] [Google Scholar]
- 62.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 63.El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27:244–252. doi: 10.1038/sj.onc.1210912. [DOI] [PubMed] [Google Scholar]
- 64.Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review) Int J Mol Med. 2006;18:1019–1023. [PubMed] [Google Scholar]
- 65.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 66.Meyer-Rosberg K, Scott DR, Rex D, Melchers K, Sachs G. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology. 1996;111:886–900. doi: 10.1016/s0016-5085(96)70056-2. [DOI] [PubMed] [Google Scholar]
- 67.Cave DR, King WW, Hoffman JS. Production of two chemically distinct acid inhibitory factors by Helicobacter pylori. Eur J Gastroenterol Hepatol. 1993;5:S23–S27. [Google Scholar]
- 68.Konturek PC, Brzozowski T, Karczewska E, Duda A, Bielański W, Hahn EG, Konturek SJ. Water extracts of Helicobacter pylori suppress the expression of histidine decarboxylase and reduce histamine content in the rat gastric mucosa. Digestion. 2000;62:100–109. doi: 10.1159/000007802. [DOI] [PubMed] [Google Scholar]
- 69.Zaki M, Coudron PE, McCuen R. Helicobacter pylori, acting via neural pathways, stimulates somatostatin and thus inhibits histamine and acid secretion in the fundus of rat stomach. Gastroenterology. 1998;114:A344. [Google Scholar]
- 70.Beales IL, Calam J. Inhibition of carbachol stimulated acid secretion by interleukin 1beta in rabbit parietal cells requires protein kinase C. Gut. 2001;48:782–789. doi: 10.1136/gut.48.6.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.El-Omar EM. Mechanisms of increased acid secretion after eradication of Helicobacter pylori infection. Gut. 2006;55:144–146. doi: 10.1136/gut.2005.071779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato S, Matsukura N, Matsuda N, Tsukada K, Naito Z, Tajiri T. Helicobacter pylori eradication therapy modulates acidity and interleukin-1β mRNA levels in un-operated stomach and in remnant stomach after gastrectomy in gastric cancer patients. Aliment Pharmacol Ther. 2006;24(Suppl 4):278–284. [Google Scholar]
- 73.Saha A, Hammond CE, Gooz M, Smolka AJ. IL-1beta modulation of H,K-ATPase alpha-subunit gene transcription in Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1055–G1061. doi: 10.1152/ajpgi.00338.2006. [DOI] [PubMed] [Google Scholar]
- 74.Fossmark R, Johnsen G, Johanessen E, Waldum HL. Rebound acid hypersecretion after long-term inhibition of gastric acid secretion. Aliment Pharmacol Ther. 2005;21:149–154. doi: 10.1111/j.1365-2036.2004.02271.x. [DOI] [PubMed] [Google Scholar]
- 75.Courillon-Mallet A, Launay JM, Roucayrol AM, Callebert J, Emond JP, Tabuteau F, Cattan D. Helicobacter pylori infection: physiopathologic implication of N alpha-methyl histamine. Gastroenterology. 1995;108:959–966. doi: 10.1016/0016-5085(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 76.Zavros Y, Rieder G, Ferguson A, Samuelson LC, Merchant JL. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology. 2002;122:119–133. doi: 10.1053/gast.2002.30298. [DOI] [PubMed] [Google Scholar]
- 77.Beales I, Blaser MJ, Srinivasan S, Calam J, Pérez-Pérez GI, Yamada T, Scheiman J, Post L, Del Valle J. Effect of Helicobacter pylori products and recombinant cytokines on gastrin release from cultured canine G cells. Gastroenterology. 1997;113:465–471. doi: 10.1053/gast.1997.v113.pm9247465. [DOI] [PubMed] [Google Scholar]
- 78.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412:99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- 79.Chen J, Liu TH, Ye SF, Gu CF, Chen SP. Gastrin and somatostatin cells in dyspeptic patients with and without duodenal ulcer: a quantitative study based on multiple biopsy specimens. J Gastroenterol Hepatol. 1989;4:41–47. doi: 10.1111/j.1440-1746.1989.tb00805.x. [DOI] [PubMed] [Google Scholar]
- 80.Burkitt MD, Varro A, Pritchard DM. Importance of gastrin in the pathogenesis and treatment of gastric tumors. World J Gastroenterol. 2009;15:1–16. doi: 10.3748/wjg.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chakravorty M, Datta De D, Choudhury A, Roychoudhury S. IL1B promoter polymorphism regulates the expression of gastric acid stimulating hormone gastrin. Int J Biochem Cell Biol. 2009;41:1502–1510. doi: 10.1016/j.biocel.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 82.Datta De D, Bhattacharjya S, Maitra M, Datta A, Choudhury A, Dhali GK, Roychoudhury S. IL1B induced Smad 7 negatively regulates gastrin expression. PLoS One. 2011;6:e14775. doi: 10.1371/journal.pone.0014775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Datta De D, Datta A, Bhattacharjya S, Roychoudhury S. NF-kappaB mediated transcriptional repression of acid modifying hormone gastrin. PLoS One. 2013;8:e73409. doi: 10.1371/journal.pone.0073409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stefani CB, de Oliveira RM, Silveira AA, Ferraz LF, Ribeiro ML, Gambero A, Pedrazzoli Júnior J. Expression of Toll-like receptors in enterocromaffin-like cells and their function in histamine release. Dig Dis Sci. 2012;57:2270–2277. doi: 10.1007/s10620-012-2176-6. [DOI] [PubMed] [Google Scholar]