

INTRODUCTION
Infection and sepsis are leading causes of morbidity and death in trauma patients that survive their initial injuries 1. Hospital-associated infections (HAI) occur after both trauma and elective surgery. The Centers for Disease Control and Prevention have estimated roughly 1.7 million HAI contribute to about 100,000 deaths per year. Thus HAI are a major public health burden. The lung acts as an interface between the body and external microbes, and pneumonia (PNA) is the most common infection in injured patients. Health care associated PNA (HCAP) occurs in up to 30% of intubated patients with an injury severity score (ISS) ≥ 16 2. Historically HCAP after injury has been attributed to chest injury, splinting and atelectasis that promote inoculation of the lung by oropharyngeal flora that are often resistant to antibiotics. But no mechanistic studies link atelectasis to inflammation and pneumonia, so this paradigm must be reevaluated 3, 4.
Although there are no proven mechanistic links between traumatic tissue injury and subsequent susceptibility to infection, tissue injury releases Damage Associated Molecular Patterns (DAMPs). Mitochondria are evolutionary endosymbionts that bear close molecular similarities to bacterial Pathogens Associated Molecular Patterns (PAMPs). Our work 3 showed that mitochondrial DAMPs (mtDAMPs) are critical contributors to systemic inflammation and our recent work shows mtDAMPs can suppress innate immune responses in the lung 4. Neutrophils (PMN) are the predominant innate immune effector. They migrate to the lung by chemotaxis (CTX) following a series of G-protein coupled chemoattractants. Trauma causes release of mtDAMPs like mtDNA and formyl peptides (FP) 4,5. We have shown mtDAMPs act like bacteria in that they activate PMN by mobilizing Ca2+ flux through formyl-peptide receptors (FPR-1/2) 6. Moreover, we have also shown that PMN chemoreceptors can be suppressed by injury.7 This suppression is effected by exposure to FP 3.
Now therefore, we hypothesized that DAMPs released acutely from fracture injuries might divert PMN to sites of injury and thus leave the lung vulnerable to bacteria. Moreover, since mtDAMPs might activate PMN prematurely while still in the circulation, we thought they might diminish PMN bactericidal functions at authentic sites of infection. Last, since mtDAMPs contain FP, we hypothesized that PMN diversion and de-activation by mtDAMPs might reflect premature activation by FP in the circulation through FPR-1 and FPR-2. We investigated these hypotheses through a number of animal and human models that are designed to model bacterial challenge to the lung after trauma together in a translational fashion.
MATERIALS AND METHODS
Materials
Heparin and 0.9% sodium chloride were purchased from Hospira (Lake Forest, IL). Ethylene glycol-bis (2-aminoethylether)-N,N,N,N-tetraacetic acid (EGTA), protease inhibitor cocktail, and dimethyl sulfoxide were purchased from Sigma (St Louis, MO). Fura-2-AM, anti-human formyl peptide receptor-1 (FPR-1) and anti-FPR-2 antibodies were purchased from R&D (Minneapolis, MN). Other supplies were obtained as below.
Research Compliance
Animal Protocols
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Beth Israel Deaconess Medical Center. Male Sprague-Dawley rats (250–350 g; Charles River, Boston, MA) were acclimatized under barrier-sustained conditions (25°C, 12 hour light/dark cycles, water and chow ad libitum) for 48 hours before experiments. Anesthesia was induced with ketamine (40 mg/kg) and xylazine (5 mg/kg).
Human specimen acquisition
The Institutional Research Board (IRB) of Beth Israel Deaconess Medical Center approved all human blood sampling. Blood specimens were collected via indwelling catheters or by venipuncture after informed consent either from the donor or their authorized representative. Human cancellous bone was obtained from the neck of femurs removed at the time of fracture repair. A portion of each specimen that was not used by Pathology was obtained and used a under waiver of consent for discarded materials.
Pseudo-fracture (PsFx)
Authentic fracture models raise animal welfare concerns in survival studies. We therefore chose to create a modification of the pseudofracture (PsFx) model described by Pape.8 This has been shown to mimic the immunologic environment of extremity fractures. Rats were sacrificed by cardiac puncture under anesthesia. Femurs were extracted under sterile conditions. The bone was weighed, and an equal amount of muscle was then removed from the thigh. The specimens were kept separately on ice. Femurs were crushed in 2 mL saline using a mortar and pestle, then homogenized for 2 minutes and finally passed through a 70 μm cell strainer (BD Biosciences, Bedford, MA). The suspension was collected and aliquotted. Thigh muscle was minced with scissors in 2 mL saline and homogenized at 1000 rpm. Specimens were put through a 70 μm cell strainer. This suspension was also collected, aliquotted and stored at −20ºC. Half an hour prior to injection, PsFx material was placed at room temperature and the suspensions of crushed femur and muscle were mixed using a 1cc syringe with an 18G needle. PsFx material (0.2 mL) was then injected into the mid-lateral portion of the thigh under general anesthetic 8–10. For [Ca2+]i measurement using human PMN, bone was collected from patients with femoral neck fractures as described above. Cancellous bone was extracted and the supernatants prepared by the same methods as the rat PsFx injectate.
Pulmonary contusion (PC)
The PC model was developed by Badami and Livingston to simulate a 70 kg man suffering a unilateral blunt lung contusion during a 20 mph motor vehicle accident 11. Rats were anaesthetized and restrained on a board in the supine position. A 12.5 g metal slug placed in a 2 cm plastic cup was fixed to the rat’s right lateral chest and the chest was percussed by firing a Craftsman® EasyFire™ Forward Action™ Stapler against the slug. The injury created was a clinically relevant, non-lethal pulmonary contusion 4, 11.
Intra-tracheal (i.t.) bacterial injection
S. aureus were grown overnight in 100 mL of Tryptic Soy Broth. The next morning 2 mL of bacteria stock was added to 100 mL of fresh broth and incubated for 90 minutes. The medium was the centrifuged at 4000 rpm for 10 minutes, the pellet was re-suspended in saline and titrated to an OD of 0.3 at 600 nM.
Rats were anaesthetized and a small incision was made through the skin of the neck. The sublingual glands, sternothyroid and sternocleidomastoid muscles were gently dissected to expose the trachea. A 30G needle was carefully inserted between the cartilaginous rings and 250 μL of bacterial suspension (mean 3.2×108 bacteria) was slowly infused. Following inoculation the neck was closed with sterile stainless steel staples 12. After 16 hours rats were anesthetized and then sacrificed by cardiac puncture. The lungs were then excised, the trachea was cannulated and the lungs were washed 5 times with one 2.5 mL aliquot of cold saline. This was then repeated with a second 2.5 mL aliquot of saline. The pooled bronchoalveolar lavage fluid (BALF) was centrifuged at 300g for 10 minutes. Supernatants were removed, and the pellets resuspended in 100 μL of saline: 20–30 μL were removed for Cytospin where the PMN were counted using a HEMA-3 stain (Fisher Scientific, Kalamazoo, MI). To count bacteria, 20 μL of the BALF was plated on Tryptic Soy agar, incubated overnight and colony forming units (CFU) counted.
PMN Isolation
Whole blood was collected from trauma patients or volunteers in two 15mL tubes pre-treated with heparin (150U/tube). PMN isolation was carried out according to our previously reported methods using a brief hypotonic lysis on ice to remove any contaminating red blood cells 3, 13. PMN were obtained from 32 major trauma patients (Mean ISS = 27±10 [SE]). Each patient was paired with an age, sex, ethnicity-matched volunteer. Samples were obtained Day 1 (14±1 h), Day 3 and Day 7 after injury. All patients survived and completed the collection protocol.
Ca2+ Spectroflurometry
Following PMN separation intracellular Ca2+ was determined by fura fluorescence at 505 nm using 340/380 nm dual wavelength excitation as per our reported methods 14. Cells were divided into aliquots and placed on ice. Cuvettes were prepared with 3.0 mL HEPES buffer solution with 0.1% bovine serum albumin, with 0.3 mmol/L EGTA added to ensure a calcium-free environment. PMN were warmed to 37ºC in the water bath, centrifuged at 5000 rpm for 30 seconds and transferred to the cuvette. PMN [Ca2+]i was measured using fura-2-AM in a spectrofluorometer (Fluromax-2; Jobin-Spex, Edison, NJ) using our modifications of the methods of Grynkiewicz et al. 12. MTD from intra-operative femur specimens was applied at 30 seconds 3, 12. [Ca2+]i was then calculated from the 340/380 nm fluorescence ratio over the next 2 minutes 15.
Chemotaxis
PMN were isolated using our standard one-step method. Chemotaxis assays were conducted in Biomatrix-coated 96-well plates with 3 μm pores (Millipore; Billerica, MA) as previously reported 16. Briefly, 106 calcein-loaded cells PMN were placed in the upper wells and chemoattractants (GRO-α, IL-8 and fMLP) in the lower wells. Blank wells (no chemoattractant) were used to determine random movement (chemokinesis). Calcein-loaded cells were used to create a standard curve. After incubation the lower chambers were read at 535 nm with results reported as the percent of cells undergoing chemotaxis. Migration in the blank was subtracted from chemoattractant wells to yield specific chemotaxis. Dose response curves to fMLP (also IL-8, GRO-α and LTB4, not shown) used PMN from 9 major trauma patients and 6 volunteers. After that all chemoattractants were used at 10 nM. For experiments exploring the effects of formyl peptides on chemotactic responses of LTB4, volunteer PMN were assayed for chemotaxis to LTB4 at 1 nM and 10 nM after 10 min pre-incubation in 10 nM fMLP or in media plus vehicle (DMSO) only.
Neutrophil Extracellular Traps (NETs)
NET formation was analyzed in fixed PMN according to the methods of Brinkmann 17, 18. Following PMN isolation, cells were resuspended in live cell medium with 2% BSA, and applied to a coverslips in a 24-well plate (0.5 x 106 per well). Unstimulated cells or cells stimulated with 100 nM PMA for 4 hours were fixed with 4% paraformaldehyde and permeabilized with 0.05% Triton X-100. PMN were then incubated with 5% donkey serum, followed by a mixture of primary antibodies to neutrophil elastase (rabbit polyclonal, EMD Millipore, Billerica, MA) and histone H1 (mouse monoclonal, Santa Cruz Biotechnology, Dallas, TX). After three washings cells were incubated with secondary antibodies (donkey anti-rabbit IgG-Alexa 488 and donkey anti-mouse IgG-Alexa 546, both from Life Technologies). Cells were then treated with ProLong Gold antifade reagent containing DAPI (Life Technologies), and the coverslips were attached to slides. NETs were analyzed using a Zeiss LSM 510 Meta Live Cell Inverted Confocal system. Representative NET images for 3 samples in each group (control, stimulated control, trauma PMN, stimulated trauma PMN) were evaluated. Images were captured from 3 representative areas on each coverslip. Nets were defined by automated co-localization of the stains.
Statistics
All results were analyzed for groups of 10 animals except where otherwise noted using ANOVA followed by Tukey’s post hoc test for intergroup variability (SigmaStat, Systat software, Inc.).
RESULTS
PsFx reduces bacterial clearance from the lung
Rats inoculated with S. aureus i.t. do not develop pneumonia and only have small numbers of residual bacteria in the BALF after 16 hours of incubation (Figure 1). Remarkably though, the addition of PsFx to bacterial inoculation increased bacteria in the overnight BALF by more than five-fold (from 200±88 to 1025±259 CFU/mL (SE), P=0.003). Thus the presence of a peripheral fracture injury, as represented here by our PsFx model, markedly decreases the ability of rats to clear inoculated S. aureus from the lung.
Figure 1.
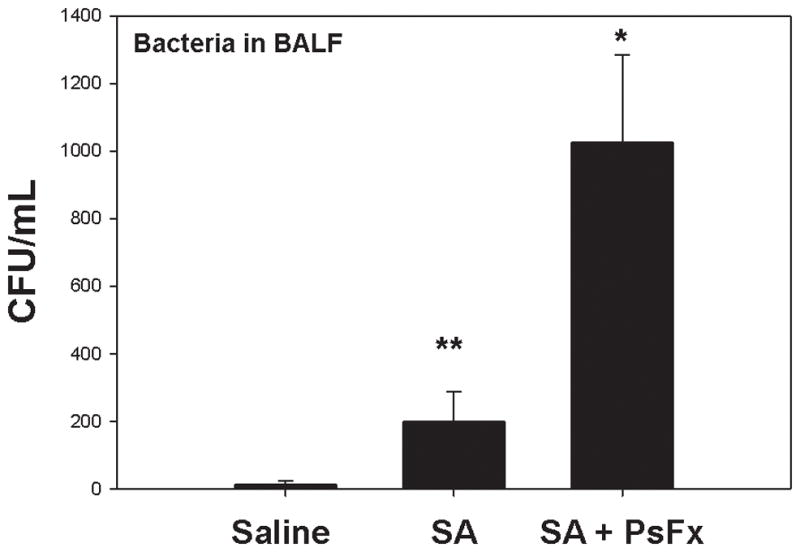
Bacterial counts in bronchoalveolar lavage fluids (BALF). Almost no bacteria were detected in saline controls. S. aureus (SA) injected i.t. were cleared overnight. BALF bacterial counts after SA plus pseudo-fracture (SA+PsFx) were more than five times the counts seen with SA alone (p=0.003).
PsFx reduces PMN migration to the lung
After injury, PMN respond to chemokines synthesized in the lung both in response to mechanical lung injury and as a result of bacterial inoculation. To investigate the mechanisms by which injuries might limit bacterial clearance in trauma, we first studied how PsFx affected PMN migration to sterile lung injury. PMN were measured in BALF after pulmonary contusion (PC),11 which elicits a sterile, chemokine dependent accumulation of neutrophils in the lung 4 Here, we found that PC elicited a 16-fold increase in alveolar PMN compared to control animals (Figure 2a, P< 0.001) but that rats sustaining PC in the presence of a PsFx accumulated far fewer alveolar PMN (P= 0.002). Inoculation with S. aureus similarly increased airway PMN and PsFx decreased PMN accumulation in the lung in response to a bacterial inoculum (Figure 2b p<0.01),
Figure 2.
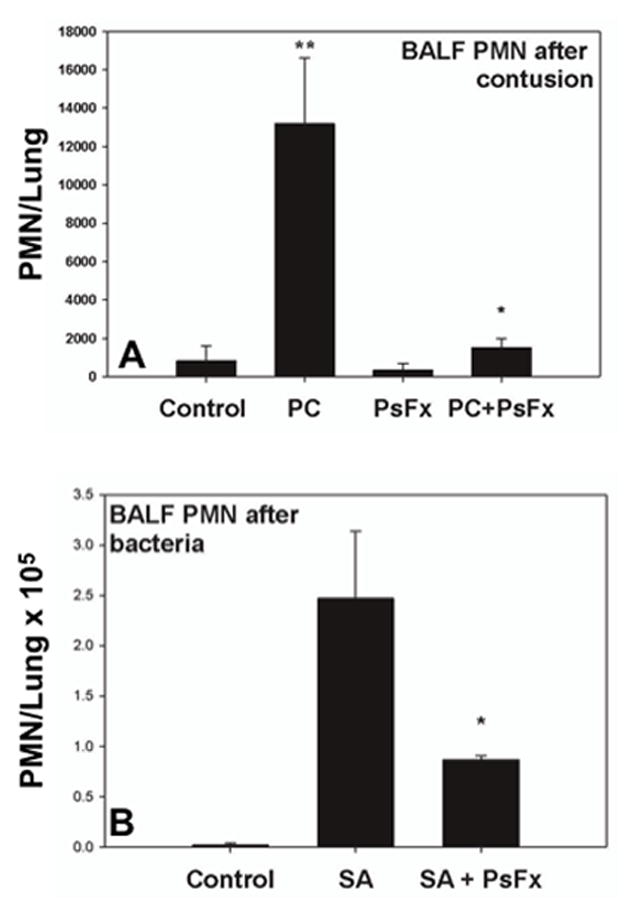
PMN in BALF. A: BALF PMN after pulmonary contusion (PC). PC induced a 16-fold increase in PMN count compared to controls (p<0.001). PsFx per se did not induce a rise in BALF PMN but when combined with PC (PC+PsFx) PMN number was significantly less than PC alone (p=0.002). B: BALF PMN after bacterial inoculation: S. aureus increased airway PMN and PsFx decreased airway PMN accumulation in response to bacterial inoculation (p<0.01).
Fractures stimulate PMN via formyl peptide receptors FPR-1 and FPR-2
To better understand why fractures decrease PMN accumulation in the lung we first determined whether fractures per se release chemoattractants. We crushed human cancellous bone from femur and made suspensions from it the same way we made PsFx supernatants from rat femur. Since formyl peptides (FP) only exist in mitochondria, we then used PMN FP-receptor mediated [Ca2+]i responses to determine whether PsFx supernatants contained mitochondrial debris. Stimulation with femur supernatant caused immediate release of Ca2+ from PMN intracellular stores (Figure 3). PMN pre-incubated with monoclonal antibodies against FPR-1 (n=5) or FPR-2 (10 min at 37ºC, n=8) showed significant reductions in [Ca2+] flux (P=0.027 for anti-FPR-1 and P=0.007 for anti-FPR-2) in comparison to controls. These results were consistent with previous studies showing that purified mitochondria activate neutrophils similarly 6.
Figure 3.
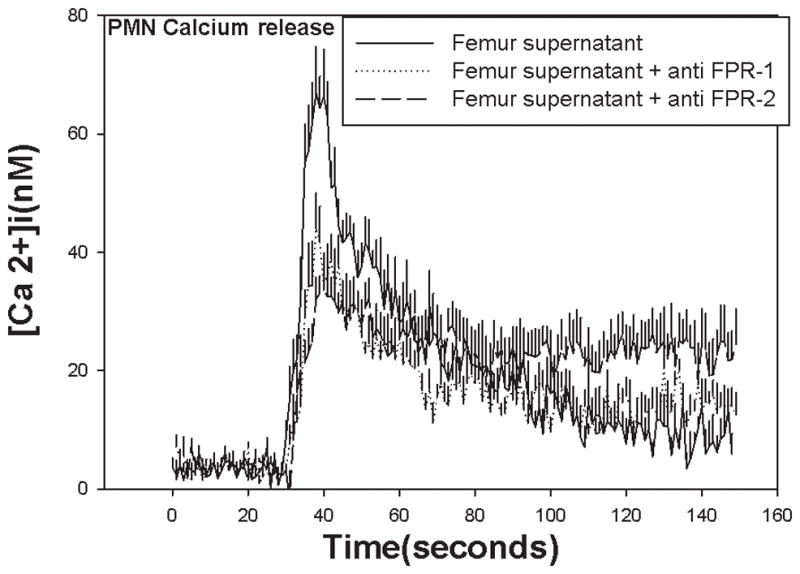
Ca2+ flux responses to bone supernatants depend on FPR-1 and FPR-2. Ca2+ flux is stimulated in human PMN at t=30 seconds by bone supernatants. There was a significant reduction in responses seen in cells pre-incubated with antibodies to FPR-1 (p = 0.027) or anti FPR-2 (p = 0.007). Incubation with both antibodies blocks better but inhibition is still incomplete. This may be due to the presence of other FPRs or FPR heterodimerization.23
Formyl Peptides suppress chemotaxis to other chemoattractants
FP are terminal chemoattractants 19 critical for wound cleansing and healing. Bone injury both produces FP and diminishes PMN recruitment to the lung. Moreover, we have previously shown clinical PMN have depressed Ca2+ mobilization in response to chemokines 7. We therefore studied the effects of PMN exposure to FP on chemotaxis to other chemoattractants ex vivo. LTB4 is an important PMN chemoattractant 20 that acts through a high affinity (BLT1) and a low affinity (BLT2) cell surface receptor. We studied human PMN chemotaxis to 1 nM LTB4 (active at BLT1) and 10 nM LTB4 (active at BLT1 and BLT2) with or without pre-treatment by 10 nM fMLP. fMLP pre-treatment markedly reduced chemotaxis to LTB4 (Figure 4) at doses that were optimal for each receptor 16. This parallels our prior findings that chemokines and leukotrienes cross-regulate PMN by suppression of each other’s receptors 16.
Figure 4.
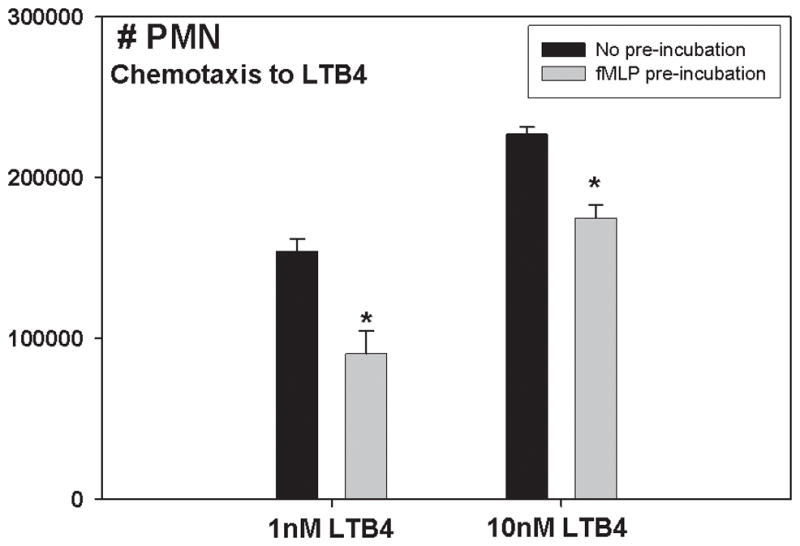
FP suppress CTX to LTB4: LTB4 stimulates CTX of human PMN at both high and low doses (1 nM and 10 nM). Both effects are markedly reduced by fMLP pre-treatment.
PMN chemotaxis is suppressed by clinical injury
Having shown FP were present in human fracture sites and regulate PMN attraction to chemokines, we reasoned that major injuries should diminish PMN chemotaxis globally. We studied PMN chemotaxis prospectively in a tightly-controlled group of major trauma patients (n=32) and compared them to PMN from age, sex, and ethnically matched volunteers. Here, we saw that responses to IL-8, GRO-α and fMLP were all suppressed (P<0.01 for all) by major injury (ISS>25). The nadir was between post-trauma Days 1 and 3 (Figure 5). Suppression was seen at all agonist concentrations, (bottom right panel) suggesting decreased receptor density rather than altered binding. FP themselves cause less chemotaxis at supra-optimal doses, suggesting homologous receptor desensitization.
Figure 5.

CTX in trauma patients (ISS 27±10[SE]) and matched volunteers (n=32/group). PMN CTX to IL-8, GRO-α and fMLP were all significantly suppressed by injury (p<0.01). Suppression was most significant on Day 3.
Trauma alters neutrophil extracellular trap (NET) formation
NETs are DNA webs PMN extrude to trap pathogens and kill them antimicrobial molecules 21, 22. Suspecting that pneumonia reflects diminished anti-bacterial function as well as diminished PMN arrival in the lung after injury, we compared NET formation by PMN sampled from trauma patients and from volunteer controls (n=3 each). We also assessed maximal NET stimulation as determined by their response to the receptor independent Protein Kinase C (PKC) activator PMA. We find after clinical trauma that unstimulated PMN circulate in a state of partial NET activation (Figure 6). Paradoxically though, PMN from injured patients then showed impaired NET formation when stimulated with PMA at a dose (20 nM) that elicits maximal responses in volunteer PMN. Together, these observations suggest that pre-activation and subsequent inability to form NETs may contribute to impaired bacterial killing in the lung by PMN after trauma.
Figure 6.
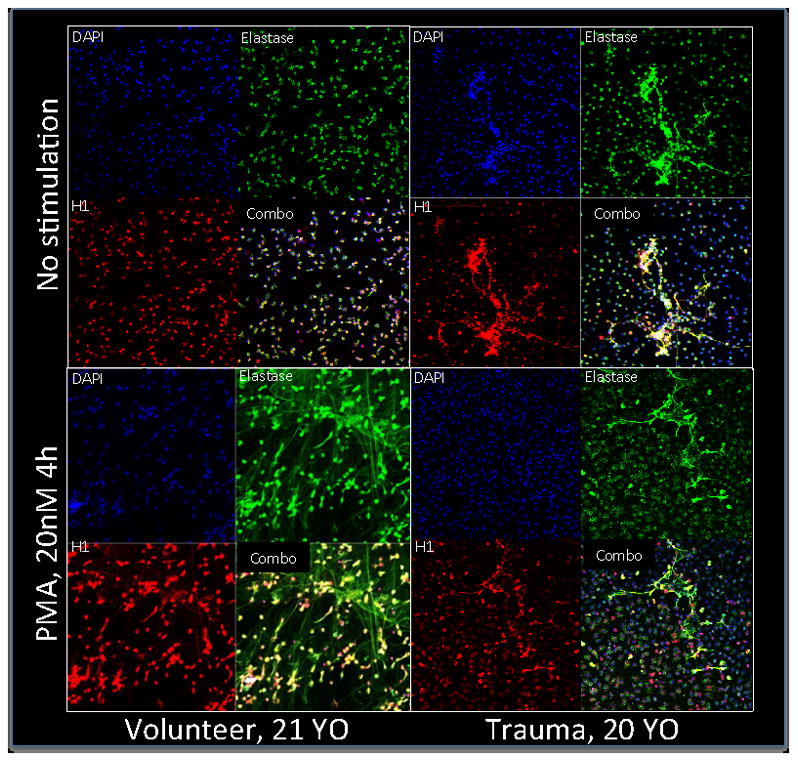
NETosis. NETosis is not seen in unstimulated volunteer PMN. Unstimulated PMN in trauma patients consistently show modest NET formation. PMA induces maximal activation of NETs in volunteers but PMN from trauma patients treated with PMA remain largely unchanged when compared to unstimulated cells.
DISCUSSION
Pneumonia (PNA) is common after major trauma and a significant contributor to morbidity and mortality 2. Pain, poor pulmonary toilet, altered mental status and decreased airway reflexes may contribute to bacterial inoculation of the lung. But even using optimal prevention strategies to alleviate these risk factors, the lung remains markedly susceptible to infection and the reasons remain poorly defined. This study was designed to examine that problem by 1) studying animal systems that model common injury patterns; 2) correlating observations made in those systems to functional studies of PMN sampled from trauma patients and 3) studying the mechanisms that underlie PMN responses to DAMPs present in the trauma environment.
Long bone fractures are common precursors of PNA that release mtDAMPs into the circulation and can modulate PMN function 13. Thus it is critically important that animals exposed to fracture DAMPs in a PsFx model showed a 5-fold decrease in the ability of their lung to clear bacteria overnight (Figure 1). Then, since PMN are a major effector cells clearing bacteria from the lung, we went on to study potential defects in PMN chemotaxis to the lung after fractures and functional deficits of the PMN that did arrive there. We could not study the entire problem in a single system though: clearly human subjects couldn’t be inoculated to study bacterial clearance and rodents do not express FPR on PMN prior to activation. These are all limitations inherent in translational studies, but by studying linked events serially we hope to have generated hypotheses that can be studied in therapeutic models.
Two major initiators of PMN chemotaxis to the lung after trauma are local lung injury and bacterial inoculation, both of which cause release of chemokines. We therefore created sterile pulmonary contusions and assayed the effect of fractures on PMN recruitment to the lung. As previously shown 4, PC by itself induces an influx of PMN and in Figure 2a we see that a simultaneous PsFx profoundly impairs PMN recruitment to the lung (>8-fold less than PC alone). S. aureus inoculation similarly induces an influx of PMN (Figure 2b) that is significantly decreased by the concomitant presence of remote injury in the form of PsFx. These findings parallel our previous work 4 where intraperitoneal mitochondria diverted PMN from the PC to the abdomen, and suggest that mtDAMPs released from fractures also attract PMN to remote sites, thus diminishing CTX to the lung and impairing host resistance to bacterial inoculation of the airway.
To compare post-traumatic PMN CTX to our laboratory model, we prospectively assayed PMN from our well-controlled series of major trauma patients. The major pulmonary chemo-attractants in humans are the chemokines GRO-α and IL-8 and the eicosanoid LTB4. We have previously shown that human injury suppresses PMN CTX to LTB416. We now show that there are also important decreases in clinical PMN CTX to the chemokines IL-8 and GRO-α after injury. Suppression was most profound on Day 3 post-injury (Figure 3). Moreover we have previously shown that chemokine receptors are suppressed by mitochondria-derived formyl peptides 3. We therefore now questioned whether fractures released formyl peptides. We assessed this by a bioassay using human PMN and human bone supernatants prepared identically to the rat PsFx. Here we found that the bone supernatants caused brisk intracellular Ca2+ flux that was suppressed by antibodies to either FPR-1 or FPR-2 (Figure 4a). We also confirmed that CTX to LTB4 was suppressed by formyl peptides (Figure 4b). Last, we also showed that CTX to formyl peptides themselves was suppressed by injury (Figure 4a, lower right). All of these events are compatible with suppression of cell surface chemotactic receptors by formyl peptides. Thus we conclude that it is likely the FP component of mtDAMPs that contribute most to suppression of PMN responses to bacterial inoculation of the lung after fracture injury. This remains to be definitively proven by studies using FPR blockade in vivo. These are planned.
Having shown that PMN arrival in the lung is diminished by distant injury we also considered whether PMN antimicrobial function was suppressed. PMN capture and kill bacteria by extending Neutrophil Extracellular Traps (NETs) in a process known as “NETosis” 17. NETs contain protein granules and chromatin, and extend out from PMN to ensnare invading pathogens and degrade them. This is a pivotal function in host-defense. Since we found PMN had impaired chemotactic ability following trauma, we also hypothesized that trauma might impair NETosis. We therefore studied PMN from trauma patients and healthy volunteers, defining the degree to which PMN exhibited NETosis at rest and when induced by PMA, a known potent inducer of NETosis. We found that healthy volunteers display little if any NETosis at rest and that PMA induced maximal NETosis at 4 hours, as expected. In trauma patients however, NETosis was present at rest. And though some PMN released NETs after PMA stimulation, maximal, diffuse NETosis was never achieved. These qualitative NET studies will require careful mechanistic re-evaluation as quantitative assays for NETosis improve. But we believe DAMPs released by trauma will prove an important agent of NET formation in circulating PMN. Also we think subsequent DAMP-induced tolerance may be important in later suppression of maximal NETosis, especially at the sites where it is needed.
In summary, we believe that the work shows fracture-derived formyl-peptide DAMPs divert PMN from ‘authentic’ bacterial foci in the lung toward remote injury sites. This likely represents the net PMN chemo-attractant effect of FP release at fracture sites and concurrent desensitization of PMN chemokine receptors needed for localization to the lung. Simultaneously, DAMPs from MT may tolerize the host to subsequent pathogenic danger signals (ie PAMPs). This appears to represent a dysfunctional form of PMN priming, and suggests that pneumonia is often neither the patient’s fault nor the doctor’s: it’s the injury’s fault. And although we can’t prove causality in these translational experiments, they do suggest therapeutic interventions which can be studied comprehensively in animal models and then if successful, in man. A better understanding of the timing, magnitude and type of DAMPs released after various injuries may allow judicious use of biologic response modification aimed at preventing the infectious sequelae of fractures while not unduly suppressing needed host responses to injury itself.
Acknowledgments
This study was funded by grant R01 GM089711 from the NIH/NIGMS (CJH).
Footnotes
Authorship
C.J.H. conceived the study. H.L., K.I., E.K., L.O. and C.J.H. designed the studies. H.L., K.I., D.G., A.G., E.K., Y.Z., Y.T.L., I.T.T and B.I. performed the studies. H.L., D.H.L., L.O., N.S. and C.J.H. analyzed and interpreted the data. N.S., H.L., EK, L.O. and C.J.H. prepared the manuscript.
References
- 1.Michetti CPFS, Ferguson PL, Cook A, Moore FO, Gross R. Ventilator-associated pneumonia rates at major trauma centers compared with a national benchmark: A multi-institutional study of the aast. The Journal of Trauma and Acute Care Surgery. 2012;72:1165–1173. doi: 10.1097/TA.0b013e31824d10fa. [DOI] [PubMed] [Google Scholar]
- 2.Eckert MJDK, Reed RL, 2nd, Santaniello JM, Poulakidas S, Esposito TJ, Luchette FA. Urgent airways after trauma: Who gets pneumonia? The journal of Trauma. 2004;57:750–755. doi: 10.1097/01.ta.0000147499.73570.12. [DOI] [PubMed] [Google Scholar]
- 3.Zhang QRM, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial damps cause inflammatory responses to injury. Nature. 2010;424:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao CIK, Gupta A, Odom S, Sandler N, Hauser CJ. Mitochondrial damage-associated molecular patterns released by abdominal trauma suppress pulmonary immune responses. Journal of Trauma and Acute Care Surgery. 2014;76:1222–1227. doi: 10.1097/TA.0000000000000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun SST, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial damps increase endothelial permeability through neutrophil dependent and independent pathways. PLOS ONE. 2013;8:e59989. doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raoof MZQ, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils via fpr-1. The Journal of Trauma. 2010;68:1328–1332. doi: 10.1097/TA.0b013e3181dcd28d. [DOI] [PubMed] [Google Scholar]
- 7.Tarlowe MHDA, Kannan KB, Itagaki K, Lavery RF, Livingston DH, Bankey P, Hauser CJ. Prospective study of neutrophil chemokine responses in trauma patients at risk for pneumonia. American journal of respiratory and critical care medicine. 2005;171:753–759. doi: 10.1164/rccm.200307-917OC. [DOI] [PubMed] [Google Scholar]
- 8.Pfeifer RKP, Darwiche SS, Billiar TR, Pape HC. Role of hemorrhage in the induction of systemic inflammation and remote organ damage: Analysis of combined pseudo-fracture and hemorrhagic shock. Journal of orthopaedic research: Official Publication of the Orthopaedic Research Society. 2011;29:270–274. doi: 10.1002/jor.21214. [DOI] [PubMed] [Google Scholar]
- 9.Kobbe PKD, Vodovotz Y, Tzioupis CH, Mollen KP, Billiar TR, Pape HC. Local exposure of bone components to injured soft tissue induces toll-like receptor 4-dependent systemic inflammation with acute lung injury. Shock. 2008;20:686–691. doi: 10.1097/SHK.0b013e31816f257e. [DOI] [PubMed] [Google Scholar]
- 10.Darwiche SS1, Kobbe P, Pfeifer R, Kohut L, Pape HC, Billiar T. Pseudofracture: an acute peripheral tissue trauma model. J Vis Exp. 2011 Apr 18;(50) doi: 10.3791/2074. pii: 2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badami CDLD, Sifri ZC, Caputo FJ, Bonilla L, Mohr AM, Deitch EA. Hematopoietic progenitor cells mobilize to the site of injury after trauma and hemorrhagic shock in rats. The Journal of trauma. 2007;63:596–600. doi: 10.1097/TA.0b013e318142d231. [DOI] [PubMed] [Google Scholar]
- 12.Czaikoski PG, Nascimento DC, Sonego F, de Freitas A, Turato WM, de Carvalho MA, Santos RS, de Oliveira GP, dos Santos Samary C, Tefe-Silva C, Alves-Filho JC, Ferreira SH, Rossi MA, Rocco PR, Spiller F, Cunha FQ. Heme oxygenase inhibition enhances neutrophil migration into the bronchoalveolar spaces and improves the outcome of murine pneumonia-induced sepsis. Shock. 2013;39:389–396. doi: 10.1097/SHK.0b013e31828bbcf9. [DOI] [PubMed] [Google Scholar]
- 13.Hauser CJST, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and p44/42 map kinase. Journal of orhtopaedic trauma. 2010;24:534–538. doi: 10.1097/BOT.0b013e3181ec4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itagaki KKK, Livingston DH, Deitch EA, Fekete Z, Hauser CJ. Store-operated calcium entry in human neutrophils reflects multiple contributions from independently regulated pathways. Journal of Immunology. 2002;168:4063–4069. doi: 10.4049/jimmunol.168.8.4063. [DOI] [PubMed] [Google Scholar]
- 15.Grynkiewicz GPM, Tsien RY. A new generation of ca2+ indicators with greatly improved fluorescence properties. The journal of biological chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 16.Tarlowe MHKK, Itagaki K, Adams JM, Livingston DH, Hauser CJ. Inflammatory chemoreceptor cross-talk suppresses leukotriene b4 receptor 1-mediated neutrophil calcium mobilization and chemotaxis after trauma. Journal of Immunology. 2003;171:2066–2073. doi: 10.4049/jimmunol.171.4.2066. [DOI] [PubMed] [Google Scholar]
- 17.Brinkmann VRU, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 18.Brinkmann VLB, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: How to generate and visualize them. Journal of visualized experiments. 2010;24:1724. doi: 10.3791/1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald BPK, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 20.Mikami ML-JC, Bayley D, Hill SL, Stockley RA. The chemotactic activity of sputum from patients with bronchiectasis. American journal of respiratory and critical care medicine. 1998;157:723–728. doi: 10.1164/ajrccm.157.3.9606120. [DOI] [PubMed] [Google Scholar]
- 21.Remijsen QKT, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: Netosis, mechanisms behind an antimicrobial cell death modality. Cell death and differentiation. 2011;18:581–588. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remijsen QVBT, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell research. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooray SN, Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJ, Flower RJ, Perretti M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A. 2013 Nov 5;110(45):18232–7. doi: 10.1073/pnas.1308253110. [DOI] [PMC free article] [PubMed] [Google Scholar]