

Abstract
Cimyunnins A–C (1–3), characterized with an unusual fused cyclopentenone ring G, together with cimyunnin D (4), possessing a highly rearranged γ-lactone ring F, were characterized from the fruit of Cimicifuga yunnanensis. Their structures were elucidated by spectroscopic analysis, X-ray diffraction, and density functional theory calculations. In addition, cimyunnin A exhibited comparable anti-angiogenic activities to those of sunitinib, a clinically-used first-line angiogenesis inhibitor, in the in vitro and ex vivo studies.
Pathological angiogenesis is a pivotal process for a broad range of diseases, including cancer, rheumatoid arthritis, age-related macular degeneration, diabetic retinopathy, psoriasis and atherosclerosis1,2. Thus, anti-angiogenesis has been identified as an attractive target in modern drug discovery. Notably, a number of anti-angiogenic agents, such as sunitinib and pegaptanib, have been approved by FDA for the treatment of cancer and age-related macular degeneration, respectively1.
Plants of Cimicifuga genus are famous herb medicines worldwide3. Up until now, more than 300 cycloartane triterpenoids (CTs) have been isolated (among them, more than 100 compounds were discovered by our research group3,4,5,6,7,8,9,10,11,12,13,14,15,16,17) from the genus3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21. These secondary metabolites showed divers bioactivities, such as cytotoxicity3,4,5,6,7,8,9,10,11,12,13,14,15,16, antiosteoporotic22, anti-AIDS23, anti-Alzheimer24, and immunosuppression25. To date, however, anti-angiogenic knowledge of CTs from Cimicifuga spp is mainly not yet involved.
C. yunnanensis is a rare species distributed in the southwest region of China8. Our previous studies on the roots of C. yunnanensis led to the discovery of three cytotoxic CTs, which showed as potent activities as taxol against drug resistant breast cancer cell line R-MCF-7. Furthermore, it was found that the p53-dependent mitochondrial signaling pathway contributed to the apoptosis activities induced by these compounds26. More recently, a series of active CTs against p53N236S mouse embryonic fibroblasts were obtained from the overground parts of this plant3. However, there is no current literature reported the chemical components and their bioactivities of the fruit of C. yunnanensis. This study aimed to fill this gap in the knowledge and has subsequently isolated and identified, two unprecedented types of CTs, cimyunnins A–C (1–3) and cimyunnin D (4). Compounds 1–3 contains a fused cyclopentenone ring G, which formed by direct carbon-carbon linkage between C-22 and C-26. While, 4 features a highly rearranged γ-lactone ring F among C-23 to C-27 (Fig. 1). Biological assay revealed that compound 1 showed significant anti-angiogenic properties both in vitro and ex vivo. Moreover, a mechanism study indicated that 1 exerted its anti-angiogenic activities by directly targeting VEGFR2-signaling pathways.
Figure 1. Chemical structures of compounds 1–4.

Results and Discussion
Structural Elucidation of Compounds 1–4
Cimyunnin A (1), a white powder, with a molecular formula of C30H44O4, as established by HREIMS (m/z 486.3235, calcd for [M]+ 486.3240), requires nine sites of unsaturation. The 1H-NMR spectrum (Table S1) displayed the presence of characteristic cyclopropane methylene signals at δH 0.26 (d, J = 3.8 Hz) and 0.67 (d, J = 3.4 Hz). The 13C NMR and DEPT (Table S1) spectra showed the existence of one ketone (δC 203.5), and one tetrasubstituted double bond (δC 148.9 and 149.3). Aforementioned data suggested that 1 was a CTs with a seven-ring skeleton3.
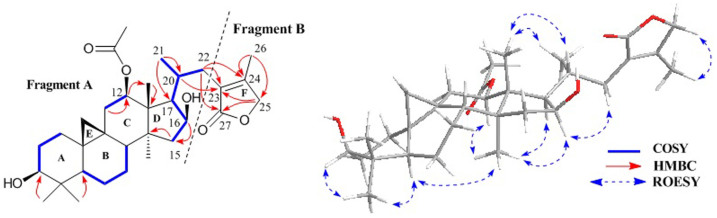
The NMR data for rings A, B, C, D, and E of 1 were similar to those of asiaticoside A (5)18, except that the sugar unit at C-3 and the acetoxy group at C-12 were replaced by two hydroxy groups, respectively. This deduction was confirmed by the 1H-1H COSY correlations (Figure 2) of hydroxymethine protons at δH 3.50 (H-3), and δH 4.23 (H-12) with H-2 (δH 1.81 and 1.93) and H-11 (δH 1.42 and 2.62), respectively. Further study of the 1H-1H COSY spectrum starting from the H-16 at δH 4.52 revealed the presence of a spin system - CHCHCH-(CH3)- (for C-16, C-17, C-20, and CH3-21) in 1. Thus, the fragment A of 1 was constructed as shown (Figure 2).
Figure 2. Fragments, key COSY and HMBC correlations of 1.

The spin system -CH2CHCH3- due to C-26, C-25, and C-27 in fragment B was also deduced from 1H-1H COSY correlations (Fig. 2). Besides, HMBC correlations (Fig. 2) of H-20 (δH 3.30) and H-26 (δH 2.67, 1.73) with the quaternary olefinic carbon at δC 148.9 (C-22), required the connections of C-20 and C-26 to C-22. Further analyses of the HMBC spectrum showed the correlations from H-25 (δH 2.32) to the carbonyl group at δC 203.5 (C-24) and the olefinic carbon at δC 149.3 (C-23). This information coupled with the UV and IR absorptions of λmax 266 nm, and νmax 1702 and 1655 cm−1, indicated the existence of a cyclopentenone ring G (C-22 to C-26). Therefore, to fulfill the molecular formula and unsaturation requirement, C-16 and C-23 should be connected by an oxygen atom, which also supported by similar chemical shifts of C-22 (δC 148.9) and C-23 (δC 149.3) due to the electronic effect and conjugative effect from the oxygen atom at C-16 and carbonyl group at C-24, repectively. Finally, fragment B and planar structure of 1 were established.
In the ROESY spectrum (Figure 2), correlations of H-5 (biogenetically α-oriented)/H-3, Me-28 (biogenetically α-oriented)/H-17, Me-28/H-16, H-17/H-12, H-17/Me-21, H-8/Me-18 (biogenetically β-oriented), Me-18/H-20, H-20/H-26β, H-26β/Me-27, and H-26α/Me-21 were observed, which helped to establish the relative configuration of 1. Our efforts to make fine crystals from 1 failed which precluded the possibility to determine the absolute configuration directly by X-ray crystallography. Therefore, quantum chemical CD calculations were applied27. The results showed that spectrum calculated for 1A was nearly identical with the experimental data of 1 (Fig. 3, left) over the whole range of wavelengths under investigation, whereas the spectrum simulated for 1B exhibited very different CD behavior compared with the experimental CD curve of 1 (Fig. 3, right). Therefore, the absolute configuration of 1 was determined as shown.
Figure 3. Assignment of the absolute configuration of 1 by comparison of the experimental CD spectra with the spectra calculated for 1A and 1B using TDDFT methods.

Cimyunnins B (2) and C (3), obtained as an inseparable mixture. Side-by-side comparison of their NMR data indicated that 2 and 3 were a pair of C-25 epimers and most signals were exchangeable between the two compounds (Table S2). Whereas, the signals of Me-27 (δH 1.03 in compound 2 and δH 1.00 in compound 3) and H-24a (δH 2.68 in compound 2 and δH 2.65 in compound 3) showed clear differences between compounds 2 and 3. In the ROESY spectrum, the correlations of H-20 (δH 2.83) with Me-18 (δH 0.76, biogenetically β-oriented) and Me-27 (δH 1.03) were observed, indicating the β-orientation of Me-27 in compound 2. Therefore, the configuration of Me-27 in compound 3 was α-orientation. 13C NMR signals for C-24 (δC 34.52), C-25 (δC 38.04) and C-26 (δC 205.13) were able to ascribed to compound 2 on the basis of their HMBC correlations with Me-27 at δH 1.03. Similarly, the 13C NMR signals for C-22 and C-23 were successfully assigned based on HMBC correlations between H-24a of compounds 2 and 3 and corresponding carbons.
The 1H and 13C NMR spectra (Table S2) of 2 and 3 were very similar to those of 1 with the major differences for signals of ring G. A spin system -CH2CHCH3- in the ring G was established by the 1H-1H COSY spectrum, which coupled with extreme downfield double bond (C-23) at δC 180.15 (180.20) positioned the cabonyl carbon at C-26. Fortunately, crystals of the epimers (1:1 mixsture) were obtained and X-ray diffraction analysis (Figure S1) allowed to establish the structures of 2 and 3 and further confirm the new skeleton of this type of triterpenoid (1–3).
The molecular composition of cimyunnin D (4), C32H48O6, was deduced from HR-EIMS ([M]+, m/z 528.3449), indicating 9 degrees of unsaturation. In the 1H NMR spectrum (Table S3) of 4, signals due to a typical cyclopropane methylene at δH 0.27 (d, J = 4.1 Hz) and 0.46 (d, J = 3.7 Hz), an acetoxy methyl group at δH 2.03, a secondary methyl resonance at δH 0.86 (d, J = 6.8 Hz), and five tertiary methyl groups at δH 0.85–1.84 (each 3H, s) were observed, in agreement with a structure of a CTs with an acetoxy group3. The 1H-1H COSY correlations (Fig. 4) of H-2 (δH 1.19 and 1.80) with the proton (δH 3.39) of the hydroxymethine at δC 78.3 and H-15 (δH 1.77 and 2.06) with the proton (δH 4.67) of the hydroxymethine at δC 72.1, located the hydroxy groups at C-3 and C-16, respectively. In the HMBC spectrum, correlation of H-12 at δH 5.09 and the carbonyl carbon at δC 171.2 was observed, indicating an acetoxy group were connected to C-12. Further analyses of 1H-1H COSY associations disclosed that 4 had a partial structure of -CH2CHCHCH-(CH3)CH2- (for C-15 to C-17, C-20 to C-22). Thus, fragment A was constructed as shown (Fig. 4).
Figure 4. Fragments, key COSY and HMBC correlations of 4.
Apart from fragment A, an isolated methyl (Me-26, δH 1.84, s), an oxygenated methylene (C-25, δH 4.55 and 4.59, each 1H, d, J = 17.0 Hz), an ester carbonyl carbon (C-27, δC 177.3), and two quaternary olefinic carbons (C-23, δC 127.1; and C-24 δC 159.7) were observed in 4. Thus, there was still one degree of unsaturation unaccounted for, requiring another ring in the final structure. The existence of an oxygenated methylene indicating the esterification of C-25 by C-27, which allowed to construct two possible five-membered ring F of 4, lactone A and lactone B (Fig. 5A). In addition, significant HMBC correlations from H2-22 (δH 1.93 and 2.71) to the carbonyl carbon at C-27 (δC 177.3) and ROESY associations of H-25 (δH 4.55 and 4.59) with CH3-26 (δH1.84) indicated that the strucutre of lactone B is reasonable.
Figure 5. Structure elucidation of 4.
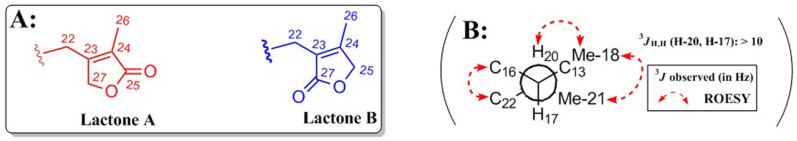
(A) The possible structures of ring F for 4; (B) Determination of relative stereochemistry of coupling systems of C-17/C-20 in 4.
On the basis of similar ROESY correlations (Fig. 4), same relative stereochemistries of C-3, C-5, C-8, C-9, C-10, C-12, C-13, C-14, C-16, and C-17 in 4 as in 1 were elucidated. Figure 5B depicted the relative stereochemistry of the C-17/C-20 coupling system by Newman projection. The anti-orientation of H-17/H-20 was suggested by a large 3JH,H (>10.0 Hz), while the gauche orientations of H-17/CH3-21 and H-20/C-16 were supported by the ROESY correlations of H-20/CH3-18, CH3-21/CH3-18, and H-16/H-22b. Finally, the 13C NMR and OR data of 4 were compared to the calculated results of lactone A (4A) and lactone B (4B) (Table S9, S10, and S11), which further confirmed the absolute configuration of 4.
To the best of our knowledge, compounds 1–4 stand for two unprecedented classes of CTs. Biosynthetically, 1–3 probably originate from the cycloartane precursors asiaticoside A (5)18. The key steps are the formation of intermediates 5A, and 5B through nazarov reaction28, and [2 + 2] cycloaddition29, respectively. While, compound 4 may derive from 1 through a series of oxidation rearrangements (see Scheme 1 in the Supporting Information).
In vitro and Ex vivo Anti-angiogenic Activities of Compound 1
Although CTs from Cimicifuga spp showed various bioactivities, anti-angiogenic activities were still unknown. In the present study, compound 1 notably inhibited VEGF-induced proliferation of HUVECs at 5.0 μM (due to sample quantity limitation, the activities of compounds 2–4 were not studied), without obvious cytotoxicity against human umbilical vein endothelial cells (HUVECs) (Fig. 6A). Then, the effect of 1 on the motility of HUVECs was studied. As shown in Figure 6B, HUVECs migrated into the clear area when stimulated with 25 ng/ml VEGF. Conversely, compound 1 significantly inhibited the VEGF-induced migration of HUVECs in a time dependent manner at concentration of 2.5 μM. This effect of 1 as strong as that of sunitinib, a clinically-used first-line angiogenesis inhibitor. Subsequently, 1 drastically reduced the new vascular growth density at a dose-dependent manner in the chick chorioallantoic membrane (CAM) assay. The maximum reduction of new vascular density was observed at concentration of 10.0 nmol/egg (Figure 6C), which was comparable to that of sunitinib at the same concentration. The aforementioned results indicated the anti-angiogenic potential of 1 both in vitro and ex vivo.
Figure 6. In vitro and ex vivo anti-angiogenic activities of compound 1.

(A) The proliferation inhibitory effect of 1 against normal and VEGF-induced HUVECs; (B) Compound 1 inhibited the VEGF-induced migration of HUVECs. Cells were wounded with a pipette then treated with vehicle or 2.5 μM sunitinib and compound 1. After 6, 12 and 24 h, the migrated cells were quantified by the Image Pro Plus software; (C) Compound 1 inhibited ex vivo angiogenesis in CAM assay, quantification of the number of new vascular growth counts was performed with Image Pro Plus software. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle control, data were analyzed by using Graphpad student t test (n = 3).
VEGFR2-signaling pathways is essential for the function of vascular endothelial cells30. Finally, to understand the molecular mechanism of 1, we examined the pathways and signaling molecules using western blot. As shown in Figure 7, phosphorylation of VEGFR2 was suppressed by 1 in a dose-dependent manner. Dramatic downregulations of phosph-AKT (Ser473) and phospho-ERK (Thr202/Tyr204), well-known downstream targets of VEGFR2, were observed at 5.0 and 10.0 μM of 1. However, total VEGFR2, ERK, and AKT remain unchanged. Therefore, compound 1 exerted its anti-angiogenic effect through directly targeting VEGFR2 on the surface of endothelial cells and further antagonizing VEGFR2-mediated downstream signaling cascade.
Figure 7. Compound 1 inhibited the activation of VEGFR2-mediated signaling pathways in HUVECs.

Proteins (12 μg) from the whole cell lysates were separated by 12% SDS-polyacrylamide gel electrophoresis. The full-length gel and blots were in supplementary materials.
Natural products, due to unrivaled chemical diversity and structural plasticity, are rich sources of novel leading structures for drug discovery31. So far, diverse natural compounds, such as anthocyanins, genistein, resveratrol, curcumin, taxol, betulinic acid, and squalamine, showed anti-angiogenic activities both in vitro and in vivo32. However, few studies about the anti-angiogenic CTs were reported33. Herein, compound 1, with an unprecedented skeleton, showed same level of anti-angiogenic activities as sunitinib both in vitro and ex vivo. Taken together, compound 1 stands for a new type of leading structure of anti-angiogenesis.
Methods
General Experimental Procedures
Optical rotations were obtained with a JASCO P-1020 digital polarimeter, using MeOH as solvent. UV spectra were taken on Shimadzu 2401PC spectrophotometer. CD spectra were obtained by a Chirascan instrument. 1H, 13C and 2D NMR experiments were measured on Bruker DRX-500 and Avance III-600 MHz spectrometers (Bruker, Zürich, Switzerland) with the solvent signal as internal reference. Mass spectra were collected from a VG Autospec-3000 spectrometer. BRUKER Tensor-27 instrument were used to record infrared spectra (with KBr pellets). X-ray diffraction was realized on a Bruker SMART APEX CCD crystallography system. Precoated TLC plates (200–250 μm thickness, silica gel 60 F254, Qingdao Marine Chemical, Inc.) were used for the thin-layer chromatography. Semipreparative HPLC was taken on an Agilent 1100 liquid chromatography, the column used was an YMC-Pack 10 mm × 250 mm column (Pro C18 RS). Column chromatography (cc) was performed on silica gel (200–300 mesh; Qingdao Marine Chemical Inc., P. R. China), on C-18 silica gel (40–60 μm; Merck), and on Sephadex LH-20 (Amersham Pharmacia, Sweden).
Plant Material
The fruit of Cimicifuga yunnanensis (234 g) were collected during September 2012 from Daocheng County, Sichuan Province, China. The herbarium specimen was authenticated by Prof. Shengji Pei, Kunming Institute of Botany, CAS. A voucher specimen (KUN No. 201209002) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, CAS, P. R. China.
Extraction and Isolation
The powdered and air-dried fruit of C. yunnanensis (234 g) were soaked in methanol at room temperature (1.5 L × 24 h × 3). The combined extracts were concentrated under reduced pressure to afford a dark brown residue (24.2 g). The extract was fractionated by cc (silica gel 600 g, CHCl3/MeOH step gradients: 100:0, 50:1, 20:1, 10:1, 5:1, and 0:100) to afford fractions A-U. Fraction G (0.9 g) was divided into ten sub-fractions (Fractions G.1–G.10) by RP-18 cc (200 g, MeOH/H2O gradient from 50:50 to 100:0). Fraction G.7 (25 mg) was purified by semipreparative HPLC (eluted with CH3CN/H2O, gradient from 60:40 to 85:15) to yield compounds 1 (2.4 mg), and 4 (1.2 mg). Compounds 2 and 3 (1.3 mg) were isolated from fraction G.6 (40 mg) by semipreparative HPLC (eluted with CH3CN/H2O, gradient from 60:40 to 85:15).
Cimyunnins B and C Single Crystal Cultivation
The clear solution of the pair of epimers (Cimyunnins B and C, 1.2 mg) in MeOH (5 mL) was added several drops of water, and then was kept at ambient temperature for slow evaporation to cultivate a single crystal suitable for X-ray crystallographic measurement.
X-ray Crystallographic Analysis of the pair of epimers (Cimyunnins B and C)
The crystal data: C30H44O4, M = 468.65, orthorhombic, a = 11.6984(4) Å, b = 14.5084(5) Å, c = 15.4962(6) Å, α = 90.00°, β = 90.00°, γ = 90.00°, V = 2630.09(16) Å3, T = 100(2) K, space group P212121, Z = 4, μ(CuKα) = 0.599 mm−1, d = 1.184 mg/m3, crystal dimensions 0.36 × 0.36 × 0.10 mm, were collected from a Bruker APEX DUO diffractometer with a graphite monochromator (Φ/ω scans, 2θmax = 70.01°), Cu Kα radiation. The total number of independent reflections measured was 29685, of which 4478 were observed (|F|2 ≥ 2σ|F|2). The final R1 = 0.0610 (I > 2σ(I)), and wR(F2) = 0.1612 (I > 2σ(I)). The final R1 = 0.0616 (all data), and wR(F2) = 0.1619 (all data). The goodness of fit F2 = 1.047. Flack parameter = 0.6(3). The crystal structure of 1 was solved by direct method SHELXS-97 and the full-matrix least-squares deposited in the Cambridge Crystallographic Data Centre (deposition number: 937160).
ECD Calculation
The theoretical calculations of compound 1 were performed using Gaussian 0934. Conformational analysis was initially carried out using Maestro7.5 conformational searching, together with the OPLS_2005 molecular mechanics methods. The optimized conformation geometries and thermodynamic parameters of the predominant conformations were provided (see computational data for 1 in Supplementary Information). The OPLS_2005 conformers were optimized at B3LYP/6-31G(d,p) level. The theoretical calculation of ECD was performed using time dependent Density Functional Theory (TDDFT) at B3LYP/6-31G(d,p) level in methanol with PCM model. The ECD spectra of compound 1 were obtained by weighing the Boltzmann distribution rate of each geometric conformation35.
The ECD spectra were simulated by overlapping Gaussian functions for each transition according to:
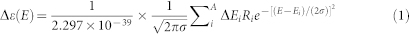 |
The σ represented the width of the band at 1/e height, and ΔEi and Ri were the excitation energies and rotational strengths for transition i, respectively. σ = 0.30 eV and Rvel had been used in this work.
NMR calculation
For the calculations of 13C NMR chemical shifts, B3LYP/6-31G(d,p) method was used to optimize the selected conformations. For all optimized structures, vibrational spectra were calculated to ensure that no imaginary frequencies for energy minimum were obtained. NMR calculations were performed at the levels of B3LYP/6-31G(d,p) with the gauge-independent atomic orbital (GIAO) method36,37,38. The solvent effect was considered by using pyridine in the calculations to resemble the experimental condition. The polarized continuum model (PCM) of Tomasi et al. was used39,40,41. The calculated 13C NMR chemical shifts were analyzed by subtracting the isotopic shifts for TMS calculated with the same methods36,37,38. Different conformers for Lactone A and Lactone B were considered. The 13C NMR chemical shifts in each compound were considered as the average values of the same atoms in the different conformers (see computational data for 4 in Supplementary Information). The average values were obtained by the Boltzmann distributions, using the relative Gibbs free energies as weighting factors35. The differences Δδ were determined by subtracting the experimental chemical shifts δexptl from the calculated chemical shiftsδscal.calc. All calculations were performed using Gaussian 0934.
OR calculation
For optical rotation, compounds 1A, lactone A (4A) and Lactone B (4B) were obtained in the gas phase at the B3LYP/6-31G(d,p) level of theory. The optical rotation values were calculated using B3LYP/6-31G(d,p) theory. The solvation effect was considered using methanol in the calculations to resemble the experimental conditions. The polarized continuum model (PCM) of Tomasi et al. was used39,40,41. The OR spectra of compounds 1A, lactone A and Lactone B were obtained by weighing the Boltzmann distribution rate of each geometric conformation35. All calculations were performed using Gaussian 0934.
Cell Growth Inhibition Assay
Growth inhibition of human cancer cells by compound 1 was assessed by the MTT assay, along with DMSO as a control. HUVECs were grown in DMEM media with 10% FBS. HUVECs were treated with compound 1 at various concentrations. After a 72 h incubation, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was added to the wells (50 μL; 0.4 mg/ml) and incubated another 4 h. Medium were aspirated and DMSO (150 μL) was added to each well. Absorbance was measured at 490 nm using 2030 Multi-label Reader (Perkin-Elmer Victor X5, US). Compound concentrations causing 50% growth inhibition (IC50) were calculated.
Cell Proliferaion Assay
HUVECs were seeded in 96-well plates and incubated for 24 h, cells were then starved in M200 medium containing 2% FBS for another 16 h. After starvation, cells were pretreated for 30 min with indicated concentration of compound 1 (1, 10, 20, 30 μmol/L), followed by the stimulation with VEGF (25 ng/mL) for another 24 h. Cell viability was then determined by MTT assay.
Wound-healing Migration Assay
HUVECs were seeded and grown into full confluence in 6 well plates. Cells were starved with 2% FBS M200 media for 12 h to inactivate cell proliferation and then wounded by pipette tips. Fresh M200 medium with 25 ng/mL VEGF containing vehicle or 2.5 μmol/L sunitinib and compound 1 was added to the scratched monolayers. Images were taken after 0, 6, 12, 24 hours using an inverted microscope (magnification, 10×; Nickon). Sunitinib used as a positive control.
CAM Assay in Fertilized Chicken Eggs
The effect of compound 1 on ex vivo angiogenesis was determined by CAM assay. Briefly, fertile leghorn chicken eggs (Poultry Breeding farm, Kunming) were incubated in incubator at 37.8°C with 40% humidity. A small opening was made at the top of live eggs on day 7 under aseptic conditions. Indicated concentrations of compound 1 and sunitinib was mixed with DMSO and tipped on the filter paper, then gently placed on the CAM. The eggs were incubated for 48 h, then fixed with methanol and photographed.
Western Blot Assay
To determine the effects of compound 1 on VEGFR-2 dependent signaling pathway, HUVECs were serum-starved overnight, then pretreated with or without compound 1 (1, 2.5, 5, 10 μM) for 2 h, followed by the stimulation with 50 ng/mL VEGF165 for 15 min. cells were lysed with buffer containing 20 mmol/L Tris, 2.5 mmol/L EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 40 mmol/L NaF, 10 mmol/L Na4P2O7, proteinase inhibitor cocktail and 1 mmol/L phenylmethylsulfonyl fluoride. Protein concentrations were determined by Bradford assay and equalized before loading. About 20 μg cellular proteins were separated using gradient SDS-PAGE gels and probed with specific antibodies (Cell Signaling Technology) including phospho-VEGFR2(p-VEGFR2; Tyr1175), VEGFR2, phospho-ERK1/2 (p-ERK1/2; Thr202/Tyr204), ERK, phospho-AKT (p-AKT; Ser473), AKT and actin. Blots were developed by incubating with horseradish peroxidase-conjugated antibodies (GE health care UK) and visualized with enhanced chemiluminescene reagent (Thermo).
Cimyunnin A (1): white powder; [α]D25 = −52.23 (c 0.11, MeOH); UV (MeOH) λmax (log ε): 202 (0.32), 266 (0.51). IR (KBr): νmax 3442, 2959, 2868, 1702, 1650, 1456, 1374, 1138, 1024, 986 cm−1; 1H (C5D5N, 600 MHz) and 13C NMR (C5D5N, 150 MHz) spectra see Table S1; positive ESIMS: m/z 491 [M + Na]+; HR-EIMS: m/z 486.3235 (calc. for C30H46O5, 486.3240).
Cimyunnins B and C (2 and 3): colorless crystals; [α]D25 = −105.99 (c 0.15, MeOH); UV (MeOH) λmax (log ε): 200 (0.28), 252 (0.79); IR (KBr): νmax 3440, 2957, 2867, 1691, 1627, 1415, 1383, 1130, 1024, 990 cm−1; 1H (C2D6OS, 500 MHz) and 13C NMR (C2D6OS, 150 MHz) spectrum see Table S2; positive ESIMS: m/z 491 [M + Na]+; HREIMS: m/z 468.3249 (calc. for C30H44O4, 468.3240).
Cimyunnin D (4): white powder; [α]D25 = −51.22 (c 0.11, MeOH); UV (MeOH) λmax (log ε): 215 (0.53). IR (KBr): νmax 3442, 2932, 2869, 1734, 1631, 1452, 1384, 1248, 1098, 1028, 986 cm−1; 1H (C5D5N, 600 MHz) and 13C NMR (C5D5N, 150 MHz) spectrum see Table S3; positive ESIMS: m/z 551 [M + Na]+; HR-EIMS: m/z 528.3449 (calc. for C30H46O5, 528.3451).
Author Contributions
Q.M.H., Z.J.H. and N.Y. designed the phytochemical and biological experiments. Y.J. designed and conducted the density functional theory calculations. N.Y. and Z.J.H. analyzed data. N.Y., L.Y. and Z.J.H. wrote the paper. N.Y. and L.T.Y. conducted the phytochemical and biological experiments, repectively. N.Y. and Y.J. were designated as co-first authors.
Supplementary Material
Supplementary Information for New Anti-angiogenic Leading Structure Discovered in the Fruit of Cimicifuga yunnanensis
Acknowledgments
This project was supported by the National Natural Science Foundation of China (U1132604 and 81302670) and China Postdoctoral Science Foundation (2013M531996).
References
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 6, 273–286 (2007). [DOI] [PubMed] [Google Scholar]
- Schoors S. et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 19, 37–48 (2014). [DOI] [PubMed] [Google Scholar]
- Nian Y. et al. Triterpenes from the aerial parts of Cimicifuga yunnanensis and their antiproliferative effects on p53N236S mouse embryonic fibroblasts. J. Nat. Prod. 76, 896–902 (2013). [DOI] [PubMed] [Google Scholar]
- Nian Y. et al. Cytotoxic cycloartane triterpenes of the traditional chinese medicine “Shengma” (Cimicifuga dahurica). Planta Med. 79, 60–69 (2013). [DOI] [PubMed] [Google Scholar]
- Nian Y. et al. Cytotoxic cycloartane triterpenes from the roots of Cimicifuga heracleifolia. Tetrahedron. 68, 6521–6527 (2012). [Google Scholar]
- Nian Y. et al. Cycloartane triterpenoids from the aerial parts of Cimicifuga foetida Linnaeus. Phytochemistry. 72, 1473–1481 (2011). [DOI] [PubMed] [Google Scholar]
- Nian Y. et al. Cytotoxic chemical constituents from the roots of Cimicifuga foetida. J. Nat. Prod. 73, 93–98 (2010). [DOI] [PubMed] [Google Scholar]
- Nian Y. et al. Four new 9,19-cyclolanostane derivatives from the rhizomes of Cimicifuga yunnanensis Hsiao. Helv. Chim. Acta. 92, 112–120 (2009). [Google Scholar]
- Lu L. et al. Studies on the constituents of Cimicifuga foetida collected in Guizhou province and their cytotoxic activities. Chem. Pharm. Bull. 60, 571–577 (2012). [DOI] [PubMed] [Google Scholar]
- Lu L. et al. Five new triterpene bisglycosides with acyclic side chains from the rhizomes of Cimicifuga foetida L. Chem. Pharm. Bull. 58, 729–733 (2010). [DOI] [PubMed] [Google Scholar]
- Lu L. et al. Trinor-cycloartane glycosides from the rhizomes of Cimicifuga foetida. Molecules. 14, 1578–1584 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. R. et al. Two new triterpene glycosides with monomethyl malonate groups from the rhizome of Cimifuga foetida L. Molecules. 16, 5701–5708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. R. et al. New triterpene diglycosides from the rhizome of Cimifuga foetida. Molecules. 13, 1712–1721 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. R. et al. Cimicifine A: a novel triterpene alkaloid from the rhizomes of Cimicifuga foetida. Helv. Chim. Acta. 90, 1313–1318 (2007). [Google Scholar]
- Sun L. R. et al. Cimicifoetisides A and B, two cytotoxic cycloartane triterpenoid glycosides from the rhizomes of Cimicifuga foetida, inhibit proliferation of cancer cells. Beilstein J. of Org. Chem. 3, 1–6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y. et al. Four new 9,19-Cyclolanostane triterpenes from the rhizomes of Cimicifuga foetida collected in Yulong. Chin J. Chem. 30, 1265–1268 (2012). [Google Scholar]
- Li D. S., Nian Y., Sun Y. & Qiu M. H. Three new cycloartane (9,19-cyclolanostane) glycosides from Cimicifuga foetida. Helv. Chim. Acta. 94, 632–638 (2011). [Google Scholar]
- Gao J. C. et al. Cytotoxic cycloartane triterpene saponins from Actaea asiatica. J. Nat. Prod. 69, 1500–150 (2006). [DOI] [PubMed] [Google Scholar]
- Li J. X. & Yu Z. Y. Cimicifugae rhizoma: from origins, bioactive constituents to clinical outcomes. Curr. Med. Chem. 13, 2927–2951 (2006). [DOI] [PubMed] [Google Scholar]
- Ali Z., Khan S. I., Ferreira D. & Khan I. A. Podocarpaside, a triterpenoid possessing a new backbone from Actaea podocarpa. Org. Lett. 8, 5529–5532 (2006). [DOI] [PubMed] [Google Scholar]
- Dan C. et al. Cimicifugadine from Cimicifuga foetida, a new class of triterpene alklaoids with novel reactivity. Org. Lett. 9, 1813–1816 (2007). [DOI] [PubMed] [Google Scholar]
- Li J. X. et al. Triterpenoids from Cimicifuga rhizoma, a novel class of inhibitors on bone resorption and ovariectomy-induced bone loss. Maturitas. 58, 59–69 (2007). [DOI] [PubMed] [Google Scholar]
- Sakurai N. et al. Anti-AIDS agents. part 57: Actein, an anti-HIV principle from the rhizome of Cimicifuga racemosa (black cohosh), and the anti-HIV activity of related saponins. Bioorg. Med. Chem. Lett. 14, 1329–1332 (2004). [DOI] [PubMed] [Google Scholar]
- Lee J. H. et al. Cycloartane-type triterpene glycosides from the rhizomes of Cimicifuga heracleifolia and their anticomplementary activity. Planta Med. 78, 1391–1394 (2012). [DOI] [PubMed] [Google Scholar]
- Findeis M. A. et al. Discovery of a novel pharmacological and structural class of gamma secretase modulators derived from the extract of Actaea racemosa. ACS Chem. Neurosci. 3, 941–951 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z. Z. et al. Cycloartane triterpenoids from Cimicifuga yunnanensis induce apoptosis of breast cancer cells (MCF7) via p53-dependent mitochondrial signaling pathway. Phytother Res. 25, 17–24 (2011). [DOI] [PubMed] [Google Scholar]
- Huang S. X. et al. Structural characterization of schintrilactone, a new class of nortriterpenoids from Schisandra chinensis. Org. Lett. 9, 4175–4178 (2007). [DOI] [PubMed] [Google Scholar]
- Magnus P., Freund W. A., Moorhead E. J. & Rainey T. Formal synthesis of (±)-methyl rocaglate using an unprecedented acetyl bromide mediated nazarov reaction. J. Am. Chem. Soc. 134, 6140–6142 (2012). [DOI] [PubMed] [Google Scholar]
- Tan H. B., Zheng C., Liu Z. & Wang D. Z. Biomimetic total syntheses of linderaspirone A and Bi-linderone and revisions of their biosynthetic pathways. Org. Lett. 9, 2192–2195 (2011). [DOI] [PubMed] [Google Scholar]
- Chong C. R. & Jane P. A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 19, 1389–1400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. H. & Vederas J. C. Drug discovery and natural products: end of an era or an endless frontier? Science. 325, 161–165 (2009). [DOI] [PubMed] [Google Scholar]
- Neal C. P. et al. Clinical aspects of natural anti-Angiogenic drugs. Curr Drug Targets. 7, 371–383 (2006). [DOI] [PubMed] [Google Scholar]
- Pudhom K., Nuanyai T. & Matsubara K. Cytotoxic and anti-angiogenic properties of minor 3,4-seco-cycloartanes from Gardenia sootepensis exudate. Chem. Pharm. Bull. 60, 1538–1543 (2012). [DOI] [PubMed] [Google Scholar]
- Frisch M. J. et al. Gaussian 09. [Gaussian, Inc. (ed.)] (Wallingford CT, USA, 2010). [Google Scholar]
- Tähtinen P., Bagno A., Klika K. D. & Pihlaja K. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. Modeling NMR parameters by DFT methods as an aid to the conformational analysis of cis-Fused 7a(8a)-Methyl Octa(hexa)hydrocyclopenta[d][1,3]oxazines and [3,1]benzoxazines. J. Am. Chem. Soc. 125, 4609–4618 (2003). [DOI] [PubMed] [Google Scholar]
- Ditchfield R. Self-consistent perturbation theory of diamagnetism: I. A gauge-Invariant LCAO (Linear Combination of Atomic Orbitals) method for NMR chemical shifts. Mol. Phys. 27, 789–807 (1974). [Google Scholar]
- Rohlfing C. M., Allen L. C. & Ditchfield R. Proton and 13C chemical shifts: comparison between theory and experiment. Chem. Phys. 87, 9–15 (1984). [Google Scholar]
- Wolinski K., Hinton J. F. & Pulay P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 112, 8251–8260 (1990). [Google Scholar]
- Miertus S., Scrocc E. & Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55, 117–129 (1981). [Google Scholar]
- Miertus S. & Tomasi J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 65, 239–241 (1982). [Google Scholar]
- Cossi M., Barone V., Cammi R. & Tomasi J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255, 327–335 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information for New Anti-angiogenic Leading Structure Discovered in the Fruit of Cimicifuga yunnanensis