

Abstract
Several studies have demonstrated that mild hypothermia exhibits a neuroprotective role and it can inhibit endothelial cell apoptosis following ischemia/reperfusion injury by decreasing caspase-3 expression. It is hypothesized that mild hypothermia exhibits neuroprotective effects on neurons exposed to ischemia/reperfusion condition produced by oxygen-glucose deprivation. Mild hypothermia significantly reduced the number of apoptotic neurons, decreased the expression of pro-apoptotic protein Bax and increased mitochondrial membrane potential, with the peak of anti-apoptotic effect appearing between 6 and 12 hours after the injury. These findings indicate that mild hypothermia inhibits neuronal apoptosis following ischemia/reperfusion injury by protecting the mitochondria and that the effective time window is 6–12 hours after ischemia/reperfusion injury.
Keywords: nerve regeneration, mild hypothermia, oxygen-glucose deprivation, cell apoptosis, neurons, mitochondrial membrane potential, Bax, ischemia/reperfusion, neural regeneration
Introduction
Therapeutic hypothermia was reported to be an effective treatment for refractory intracranial hypertension following severe traumatic brain injury and stroke in the 1950s (Bigelow et al., 1950). Since then, therapeutic hypothermia has been intensively studied at both the experimental and clinical levels (Buchan and Pulsinelli, 1990; Georgiadis et al., 2002). For unconscious adult patients with return of spontaneous circulation after out-of-hospital cardiac arrest, 12–24-hour hypothermia is strongly recommended by the International Liaison Committee on Resuscitation (Nolan et al., 2003). Therapeutic hypothermia is also beneficial to full-term newborns with hypoxic-ischemic encephalopathy (Gluckman et al., 2006). For traumatic brain injury, hypothermia may increase the chance of a positive outcome but does not change the risk of mortality (Brain Trauma Foundation, 2007). The efficacy of hypothermia for the treatment of ischemic stroke has yet to be determined by prospective randomized clinical trials. However, the neuroprotective role of hypothermia is well established in experimental animals (Green et al., 1992). A previous study has demonstrated that hypothermia can inhibit ischemia/reperfusion injury and apoptosis through reducing the expression of cleaved caspase-3 and poly(ADP-ribose) polymerase-1 (PARP) in endothelial cells (Yang et al., 2009). However, the precise details regarding the neuroprotective mechanism of hypothermia largely remain to be elucidated. Therefore, in the present study, we focused on the relationship between apoptosis, mitochondrial membrane potential (Δψm), and Bax expression in neurons exposed to oxygen-glucose deprivation (OGD) followed by reperfusion. We also investigated the effective time window of mild hypothermia in oxygen-glucose deprived neurons.
Materials and Methods
Primary culture of neurons
The study procedures involving animals were approved by the Ethics Committee of Southern Medical University, China and all efforts were made to minimize the number of animals used and their suffering. Cortical neurons were isolated and primary cultured according to the protocol described previously (Xu et al., 2012; Sendrowski et al., 2013). Briefly, cerebral cortical neurons were prepared from the cerebrum of fetuses, obtained from the pregnant SPF-class E18 Sprague-Dawley rats provided by the Experimental Animal Center of Southern Medical University, China and dissected in Hanks’ balanced salt solution (HBSS) on ice. The cortex was put into a sterile 35-mm petri dish (containing cold FBS-free HG-DMEM) and cut into small pieces approximately 1 mm in length. After digestion, the cell suspension was collected. Cortical neurons were seeded onto plates pre-coated with poly-L-lysine (25 mg/mL) at a density of 50,000 cells per cm2 and were placed in a humidified incubator (37°C with 5% CO2). HG-DMEM was replaced by a neurobasal medium (containing 10% B27 supplement and 100 U/mL penicillin-streptomycin) after 4 hours of incubation. The neurobasal medium was refreshed by exchanging half of the volume with fresh media 3 days later. Under these culture conditions, high-purity neurons can be harvested (Xu et al., 2012; Sendrowski et al., 2013). Primary antibody (1:100 rabbit anti-rat β-tubulin III containing BSA and triton X-100; Abcam, Shanghai, China) was added to the vascular surface for 2-hour incubation at 37°C, and then the cells were washed three times with HBSS. An Alexa Fluor 488-labeled goat anti-rabbit IgG (1:200) and the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) were added and incubated for 1 hour at 37°C in the dark. Fluorescence images were acquired with an inverted microscope equipped with a CoolLED fluorescent light source (Olympus, Tokyo, Japan). Nearly all of the cells were DAPI-(Figure 1A) and β-tubulin III-positive (Figure 1B). The following experiments were performed with cells cultured for 5 days.
Figure 1.
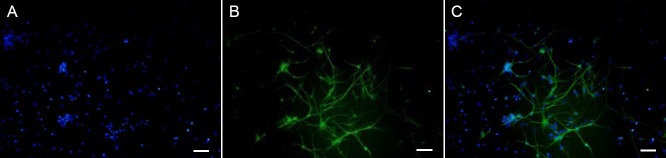
Identification of primary cultured neurons using a fluorescence microscope.
(A) DAPI-positive cells. (B) β-Tubulin III-positive cells. (C) DAPI-positive neurons in the nucleus (blue). β-Tubulin III positive cells exhibit green neuronal axons and dendrites. Scale bars: 50 μm.
OGD/reperfusion and mild hypothermia
Cell cultures were subjected to OGD injury using a protocol described previously (Huang et al., 2010). In brief, culture medium was replaced with a glucose-free balanced salt solution (BSS) and was bubbled with 95% N2/5% CO2 (OGD solution). An anaerobic glove box (Thermo, Boston, MA, USA) was equilibrated for 10 minutes with a continuous flux of gas (95% N2/5% CO2), and the oxygen-glucose deprived cells were then transferred into the chamber. After 90 minutes of anaerobic culture, the culture medium was replaced completely by the neurobasal medium (reperfusion). Then the cells were incubated at 33°C for different periods of time (6, 12, 18, 24 hours) in 95% O2/5% CO2. Cells in the control group were incubated with neurobasal medium after OGD injury process at normal temperature (37°C) for 24 hours. In the normal group, the culture medium was exchanged with neurobasal medium completely at the same time without an OGD injury process for 24 hours. When the mild hypothermia periods finished, cell cultures were transferred to normal temperature. When all the hypothermia groups were terminated, the cells were preserved for the following experiments.
Apoptosis measurement
In apoptotic cells, phosphatidylserine is translocated from the inner to the outer leaflet of the plasma membrane, allowing for the detection of phosphatidylserine on the cell surface. In the presence of Ca2+, Annexin V exhibits a high affinity for phosphatidylserine and binds to cells with exposed phosphatidylserine. Annexin V, when conjugated to a fluorophore, can be used to monitor apoptotic cells using flow cytometry or immunofluorescence microscopy (Pozarowski et al., 2004). Flow cytometric analysis with an apoptotic kit (Merk, Germany) was used for counting and distinguishing necrotic from apoptotic cell death. Cells undergoing apoptosis were detected with the use of double staining with Annexin V fluorescein isothiocyanate (Annexin V-FITC)/propidium iodide in the dark according to the manufacturer's instructions. After 15 minute incubation in the dark at room temperature, the cells were analyzed within 1 hour using a flow cytometer (BD Biosciences, San Jose, CA, USA). Annexin V-FITC selectively passed through the plasma membranes of apoptotic cells and stained them with green fluorescence. Necrotic cells were stained fluorescent red with propidium iodide. A flow cytometry instrument was used to measure the cells in each group (about 10,000 cells).
Western blot analysis
Protein level of Bax in neurons was examined by western blot analysis according to the product protocol. Briefly, neurons of each group were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (Beyotime Inc., Shanghai, China) which contained protease inhibitors to obtain the total protein from the cells. Protein concentration was determined using the bicinchoninic acid protein assay kit (Beyotime Inc.). 40 μg of protein from the cell lysate in each group was run per lane on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, electrophoresed, and transferred to a nitrocellulose filter membrane. After the membranes were blocked with 5% nonfat dry milk, they were incubated overnight at 4°C with rabbit anti-β-actin (1:1,000; Abcam, Hong Kong, China) and rabbit anti-Bax (1:1,000; Abcam). Horseradish peroxidase-conjugated goat anti-mouse IgG was used as secondary antibody (1:2,500; Merck, Darmstadt Germany). The membranes were incubated for 2 hours at 25°C. The immunoreactive bands were detected by the SuperSignal West Pico chemiluminescence detection system (Pierce, Rockford, IL, USA). Densitometric analysis was performed by NIH Image J analysis software (National Institutes of Health, Bethesda, MD, USA). The values were normalized with β-actin.
Measurements of mitochondrial membrane potential (Δψm)
The cyanine dye 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolocarbocyanine iodide (JC-1) has been widely used for microscopic and cytometric estimation and measurement of Δψm (Spinaci et al., 2005), since JC-1 forms J-aggregates spectrally distinguishable from dye monomers at the high concentrations reached in energized mitochondria of cells exposed to near-micromolar external concentrations of the dye (Yokosuka et al., 2013). The JC-1 (Beyotime Inc., Shanghai, China) was diluted 1 in 200 with the ultrapure water to give a working solution. Cells were harvested as mentioned above and collected in the centrifuge tube, and resuspended with 1mL diluted JC-1 per tube. Finally, the cells were resuspended with 1 mL JC-1 (1×) dyeing buffer, then processed by flow cytometry instrument in dual wavelength as soon as possible. 525-nm excitation wavelength and 590-nm emission wavelength can be used to detect JC-1 aggregates, and the corresponding 490-nm and 530-nm wavelengths can be used to detect the monomer (automatic quantitative count 10,000 neurons). Then the fluorescence ratio (JC-1 aggregates/monomer) was used to determine the extent of the mitochondrial depolarization.
Statistical analysis
Quantitative results were expressed as the mean ± SD. Statistical analysis was performed by employing SPSS16.0 software (SPSS, Chicago, IL, USA). One-way analysis of variance followed by the least significance difference test was performed to compare the difference between each group. Differences were considered to be significant when P values were less than 0.05.
Results
Effect of mild hypothermia on apoptosis in neurons subjected to OGD/reperfusion
Total apoptosis percentage (Q2 + Q4) was significantly higher in the control group than that in the 6-, 12- and 18-hour hypothermia and normal groups (P < 0.05), but not in the 24-hour hypothermia group. In the early (Q4) and later (Q2) apoptosis stages, apoptosis percentage was significantly higher in the control group than that in the 6-, 12- and 18-hour hypothermia and normal groups, but not in the 24-hour hypothermia group. These results indicate that mild hypothermia can reduce the apoptosis of neurons induced by OGD/reperfusion (Figure 2).
Figure 2.

Mild hypothermia reduced apoptosis in neurons subjected to oxygen-glucose deprivation/reperfusion.
(A) Under the immunofluorescence microscope, PI-positive cells appear red (the left image); FITC-positive cells exhibit green fluorescence (the middle one); the right image is the merge of the left one and the middle one. Scale bars: 50 μm. (B) Quantification of apoptosis cells determined by flow cytometric analysis. Quadrant 3 (Q3) represented the unchanged cells, quadrant 2 (Q2) represented death or late apoptosis of the cells, and quadrant 4 (Q4) represented early apoptosis of the cells. The sum of Q2 + Q4 indicates the total apoptosis in the group. *P < 0.05, vs. C group. Each experiment was repeated three times. One-way analysis of variance followed by the least significant difference test was used for difference comparison between each group. 24, 18, 12, 6 h: 24, 18, 12, 6 hours of hypothermia treatment; N: normal; C: control.
Effect of mild hypothermia on pro-apoptotic protein Bax expression in neurons subjected to OGD/reperfusion
The Bax protein expression in the control group was significantly higher than that in the 6- and 12-hour hypothermia and normal groups (P < 0.05). This indicates that mild hypothermia can reduce Bax expression in neurons induced by OGD/reperfusion (Figure 3).
Figure 3.
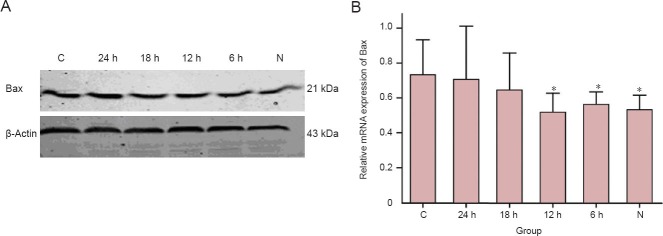
Mild hypothermia decreased pro-apoptotic protein Bax expression in neurons subjected to oxygen-glucose deprivation/reperfusion.
(A) Western blot analysis demonstrating expression patterns of indicated protein bax in the lysate of each cell group, with β-actin used as a loading control. (B) Data presented in the graph. The values were normalized with β-actin. *P < 0.05, vs. C group. Each experiment was repeated three times, and one-way analysis of variance followed by the least significant difference was used for difference comparison between each group. 24, 18, 12, 6 h: 24, 18, 12, 6 hours of hypothermia treatment; N: normal; C: control.
Effect of mild hypothermia on mitochondrial membrane potential of neurons following OGD/reperfusion
Mitochondrial membrane potentials were significantly higher in the hypothermia treated groups than that in the control group (P < 0.05), and there may be significant difference between 24-hour hypothermia group and each of the other groups but not between 6-, 12-, or 18-hour hypothermia groups. All hypothermia treated groups look significantly different from the normal group. These results suggest that mild hypothermia can stabilize mitochondrial membrane potentials and inhibit the reduction of mitochondrial membrane potential of neurons, which was induced by OGD/reperfusion (Figure 4).
Figure 4.

Mild hypothermia increased mitochondrial membrane potential (MMP) of neurons subjected to oxygen-glucose deprivation.
(A) If MMP depolarizes, JC-1 (Reers et al. (1991), a useful tool used for MMP), becomes a monomer (green), and if it polarizes, it becomes a compound (red). This voltage-sensitive dye follows the Nernst behavior, and increased uptake of this probe is caused by MMP. When illuminated with 490-nm and 525-nm light, JC-1 emits peaks at 530 nm (green) and 590 nm (red) respectively. The ratio between green and red depends on MMP. Scale bars: 50 μm. (B) Sequential change of normalized JC-1 fluorescence after 90 minutes of oxygen-glucose deprivation and reperfusion. The ratio was obtained by comparison with the value of the compound (red)/monomer (green) ratio in the group. *P < 0.05, vs. C group, one-way analysis of variance followed by the least significant difference test was performed to compare the difference between each group. 24, 18, 12, 6 h: 24, 18, 12, 6 hours of hypothermia treatment; N: normal; C: control.
Discussion
Apoptosis is a slowly progressive cell death and usually appears in the peri-infarct zone or transient global ischemia, which can cause ischemia/reperfusion damage (Tan et al., 2014; Wang et al., 2014). In the current study, OGD mimicked the ischemic condition (Zhou et al., 2013). Results from this study demonstrated that treatment with mild hypothermia decreased pro-apoptotic protein Bax expression levels, restored mitochondrial membrane potentials, and attenuated the apoptosis of cerebral cortex neurons. We compared the effectiveness of mild hypothermia for different time periods. The present results showed that after 6 and 12 hours of treatment with mild hypothermia, cell apoptosis was effectively reduced, suggesting that the effective time window might be 6 to 12 hours.
Mitochondria are key organelles in apoptosis and are necessary for many stimuli that trigger apoptosis. Mitochondrial membrane potential may play an important role in intracellular ionic homeostasis (Zhang et al., 2013). Mitochondria have been shown to act as a Ca2+ buffer (Green and Kroemer, 2004), tightly regulating the cytoplasmic Ca2+ concentration. Disruption of this buffer system may trigger a pathological increase in Ca2+. This could occur as the result of depolarization of mitochondrial membrane and trigger Ca2+ efflux from the matrix to the cytoplasm, the subsequent initiation of the apoptotic cascade. The reduction in Δψm is probably due to the activation of Bax proteins that are involved in mediating permeabilization of the mitochondrial outer membrane (Ghibelli and Diederich, 2010). Bax is a key component for induced cellular apoptosis through mitochondrial stress (Wei et al., 2001). Upon apoptotic stimulation, Bax forms oligomers and translocates from the cytosol to mitochondrial membranes (Jurgensmeier et al., 1998). Through interactions with pore proteins on the mitochondrial membrane, Bax increases the membrane's permeability, which leads to the release of cytochrome c from mitochondria, activation of caspase-9 and initiation of the caspase activation pathway for apoptosis (Narita et al., 1998; Ghibelli and Diederich, 2010). Results from this study demonstrated that treatment with mild hypothermia for 6 and 12 hours significantly inhibited the expression of Bax, and the corresponding mitochondrial membrane potential levels in the mild hypothermia-treated groups were higher than in the control group. These findings indicate that mild hypothermia may inhibit the expression of Bax, attenuate the reduction of mitochondrial membrane potential of neurons, inhibit mitochondrial mediated apoptosis pathway, and finally reduce the neuronal apoptosis which was induced by OGD/reperfusion.
These findings indicate that mild hypothermia is beneficial to attenuate apoptosis in neurons induced by OGD/reperfusion through reducing Bax expression and inhibiting mitochondrial mediated apoptosis pathway. Previous studies have shown that optimal duration of mild hypothermia was at least 3 hours in rats (Markarian et al., 1996; Ohta et al., 2007), but hypothermia was not maintained for more than 6 hours. In our study, we prolonged the mild hypothermia time to 24 hours to find the ceiling effect and acquired different results. We found that the effective time window may be 6 to 12 hours which gives the opportunity to initiate alternative long term therapy. More studies should be undertaken to further explore mechanisms and signaling pathways that are involved in the anti-apoptosis actions of mild hypothermia that may lead to a prospective neuroprotective therapy.
Footnotes
Conflicts of interest: None declared.
Copyedited by Dawes EA, Raye W, Li CH, Song LP, Zhao M
References
- Bigelow WG, Callaghan JC, Hopps JA. General hypothermia for experimental intracardiac surgery; the use of electrophrenic respirations, an artificial pacemaker for cardiac standstill and radio-frequency rewarming in general hypothermia. Ann Surg. 1950;132:531–539. doi: 10.1097/00000658-195009000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain Trauma Foundation. 2007. Guidelines for the management of severe traumatic brain injury [Google Scholar]
- Buchan A, Pulsinelli WA. Hypothermia but not the N-methyl-D-aspartate antagonist, MK-801, attenuates neuronal damage in gerbils subjected to transient global ischemia. J Neurosci. 1990;10:311–316. doi: 10.1523/JNEUROSCI.10-01-00311.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis D, Schwarz S, Aschoff A, Schwab S. Hemicraniectomy and moderate hypothermia in patients with severe ischemic stroke. Stroke. 2002;33:1584–1588. doi: 10.1161/01.str.0000016970.51004.d9. [DOI] [PubMed] [Google Scholar]
- Ghibelli L, Diederich M. Multistep and multitask Bax activation. Mitochondrion. 2010;10:604–613. doi: 10.1016/j.mito.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Gunn AJ, Wyatt JS. Hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2006;354:1643–1645. [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Green EJ, Dietrich WD, van Dijk F, Busto R, Markgraf CG, McCabe PM, Ginsberg MD, Schneiderman N. Protective effects of brain hypothermia on behavior and histopathology following global cerebral ischemia in rats. Brain Res. 1992;580:197–204. doi: 10.1016/0006-8993(92)90945-6. [DOI] [PubMed] [Google Scholar]
- Huang WC, Qiao Y, Xu L, Kacimi R, Sun X, Giffard RG, Yenari MA. Direct protection of cultured neurons from ischemia-like injury by minocycline. Anat Cell Biol. 2010;43:325–331. doi: 10.5115/acb.2010.43.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markarian GZ, Lee JH, Stein DJ, Hong SC. Mild hypothermia: therapeutic window after experimental cerebral ischemia. Neurosurgery. 1996;38:542–550. doi: 10.1097/00006123-199603000-00024. 551. [DOI] [PubMed] [Google Scholar]
- Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci U S A. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan JP, Morley PT, Vanden HT, Hickey RW, Kloeck WG, Billi J, Bottiger BW, Morley PT, Nolan JP, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Hickey RW, Carli P, Vanden HT, Atkins D. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- Ohta H, Terao Y, Shintani Y, Kiyota Y. Therapeutic time window of post-ischemic mild hypothermia and the gene expression associated with the neuroprotection in rat focal cerebral ischemia. Neurosci Res. 2007;57:424–433. doi: 10.1016/j.neures.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Pozarowski P, Grabarek J, Darzynkiewicz Z. Flow cytometry of apoptosis. Curr Protoc Cytom. 2003;(Chapter 7: Unit 7.19) doi: 10.1002/0471142956.cy0719s25. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Sendrowski K, Rusak M, Sobaniec P, Ilendo E, Dabrowska M, Bockowski L, Koput A, Sobaniec W. Study of the protective effect of calcium channel blockers against neuronal damage induced by glutamate in cultured hippocampal neurons. Pharmacol Rep. 2013;65:730–736. doi: 10.1016/s1734-1140(13)71052-1. [DOI] [PubMed] [Google Scholar]
- Spinaci M, De Ambrogi M, Volpe S, Galeati G, Tamanini C, Seren E. Effect of staining and sorting on boar sperm membrane integrity, mitochondrial activity and in vitro blastocyst development. Theriogenology. 2005;64:191–201. doi: 10.1016/j.theriogenology.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Tan RF, Xia AH, Wu XG, Cao NN, Li MM, Zhang TG, Wang YR, Yue ZL. Total Flavone of Hawthorn Leaf inhibits neuronal apoptosis in brain tissue of rat models of chronic cerebral ischemia. Zhongguo Zuzhi Gongcheng Yanjiu. 2014;18:7879–7883. [Google Scholar]
- Wang YR, Mao HF, Chen JQ. Effects of huwentoxin on tumor necrosis factor apoptotic pathway in the hippocampus of a rat model of cerebral ischemia. Zhongguo Zuzhi Gongcheng Yanjiu. 2014;18:5813–5818. [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SY, Wu YM, Ji Z, Gao XY, Pan SY. A modified technique for culturing primary fetal rat cortical neurons. J Biomed Biotechnol 2012. 2012 doi: 10.1155/2012/803930. 803930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Guo S, Zhang T, Li H. Hypothermia attenuates ischemia/reperfusion-induced endothelial cell apoptosis via alterations in apoptotic pathways and JNK signaling. FEBS Lett. 2009;583:2500–2506. doi: 10.1016/j.febslet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Goto H, Fujii H, Naruto T, Takeuchi M, Tanoshima R, Kato H, Yanagimachi M, Kajiwara R, Yokota S. Flow cytometric chemosensitivity assay using JC-1, a sensor of mitochondrial transmembrane potential, in acute leukemia. Cancer Chemother Pharmacol. 2013;72:1335–1342. doi: 10.1007/s00280-013-2303-x. [DOI] [PubMed] [Google Scholar]
- Zhang ZQ, Song JY, Jia YQ, Li PT, Pan YS. Pathological changes of brain tissue in a rat model with coexistence of hyperlipidemia and cerebral ischemia. Zhongguo Zuzhi Gongcheng Yanjiu. 2013;17:5981–5987. [Google Scholar]
- Zhou T, Jiang J, Zhang M, Fu Y, Yang Z, Jiang L. Protective effect of mild hypothermia on oxygen-glucose deprivation injury in rat hippocampal neurons after hypoxia. Mol Med Rep. 2013;7:1859–1864. doi: 10.3892/mmr.2013.1410. [DOI] [PubMed] [Google Scholar]