

INTRODUCTION
Sepsis, a systemic inflammatory response syndrome caused by severe infection, is still a tremendous burden for health-care systems and results in more than 225,000 deaths annually in the United States1. No pharmacological agents have been shown to effectively change the outcomes2.
Acetylation of histone is an important epigenetic mechanism that governs amplitude of the immune signaling by controlling the chromatin structure, accessibility of transcription factors to DNA, and gene transcription. Regulation of this process needs the opposing actions of two families of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). It is found that dysregulated HDAC activity is linked to the pathogenesis of inflammatory and autoimmune diseases3. So far, 18 HDAC isoforms have been identified in humans and mice and grouped into four classes 4. Classical HDACs (class I, II and IV) are Zn2+ dependent hydrolases, while the class III sirtuins are NAD+-dependent. Class I HDACs (HDAC1, 2, 3 and 8) play a role in cell survival and proliferation. Class II HDACs, subdivided into class IIa (HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10) based on domain organization 5, may have tissue-specific roles 6. Recently, HDAC6 has become an important target for anti-cancer drug development, and inhibition of HDAC6 was also shown to have therapeutic potential to ameliorate injury of central nervous system7.
MS-275 is a HDAC class I inhibitor with selectivity for HDAC1, 2, and 3. Tubastatin A is a newly synthesized selective inhibitor of HDAC class IIb with high selectivity for HDAC6 8. Suberoylanilide hydroxamic acid (SAHA, vorinostat) is a broad-spectrum histone deacetylase inhibitor (HDACI) with a selectivity for HDAC1, 2, 3 and 6 9. Our laboratory has previously demonstrated that administration of SAHA improves survival in rodent models of lipopolysaccharided (LPS)-induced endotoxemia and cecal ligation and puncture (CLP)-induced septic shock 10, 11. However, this HDACI was also found to increase host susceptibility to bacterial infection due to cell apoptosis 12. We hypothesized that targeting different HDACs could have different effect on animal survival in a mouse model of cecal ligation and puncture (CLP)-induced sepsis. In the present study, we first determined that inhibitor of HDAC6 (Tubastatin A) rather than that of HDAC1, 2, and 3 significantly prolonged animal lives in the CLP model. We then assessed impact of HDAC6 inhibition on production of some key pro-inflammatory cytokines, organ (liver) injury, and immune cell apoptosis. Our findings suggested that selective inhibition of HDAC6 has a substantial advantage for sepsis treatment.
METHODS
Cells Culture and Reagents
Mouse primary splenocytes and RAW 264.7 murine macrophages (American Type Culture Collection, Manassas, VA) were cultured in Dubelcco’s modified Eagle’s medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin (Invitrogen, Grand Island, NY) at 37 °C and 5% CO2.
Sepsis Model: Cecal Ligation and Puncture (CLP)
Male C57BL/6J mice (about 18–26 g), purchased from the Jackson Laboratory, were housed for 3 days before manipulations. The murine CLP model was used to induce fecal peritonitis as described previously 11. Sham-operated animals were handled in the same manner without the cecum ligation and puncture. This protocol was approved by the Animal Review Committee in our institute.
Administration of HDACI and Experimental Design
In the survival experiment, mice were randomly subjected to three groups, and received the following treatment: (1) intra-peritoneal MS-275 (70 mg/kg) dissolved in dimethyl sulfoxide (DMSO) (1 μl/g), (2) Tubastatin A (70 mg/kg) dissolved in DMSO, or (3) vehicle DMSO, 1 h after CLP (n=7–12/group). All mortality or survival was recorded for up to 10 days post-procedure.
In the non-survival experiment, animals were randomly assigned to the following three groups (n = 24/group): (a) Sham-operated animals (SHAM); (b) vehicle treated animals after CLP (CLP+DMSO), and (c) Tubastatin A treated animals after CLP (CLP + Tubastatin A). Sham-operated animals were subjected to laparotomy and intestinal manipulation, but the cecum was neither ligated nor punctured. At the time of sacrifice [3h, 24 h, 48 h, and 10 d after CLP (n= 4–7/group/time point)], abdominal cavity was opened and irrigated with 1 mL normal saline, which was collected for analysis, and blood samples were collected by cardiac puncture. Liver tissue was harvested 24 h after CLP and fixed in 10% buffered formalin for histological analysis.
Cytokine Measurements
Concentrations of tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 in the peritoneal fluid, plasma, or cell culture supernatant were measured using the Quantikine Enzyme-Linked Immunosorbent Assay (ELISA) Kit (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions.
Histological Analysis
Twenty-four hours after CLP, tissue samples of liver were harvested for histological analysis as our previous study 11. Briefly, the liver tissue was embedded in paraffin, sliced into 5-μm sections and stained with hematoxylin and eosin (H&E). Hepatocellular necrosis, hemorrhage/congestion, parenchyma inflammation, sinusoidal inflammation, and degenerative changes were assessed by a blinded pathologist. Each parameter of liver injury was graded on a scale of 0–3, with 0 meaning “absent,” 1 meaning “mild,” 2 meaning “moderate,” and 3 meaning “severe.” The total injury score was expressed as the sum of the scores for all parameters 11.
Bacteria Load Determination
Whole blood samples were obtained 48 h after sham operation or CLP by cardiac puncture. Samples were diluted in sterile saline, plated on tryptic soy agar (BD Difco, Franklin Lakes, NJ), and incubated at 37 °C. The number of bacterial colonies was assessed 24 hours later. Bacteria count was calculated as colony forming units per milliliter, and log transformation of these values was then used for further analysis 13.
Phagocytosis Assay
Mouse primary splenocytes were prepared as previously described 14. Phagocytosis was then assessed using a pHrodo™ E. coli BioParticles® Phagocytosis Kit (Invitrogen, Grand Island, NY) 15, 16. Cells were seeded in a 96-well assay plate, and treated at 37 °C in the absence or presence of LPS (1μg/mL) or Tubastatin A (10 μM) for 3 h before removing the medium and adding 100 μL uptake buffer (HBSS buffered with 20mM HEPES, pH 7.4) containing 1 mg/mL pHrodo E. coli bioparticles. The assay plate was incubated for 1 h at 37 °C in a humidified chamber, and then analyzed on a SpectraMax plate reader (Molecular Devices, Sunnyvale, CA). Net phagocytosis was calculated by subtracting the base-line fluorescence from wells containing 100 ul of 1 mg/mL pHrodo E. coli bioparticles but no cells.
Western Blot Analysis
RAW264.7 macrophages were harvested 3 and 6 h after treatment with LPS in the absence or presence of Tubastatin A. Sham (no LPS, no Tubastatin A) macrophages served as control. Equal amounts of whole cell lysate samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Western blots were employed and quantitative analysis of detected bands was performed as described previously 11.
Flow Cytometry
Cell apoptosis was measured using an annexin V-fluorescein isothiocyanate (FITC) detection kit (Abcam, Cambridge, MA). RAW264.7 Macrophages were treated with LPS in the absence or presence of another HDAC6 inhibitor Tubacin for 3, 6 and 9 h. Sham (no LPS, no Tubacin) macrophages served as control. Cells were then harvested, washed in ice-cold phosphate buffered saline, resuspended in binding buffer, and incubated with annexin V-FITC for 5 min at room temperature. Cells were analyzed by flow cytometry, and apoptotic cells were defined as positive for annexin V-FITC and negative for propidium iodide (PI) staining 17.
Statistical Analysis
Results were expressed as mean ± SEM. Student’s t-test was used to compare the differences between two groups. Differences between 3 or more groups were assessed using one way analysis of variance (ANOVA) followed by Bonferroni post hoc testing for multiple comparisons. Kaplan-Meier method was used for survival, and differences were analyzed using log-rank test. Analyses were performed using GraphPad Prism. P values of 0.05 or less were considered significant.
RESULTS
Tubastatin A significantly improves survival in CLP-induced lethal septic model
In this CLP-induced lethal sepsis model, all mice in either DMSO vehicle group or MS-275 group died within 3 days (0% survival). However, Tubastatin A-treated animals displayed significantly higher long-term survival compared to the DMSO or MS-275 group (66.7% vs. 0% survival, p < 0.001; Figure 1). We previously showed that SAHA treatment significantly improved survival (40% survival)11. Compared to this SAHA result, Tubastatin A treatment for an improvement in animal survival is as good as SAHA.
Figure 1. Tubastatin A protects mice against severe sepsis and septic shock-induced lethality.

Mice were intraperitoneally given Tubastatin A (70mg/kg), MS-275 (70mg/kg), or vehicle (DMSO) 1 h after CLP (n=7–12 animals/group). Treatment with Tubastatin A significantly improved long-term survival compared to DMSO vehicle group (66.7% vs. 0% survival, p < 0.001), while MS-275 did not show protective effects against sepsis-induced mortality. CLP: cecal ligation and puncture; Tub.A: Tubastatin A.
Tubastatin A decreases cytokine levels in peritoneal fluid and circulation
In the sham group, TNF-α levels in the peritoneal fluid and blood were low at all time points (8.7 ± 0.7 and 11.3 ± 2.3 pg/mL, respectively). Similarly, IL-6 levels in peritoneal fluid and blood were hardly detected (6.3 ± 2.0 and 14.9 ± 6.3 pg/mL, respectively). However, the levels of TNF-α at 3 and 24 h and the levels of IL-6 at 48 were increased obviously after the CLP insult.
Tubastatin A significantly attenuated the CLP-induced increase of TNF-α at 3 and 24 h in the peritoneal fluid (6.3 ± 7.7 pg/mL vs. 79.5 ± 19.0 at 3 h, p < 0.05; 53.5 ± 19.6 pg/mL vs. 463.3 ± 48.4 at 24 h, p < 0.01; Figure 2A) and plasma (7.3 ± 2.0 pg/mL vs. 9.9 ± 3.5 at 3 h, p < 0.05; 24.5 ± 5.0 pg/mL vs. 298.3 ± 24.6 at 24 h, p < 0.001; Figure 2A). Ten days after Tubastatin A treatment, TNF-α levels in the peritoneal fluid and blood returned to nearly normal (2.1 ± 1.2 and 7.3 ± 2.1 pg/mL, respectively; Figure 2A). In addition, Tubastatin A significantly suppressed an increase of IL-6 levels in the peritoneal fluid (71.7 ± 3.7 pg/mL vs. 646.4 ± 81.8, p < 0.01; Figure 2B) and plasma (88.1 ± 65.0 pg/mL vs. 521.5 ± 94.3, p < 0.05; Figure 2B) 48 h after CLP.
Figure 2. Tubastatin A decreases levels of TNF-α and IL-6 in peritoneal fluid and plasma during severe sepsis and septic shock.<.

br>The peritoneal fluid and blood were collected at different time points to measure concentrations of TNF-α (Figure 2A; at 3 h, 24 h, and 10 d after CLP) and IL-6 (Figure 2B; at 48 h after CLP) using ELISA kits (means ± SEM, n = 4 animals/group). Tub.A: Tubastatin A
Tubastatin A decreases TNF-α and IL-6 levels in culture supernatant of mouse primary splenocytes in presence of LPS ex vivo
In Sham group, only low levels of TNF-α and IL-6 were detected in cell culture supernatant of the primary splenocytes. Treatment of the cells with LPS for 6 h markedly increased secretion of these cytokines. However, Tubastatin A significantly attenuated the LPS-induced production of TNF-α and IL-6 (15.1 ± 1.4 pg/mL vs. 68.1 ± 6.4, 4.6 ± 1.4 pg/mL vs. 61.3 ± 12.1, respectively, p < 0.01; Figure 3).
Figure 3. Tubastatin A decreases TNF-α and IL-6 production in cell culture supernatant of primary splenocytes insulted by LPS.

Concentrations of TNF-α and IL-6 in culture supernatant of primary splenocytes were determined by ELISA at 6 h after LPS treatment in the absence or presence of Tubastatin A. Untreated macrophages served as control (means ± SEM, n = 4/group). Tub.A: Tubastatin A
Tubastatin A decreases acute liver injury
Sham-operated animals had normal liver histology, but the liver at 24 h after CLP showed increased parenchyma inflammation and degenerative changes. These pathological alterations were attenuated by Tubastatin A treatment, with significantly lower acute liver injury scores (2.6 ± 0.3 vs. 5.1 ± 0.8, p < 0.05; Figure 4).
Figure 4. Tubastatin A protects animals from acute liver injury 24 h after CLP (H&E, magnification 40×).
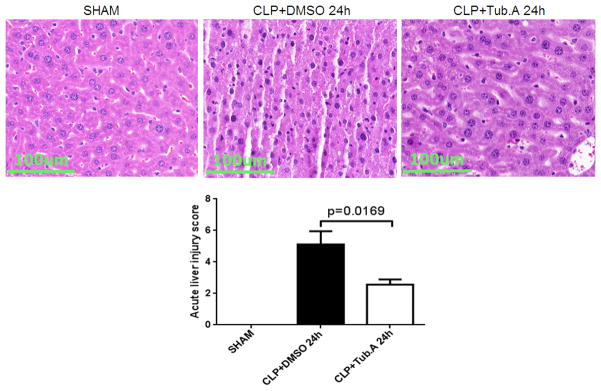
Mice were intraperitoneally administered 70mg/kg Tubastatin A, or vehicle (DMSO) 1 h after CLP. Twenty-four hours after operation, liver tissues were obtained. Representative images were chosen from different experimental groups. Semi-quantitative pathology scores were determined by a scoring system as described in Materials and Methods (means ± SEM, n = 4–6 animals/group). Tub.A: Tubastatin A
Tubastatin A increases bacteria clearance in circulation in vivo and phagocytosis of mouse primary splenocytes ex vivo
Compared with vehicle treated animals, Tubastatin A treated animals had remarkably lower bacteria load in circulation 48 h after CLP (2.5 ± 0.9 vs. 6.2 ± 0.3 log10 CFU/mL, p < 0.01; Figure 5A). Moreover, Tubastatin A treatment increased phagocytosis of splenocytes markedly (28.8 ± 1.4 RFU vs. 18.5 ± 2.2, p < 0.01; Figure 5B).
Figure 5. Tubastatin A increases blood bacteria clearance in vivo and enhances phagocytosis of mouse primary splenocytes ex vivo.

(A) Whole blood samples were obtained 48 h after operation by cardiac puncture. Samples were diluted in sterile saline, plated on tryptic soy agar, and incubated at 37 °C. The number of bacterial colonies was assessed 24 hours later. Bacteria number was calculated as colony forming units per milliliter, and data were log transformed for presentation (Means ± SEM, n = 6 animals/group). CFU: colony forming units. (B) Mouse primary splenocytes were treated as described in material and methods. Cells were then incubated with 1 mg/ml pHrodo E. coli bioparticles and the fluorescence analyzed on a plate reader. Mean values are relative fluorescence units (RFU), calculated from four wells per group subtracting the base-line fluorescence from wells containing pHrodo E. coli bioparticles but no cells. (Means ± SEM, n = 4/group). Tub.A: Tubastatin A
HDAC6 Inhibitors inhibits cell apoptosis of RAW264.7 macrophages in vitro
Tubastatin A decreases cleaved caspase-3 expression in macrophages at 3 and 6 h (23.8 ± 5.2 vs. 97.2 ± 4.2 and 52.6 ± 6.0 vs172.4 ± 9.6, respectively, p < 0.001; Figure 6).
Figure 6.

(A) Tubastatin A decreases cleaved caspase-3 protein in macrophages after LPS insult. Macrophages were harvested at 3 and 6 h after treatment with LPS in the absence or presence of Tubastatin A. Sham (no LPS, no Tubastatin A) macrophages served as a control. Whole cell lysates were subjected to Western Blot with anti-cleaved Caspase-3 and anti-actin antibodies. Specific bands were then quantified by densitometry (means ± SEM, n = 4/group). Tub.A: Tubastatin A. (B) Schematic depiction of proposed mechanism in the present study. Treatment with Tubstatin A, on one hand, could decrease production of key pro-inflammatory cytokines (TNF-α and IL-6), liver injury, and microphage apoptosis; on the other hand, it could increase bacterial clearance and splenocyte phagocytosis.
DISCUSSION
In the present study, we investigated the effects of selective HDAC inhibitors with distinct isoform selectivity, and found: (1) The HDAC6-selective inhibitor Tubastatin A significantly improves long-term survival, while MS-275, an inhibitor of HDAC1, 2, and 3, is not protective against septic shock; (2) Tubastatin A significantly attenuates local and systemic proinflammatory cytokines in vivo, as well as the LPS-stimulated cytokines production from primary splenocytes in vitro; (3) Tubastatin A treated animals display attenuated acute liver injury; (4) the HDAC6 inhibitor increases blood bacteria clearance and phagocytosis of immune cells, and (5) Tubastatin A decreases macrophage apoptosis (see Figure 6B for proposed mechanism).
It has been known that HDAC6 is a unique HDAC and predominantly localized in the cytoplasm, where it associates with non-histone substrates, such as heat shock protein (HSP) 90, α-tubulin and cortactin 7. Overexpression of HDAC6 leads to tubulin deacetylation and increased cell motility 18. Inhibition of HDAC6 activity increases acetylation of α-tubulin and HSP90, reduces cellular motility and induces degradation of client proteins of HSP90 19. So far, everal client proteins of HSP90 have been reported, such as Bcr-Abl, Raf-1, Akt, HER2/Neu (ErbB2), interleukin-1 receptor associated kinase 1(IRAK1), and hypoxia-inducible factor-1α 20. Our laboratory has recently shown that SAHA treatment increases acetylation of HSP90 in LPS-stimulated macrophages, and results in dissociation and degradation of IRAK1. The IRAK1 degradation then decreases nuclear translocation of Nuclear Factor-κB (NF-κB), leading to attenuation of key pro-inflammatory cytokines 21. Triantafilou in 2004 reported that HSPs are involved in the innate recognition of bacterial products 22. The association of TLR4 with HSP70 and HSP90 following LPS stimulation was found both on the cell surface and in the cells. HSP70 and HSP90 form a cluster with TLR4 within lipid micro-domains. In addition, HSP70 and HSP90 seem to be involved in TLR4/LPS trafficking and targeting to the Golgi apparatus 22. Therefore, instability of HSP90 after acetylation modulated by HDAC6 inhibitors may impair pathogen recognition as well.
Inhibition of HDAC6 by Tubastatin A could reduce expression of hypoxia-inducible factor-1α (HIF-1α). We have recently demonstrated that SAHA attenuates HIF-1α-mediated inflammatory pathway in macrophages, and suppresses hypoxia-induced release of pro-inflammatory nitric oxide and TNF-α 23. HIF-1α is a key pro-inflammatory transcription factor in TLR4-dependent inflammatory responses in macrophages, and is essential for myeloid-dependent inflammation in vivo 24. HDAC6 has been shown to interact with HIF-1α and regulate its function. Inhibition of HDAC4 and HDAC6 by small interfering RNA can reduce protein expression and transcriptional activity of HIF-1α in a renal cell carcinoma cell line 25.
The inhibition of macrophage apoptosis may be another explanation for the improved survival outcomes after treatment with HDAC6 inhibitors in our lethal septic model. Macrophages, as the most efficient pathogen scavengers and the predominant source of inflammatory cytokines, are critical effector cells contributing to the altered innate immune response against infection. Severe sepsis has been found to be associated with progressing macrophage dysfunction and cell death 26. It is not clear how HDAC6 affects phagocytosis and bacteria clearance. Inhibition of HDAC6 likely reduces the apoptosis of macrophages, improves their ability to phagocytize foreign pathogens and decreases blood bacteria load. HDAC6 inhibitors might also decrease bacteria translocation from ischemia/necrosis sites, by decreasing systemic pro-inflammatory chemo-attractants.
During sepsis, excessive production of pro-inflammatory cytokines promotes migration of leukocytes, lymphocytes, and platelets to the infected areas, leading to endothelial damage, increased micro-vascular permeability, platelet aggregation, local blood flow reduction, and ischemia/reperfusion (I/R) injury, finally results in multiple organ damage. We previously found that SAHA treatment attenuates acute liver injury during severe sepsis 11. We also demonstrated that SAHA significantly decreases LPS-induced expression of the pro-inflammatory MAPK, phosphorylated p38, phosphorylated extracellular signal-regulated kinase, myeloperoxidase, and IL-6 in the liver, while increasing levels of the anti-inflammatory IL-10 27, 28. The attenuation of activation of MAPK and phosphorylated extracellular signal-regulated kinase, subsequent decrease in NF-κB-dependent gene transcription, and alteration of inflammatory markers might account for the protective role of HDAC6 inhibition in reducing acute liver injury in the lethal septic model.
Notably, enthusiasm for pan-HDAC inhibition has been tempered by its toxicity toward host 12. For example, HDAC1 and HDAC2 can promote B cell proliferation 29. When HDAC1 and HDAC2 are ablated, B cell development is blocked. The pre-B cells are then stopped in G1 phase accompanied by the induction of apoptosis 29. In addition, HDAC1 and HDAC2 are essential for normal T cell development and genomic stability in mice. Conditional deletion of HDAC1 in T cells results in increased Th2 cytokine production as well as enhanced airway inflammation 30. Meanwhile, Class I HDACs repress TNF-induced NF-kB-dependent gene expression 31, and promote interferon signaling. Therefore, inhibitors of Class I HDAC may have cytotoxicity for immune cells, impair lymphocyte development, amplify production of TLR/NF-kB-inducible inflammatory mediators, and compromise anti-microbial responses. In contrast, HDAC6-deficient mice are viable and develop normally 31. Lymphoid development is normal after HDAC6 deletion, with moderately-affected immune responses. HDAC6-deficient mouse embryonic fibroblasts have apparently normal microtubule organization and stability, and the acetylation of HSP90 is increased correlating with its impaired function. Accordingly, this data demonstrate that HDAC6 deletion is not detrimental to normal mammalian development. Although the mechanism how HDAC6 inhibitors prevent immune cell apoptosis is not clear, antiapoptotic property of these inhibitors may render them advantageous to treat severe sepsis and septic shock, compared to the first generation non-selective HDACI.
The present study has demonstrated several possible mechanisms of Tubastatin A action (see Figure 6B for summary). Treatment with Tubstatin A could down-regulate key pro-inflammatory cytokines, prevent liver damage, and decrease macrophage apoptosis. Moreover, Tubastatin A can also enhance bacterial clearance and splenocyte phagocytosis. We must acknowledge that the study has certain limitations. For logistical reasons, we only measured selected cytokines and explored limited pathways. Many more mechanisms are likely to be influenced by Tubastatin A treatment. Similarly, we only studied liver injury although there are other organs which are likely affected by this treatment. Mechanism of action of Tubastatin A needs to be explored.
In conclusion, we have demonstrated that Tubastatin A, an inhibitor of HDAC6, can improve survival significantly in a lethal polymicrobial sepsis model. The survival advantage is associated with an attenuation of local and systemic proinflammatory cytokines, protection against distant organ injury, enhancement in bacterial clearance and phagocytosis, and inhibition of immune cell apoptosis. Although the fundamental molecular and cellular signaling events still require further investigation, HDAC6 may represent a novel and promising therapeutic target for septic shock.
Acknowledgments
This work was funded by a grant from NIH RO1 GM084127 to HBA. Data was presented at the 99th Annual Clinical Congress of American College of Surgeons in Washington, DC (October, 2013).
Footnotes
Conflict of Interest Disclosure: R.M. has financial interests in SHAPE Pharmaceuticals and Acetylon Pharmaceuticals. R.M.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Authors’ contribution: Y.L. designed this study, for which H.B.A. secured funding. T.Z. performed experiments, collected and analyzed data. B.L. and X.D. provided experimental support. T.Z. and Y.L. wrote the manuscript, which was critically revised by Y.L., I.H., R.M., and H.B.A. All authors read and approved the final manuscript.
References
- 1.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, 2nd, Kreisel D, Krupnick AS, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA : the journal of the American Medical Association. 2011;306(23):2594–605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, et al. Drotrecogin alfa (activated) in adults with septic shock. The New England journal of medicine. 2012;366(22):2055–64. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 3.Hawtree S, Muthana M, Wilson AG. The role of histone deacetylases in rheumatoid arthritis fibroblast-like synoviocytes. Biochemical Society transactions. 2013;41(3):783–8. doi: 10.1042/BST20130053. [DOI] [PubMed] [Google Scholar]
- 4.Suliman BA, Xu D, Williams BR. HDACi: molecular mechanisms and therapeutic implications in the innate immune system. Immunology and cell biology. 2012;90(1):23–32. doi: 10.1038/icb.2011.92. [DOI] [PubMed] [Google Scholar]
- 5.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends in immunology. 2011;32(7):335–43. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Molecular cancer research : MCR. 2007;5(10):981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 7.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends in cell biology. 2008;18(6):291–7. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. Journal of the American Chemical Society. 2010;132(31):10842–6. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nature chemical biology. 2010;6(3):238–43. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, Jin G, Lu J, deMoya MA, Velmahos GC, et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;148(2):246–54. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao T, Li Y, Liu B, Liu Z, Chong W, Duan X, Deperalta DK, Velmahos GC, Alam HB. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surgery. 2013;154(2):206–13. doi: 10.1016/j.surg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117(4):1205–17. doi: 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- 13.Jung E, Perrone EE, Liang Z, Breed ER, Dominguez JA, Clark AT, Fox AC, Dunne WM, Burd EM, Farris AB, et al. Cecal ligation and puncture followed by methicillin-resistant Staphylococcus aureus pneumonia increases mortality in mice and blunts production of local and systemic cytokines. Shock. 2012;37(1):85–94. doi: 10.1097/SHK.0b013e3182360faf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao T, Li Y, Liu B, Bronson RT, Halaweish I, Alam HB. Histone deacetylase III as a potential therapeutic target for the treatment of lethal sepsis. The journal of trauma and acute care surgery. 2014 doi: 10.1097/TA.0000000000000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, Gallan AJ, Shevchenko PD, Bindokas VP, Nelson DJ. Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. The Journal of biological chemistry. 2009;284(51):35926–38. doi: 10.1074/jbc.M109.057372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger SB, Romero X, Ma C, Wang G, Faubion WA, Liao G, Compeer E, Keszei M, Rameh L, Wang N, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nature immunology. 2010;11(10):920–7. doi: 10.1038/ni.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawada J, Zou P, Mazitschek R, Bradner JE, Cohen JI. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. The Journal of biological chemistry. 2009;284(25):17102–9. doi: 10.1074/jbc.M809090200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. The Journal of biological chemistry. 2005;280(29):26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 20.Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer science. 2007;98(10):1536–9. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong W, Li Y, Liu B, Zhao T, Fukudome EY, Liu Z, Smith WM, Velmahos GC, deMoya MA, Alam HB. Histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates Toll-like receptor 4 signaling in lipopolysaccharide-stimulated mouse macrophages. The Journal of surgical research. 2012;178(2):851–9. doi: 10.1016/j.jss.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Triantafilou M, Triantafilou K. Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochemical Society transactions. 2004;32(Pt 4):636–9. doi: 10.1042/BST0320636. [DOI] [PubMed] [Google Scholar]
- 23.Chong W, Li Y, Liu B, Liu Z, Zhao T, Wonsey DR, Chen C, Velmahos GC, deMoya MA, King DR, et al. Anti-inflammatory properties of histone deacetylase inhibitors: a mechanistic study. The journal of trauma and acute care surgery. 2012;72(2):347–53. doi: 10.1097/TA.0b013e318243d8b2. discussion 53–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer research. 2006;66(17):8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 26.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6 (Suppl 1):S27–38. [PubMed] [Google Scholar]
- 27.Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, deMoya M, Alam HB. Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. The Journal of surgical research. 2010;163(1):146–54. doi: 10.1016/j.jss.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hotchkiss RS, Opal S. Immunotherapy for sepsis--a new approach against an ancient foe. The New England journal of medicine. 2010;363(1):87–9. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes & development. 2010;24(5):455–69. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. Journal of immunology. 2010;185(6):3489–97. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi YS, Jeong S. PI3-kinase and PDK-1 regulate HDAC1-mediated transcriptional repression of transcription factor NF-kappaB. Molecules and cells. 2005;20(2):241–6. [PubMed] [Google Scholar]