

Abstract
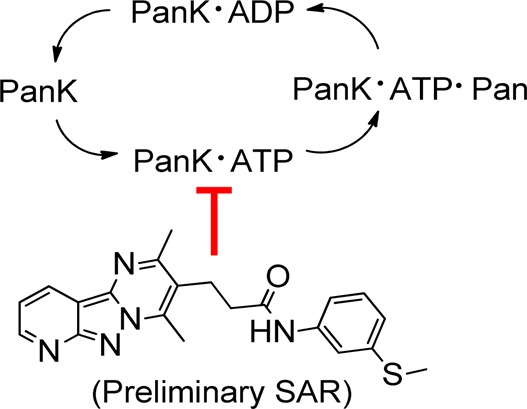
Pantothenate kinase (PanK) is a regulatory enzyme that controls coenzyme A (CoA) biosynthesis. The association of PanK with neurodegeneration and diabetes suggests that chemical modifiers of PanK activity may be useful therapeutics. We performed a high throughput screen of >520000 compounds from the St. Jude compound library and identified new potent PanK inhibitors and activators with chemically tractable scaffolds. The HTS identified PanK inhibitors exemplified by the detailed characterization of a tricyclic compound (7) and a preliminary SAR. Biophysical studies reveal that the PanK inhibitor acts by binding to the ATP–enzyme complex.
Introduction
Pantothenate kinases (PanK) catalyze the rate-limiting step in the biosynthesis of CoA and regulate the concentration of this essential cofactor.1,2 CoA is found in all living organisms, where it acts as an acyl group carrier in various synthetic and oxidative metabolic pathways such as the tricarboxylic acid cycle and fatty acid metabolism. Four closely related isoforms of PanKs have been identified in mammals: PanK1α, PanK1β, PanK2, and PanK3, which are encoded by three genes.3−5 Recently, the scientific community has shown interest in the PanK2 and PanK1 isoforms because of their role in PanK-associated neurodegeneration (PKAN) and diabetes, respectively.
PKAN is a rare and neurological disorder caused by mutations in the human PANK2 gene.3,6,7 PKAN is inherited in an autosomal recessive pattern and leads to progressive dystonia, dysarthria, parkinsonism, and pigmentary retinopathy. Classic PKAN develops around age 3, and most patients are at risk of early death because there are no FDA approved treatments for the disease. The PanK2 isoform is highly expressed in human neuronal tissues and the PANK2 mutations are predicted to result in significantly lower CoA levels, thereby reducing neuronal metabolism and function in PKAN patients. Pank2 knockout mice were generated to investigate the complex pathogenesis of PKAN but unfortunately did not reproduce the human disease.8,9 The single Pank1 and Pank2 knockout mice did not show a neurodegenerative phenotype probably due to compensation by the other PanK enzymes.9 Double knockout mice were either embryonic lethal or died in the first few weeks after birth, making potential treatments difficult to test.9 Therefore, the lack of tools to investigate the relationship between CoA levels and neurodegeneration limits our understanding of the mechanisms by which PANK2 mutations result in neurodegeneration.
Limitation of the CoA supply by genetic deletion of PanK1 activity blunts the hepatic CoA increase in response to fasting and leads to a deficit in fatty acid oxidation and impaired gluconeogenesis.10 The key role of CoA in metabolic control is highlighted by the phenotype of the Pank1–/–Lep–/– double knockout mouse.11 The abnormally high level of CoA in the Lep–/– liver is reduced by the deletion of the Pank1 gene, resulting in normalization of the hyperglycemia and hyperinsulinemia characteristic of the Lep–/– mouse. The Pank1–/–Pank2–/– double knockout mice have a severe metabolic phenotype related to decreased fatty acid and ketone oxidation and do not survive to weaning.11 Taken together, these data demonstrate the impact of reduced intracellular CoA on oxidative metabolism and, in particular, the fuel switching during the transition from the fed to the fasted state. These data and the fact that a genome-wide study12 showed an association between PANK1 variants and insulin levels in humans suggest that PanK inhibitors may be useful therapeutics for type II diabetes.
The above background and our interest in understanding CoA physiologic functions led us to hypothesize that it is possible to discover compounds acting as PanK modulators that can be used in animals to regulate CoA synthesis. One approach to PKAN treatment would be to identify PanK1 or PanK3 activators that would stimulate CoA synthesis in tissues lacking PANK2. The PanK inhibitors would serve as tools to investigate the role of CoA in adult tissues and accelerate the identification of bypass drugs to treat PKAN disease. Additionally, PanK inhibitors may have direct therapeutic application for type II diabetes.
Recently, we reported a high throughput screening (HTS) study of 3200 unique molecules with known biological activity that identified small molecule inhibitors and activators of PanK.13 However, the identified compounds could not be used as ideal probes for studying the role of PanK in a disease due to their higher affinity toward other proteins. Therefore, we decided to expand our high throughput screen to the larger, more chemically diverse compound libraries available at St. Jude Children’s Research Hospital.
Results and Discussion
We screened a total of 521133 compounds to discover novel small-molecule modulators of PanK activity using a luciferase-based method we previously reported.13 The screened compound library contained 504956 small molecules and 16177 natural products acquired from various commercial sources and collaborators or synthesized at St. Jude. The PanK3 isoform was selected to conduct the HTS because it has a wide tissue distribution in mammals and can be purified in large amounts. All PanK isoforms have homologous catalytic domains, and so we expected that the PanK3 modulators would act against other PanK isoforms in a similar fashion.
All compounds were screened at 12 μM concentration in the PanK3 HTS assay, and the data was analyzed in both activator and inhibitor modes (Figure 1). The assay provided averaged Z′ value of >0.7 for activators and >0.5 for inhibitors, demonstrating the robustness and quality of our assay (Figure 1A,D). The discriminatory power of the primary screen was assessed using receiver operating characteristic (ROC) analysis14 using 448 representative compounds. True positives were defined as any compound yielding a well-behaved, saturating sigmoidal curve in the dose–response assay as determined by fit statistics such as r2 and visual inspection. ROC analyses indicated excellent discriminatory power (AUC ≥0.9 for both assays) and suggested that a cutoff of ≥40% for activators and ≥50% for inhibitors would retain ≥80% of true positives (Figure 1C,F). On the basis of this ROC analysis, we classified 9687 compounds (2415 activators and 7272 inhibitors) as “actives” for further analysis. These active compounds were then subjected to dose response analysis for PanK1β, PanK3, luciferase interference, and cytotoxicity assays (see Supporting Information). The dose response analysis showed that the compounds identified as PanK3 modulators similarly modulated PanK1β activity. This result was consistent with the high similarity shared by the two proteins.15
Figure 1.

HTS of 521133 compounds from the St. Jude compound library against PanK3 and ROC analysis of actives. (A) Z′ factor in activator mode. (B) Scatter plot of percentage activity of each well from 1642 384-well plates analyzed in activator mode [green, the positive control for the activator screen consisted of a reaction mixture lacking ATP; red, negative control (DMSO vehicle with complete assay components); blue, compound with activity above cutoff; black, compounds with activity below cutoff. Note: Y-axis is normalized % activity, not raw count.]. (C) ROC analysis of activators showing true (y axis) versus false (x axis) positive rates of percentage compound activity. Light-gray curves represent bootstrap simulation curves. (D) Z′ factor in inhibitor mode. (E) Scatter plot of percentage activity of each well analyzed in inhibitor mode [green, the positive control for the inhibitor screen contained 60 μM acetyl-CoA; red, negative control (DMSO vehicle with complete assay components); blue, compound with activity above cutoff; black, compounds with activity below cutoff. Note: Y-axis is normalized % activity, not raw count.]. (F) ROC analysis of inhibitors.
The most promising 100 activators and 100 inhibitors were selected based on their potency, curve filter, Hill number, absence of cytotoxicity, and luciferase interference activity. These compounds were then clustered together based on their structural similarities. To ensure the synthetic tractability of the compounds, a similarity search on each of the scaffolds was performed against the initial “actives” to generate preliminary structure–activity relationships (SAR) and deprioritize singleton hits. Representative compounds of each cluster are shown in Figure 2, and the details of their dose response analysis are provided in Supporting Information, Tables S1 and S2.
Figure 2.

Structures of representative compounds with different chemical scaffolds characterized as (A) activators (1–4) and (B) inhibitors (5–8) as identified from the HTS. EC50 and IC50 values (μM) represent the activity of the compounds for PanK3 (see Supporting Information, Tables S1 and S2, for detail dose response analysis).
Several compounds with a core tricyclic scaffold (represented by compound 7) were in the curated actives list of inhibitors. Thus, we focused our efforts on the synthesis of compounds with the tricyclic scaffold to characterize an active compound from the HTS inhibitor list and to generate preliminary structure–activity relationships (SAR) for development of more advanced lead compounds.
The synthesis of tricyclic compounds is depicted in Scheme 1. Our synthetic efforts began with a microwave assisted reaction between 2-chloronicotinonitrile (9) with hydrazine.16 The reaction yielded 10, which was then reacted with methyl 4-acetyl-5-oxohexanoate to obtain tricyclic 12.17 The hydrolysis of methyl ester 12 followed by its coupling with 3-(methylthio)aniline provided the required compound 7.
Scheme 1. Synthesis of Tricyclic Compound 7.

Reagents and conditions: (a) EtOH, hydrazine (5 equiv), 30 min, 160 °C, MW, 74%; (b) EtOH, methyl 4-acetyl-5-oxohexanoate (1.5 equiv), 15 min, 80 °C, MW, 79%; (c) THF, NaOH, 2 h, rt, 99%; (d) DMF, 3-(methylthio)aniline (1.2 equiv), HBTU (1.3 equiv), Et3N (1.5 equiv), 4 h, rt, 41%.
The activity of 7 was determined for each of the principle PanK isoforms using a radiochemical enzyme assay (Figure 3).13 The IC50 calculated for compound 7 was 25 nM for PanK3, whereas the IC50s for PanK1β and PanK2 were 70 and 92 nM, respectively. These results confirmed the dose–response analysis using the HTS assay showing that compound 7 inhibited each of the PANK isoforms at about the same level. Although the radiochemical and HTS PanK assays were robust and reproducible, the HTS assay (Table 1) consistently yielded higher IC50 values than the radiochemical assay (Figure 3). The key difference between the two assays was the ATP concentration. The HTS assay employed 100 μM ATP, whereas the radiochemical assay used 2.5 mM ATP. Compound 7 appeared more potent in the radiochemical assay because the PanK was saturated with ATP4, and the inhibitor binds to the ATP–enzyme intermediate (see below).
Figure 3.

Inhibition of three PanK isoforms by compound 7. These experiments were typical for the IC50 determinations in this study. Compound 7 was equally effective against all three PanK isoforms. IC50 (PanK1β = 70 ± 1.1 nM, PanK2 = 92 ± 2.0 nM, and PanK3 = 25 ± 1.8 nM).
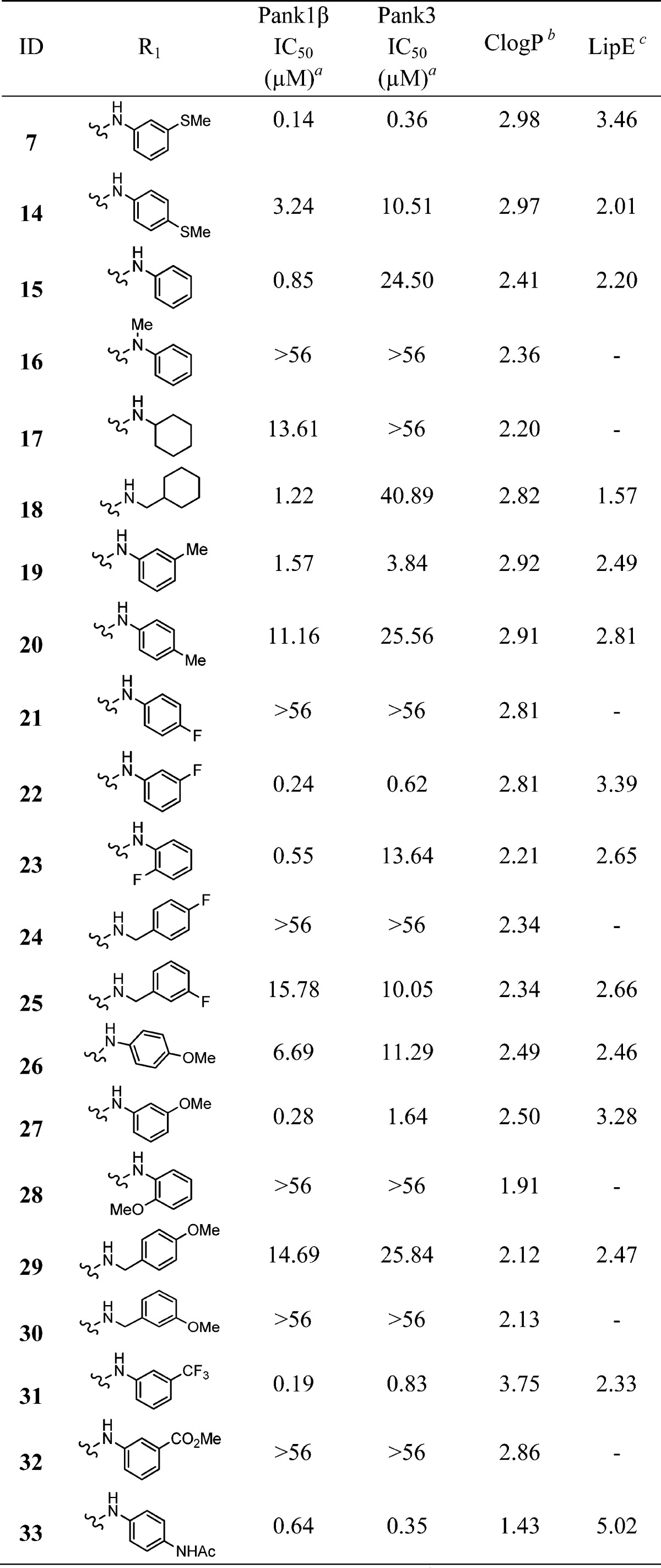
Table 1. Side Chain Structure and Inhibitory Potencies of Analogues of Tricyclic Compound 7.
Ten points dose response curve in triplicate (SD are provided in Supporting Information).
Calculated using ChemBioDraw Ultra.
LipE is calculated as LipE = pIC50(PanK3) – ClogP.
Next, we designed and synthesized several analogues of 7 to generate structure–activity relationship (SAR). Using the same approach as described for the synthesis of 7, compounds 14–33, having diversification on the right-hand side of the molecule (R1), were synthesized through intermediate 13. Table 1 illustrates the activity of these compounds against PanK1β and PanK3. Removal of the substituent on the aromatic ring (compound 15) resulted in decreased activity, indicating its importance. Interestingly, when we moved the −SMe substituent from meta- to para-position, the activity of the compound (14) was reduced by 30-fold. Replacing the aromatic ring with a cyclohexane ring in compound 17 decreased the activity against PanK1β and abrogated the activity against PanK3. Insertion of an additional methylene group between the amino moiety and the phenyl group gives rise to benzylamino derivatives, which exhibited either a complete or a significant loss of potency (compare compounds 22 and 25; 27 and 30; 26 and 29). Compound 16, where the amino group of the anilino moiety is methylated, was completely inactive against PanK1β and PanK3 (compared to 15), demonstrating the necessity of having a hydrogen bond donor at that position. By comparing the relative activities of compounds 21, 22, and 23; 26, 27, and 28; 7 and 14; and 19 and 20 suggested that the substitution at the meta-position on the aniline ring was most important for the activity observed. A small set of synthesized compounds (34–42) with the modification on the tricyclic core (R2) did not show any improved activity (<30% inhibition at 10 μM) (Figure 4). To evaluate the effect of structural modifications on overall intrinsic potential of the molecule, we used a well-accepted metric: lipophilic ligand efficiency (LipE), which combines both potency and lipophilicity.18−20 Compared to 7, compound 33 exhibited higher LipE without reducing activity significantly.
Figure 4.

Modification on the left side of the molecule (R2, tricyclic core). All these modifications resulted in inactive compounds (<30% inhibition at 10 μM).
Overall, this SAR analysis shows that the side chain of tricyclic compound 7 is tolerant to multiple modifications, providing an ample opportunity to expand the series in search of more potent, druglike molecules. Additionally, the higher potency of compound 15 and 23 toward PanK1β compared to PanK3 suggested that further medicinal chemistry efforts might ultimately lead to selective inhibitors of different PanK isoforms.
We next investigated the kinetic mechanism of compound 7 inhibition of PanK3. As shown in Figure 5A, compound 7 lowered both the Vmax and Km for ATP. The pattern of compound 7 inhibition with respect to pantothenate was mixed (Figure 5B). One interpretation of these data is that compound 7 bound to the ATP–enzyme intermediate. A thermal shift analysis was performed to confirm that compound 7 bound to the ATP–PanK3 complex (Figure 5C). Compound 7 did not stabilize the protein to thermal denaturation when ATP was absent but increased the protein’s thermal stability when added in the presence of 2 mM ATP/Mg2+. The apparent binding constant for compound 7 in these experiments was 0.3 ± 0.08 μM. These results clearly demonstrate that compound 7 binds to the ATP–PanK3 complex.
Figure 5.

Kinetic mechanism for PanK3 inhibition by 7. (A) Analysis of ATP kinetics in the presence of 0 (●), 0.038 (■), and 0.128 (▲) μM of compound 7 showing mixed-type inhibition with respect to ATP. (B) Analysis of pantothenate kinetics in the presence of 0 (●), 0.038 (■), and 0.128 (▲) μM of compound 7 showing mixed inhibition with respect to pantothenate. (C) Thermal stabilization of PanK3 by compound 7 in the presence of 2 mM ATP/Mg2+.
The ability of compound 7 to inhibit CoA biosynthesis in cultured C3A cells was evaluated in a metabolic labeling experiment. Compound 7 caused a dose-dependent decrease in [3H]pantothenate incorporation into CoA (Figure 6). There was no effect on cell viability at the concentrations of compound 7 used in this experiment. These data confirm that compound 7 acts as a PanK inhibitor in cultured cells by blocking de novo CoA biosynthesis.
Figure 6.

Inhibition of CoA biosynthesis in C3A cells. C3A cells were labeled with [3H]pantothenate for 24 h in the absence or presence of the indicated concentrations of compound 7. The cells were extracted, and the amount of radioactive CoA was determined by binding the extract to DE-81 filters as described under methods. The apparent IC50 calculated from this data was 0.9 ± 0.11 μM.
Conclusion
In summary, we conducted a HTS of 521133 compounds and identified novel PanK activators and inhibitors capable of modulating PanK activities and cellular CoA levels. The HTS correctly identifies inhibitors based on a preliminary SAR of tricyclic compound 7, and the kinetic experiments show that the inhibitor acts by binding to the ATP–enzyme complex. Improvement of the selectivity, efficacy, and other druglike properties of these PanK modulators will require extensive medicinal chemistry efforts which are beyond the scope of this work. Our future endeavors will include the further optimization of identified scaffolds which may lead to a druggable agent capable of selectively modulating PanK activity in vivo. Additional scaffolds from HTS data will be identified and developed. Indeed, the development of modulators of PanK activity could represent a promising approach to the treatment of PKAN and diabetes.
Methods
The methods are reported in the Supporting Information.
Acknowledgments
This work was supported by NIH grant GM062896 (S.J.), Cancer Center Support Grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC).
Glossary
Abbreviations Used
- PanK
pantothenate kinase
- CoA
coenzyme A
- PKAN
PanK-associated neurodegeneration
- HTS
high throughput screen
- LeP
leptin
- ROC
receiver operating characteristic
- SAR
structure–activity relationship
- LipE
lipophilic ligand efficiency
Supporting Information Available
Detailed experimental procedures and methods, full characterization data, and copies of spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ For R.L.: Department of Biochemistry, West Virginia University, 1 Medical Center Drive, Morgantown, West Virginia 26506, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Leonardi R.; Zhang Y. M.; Rock C. O.; Jackowski S. Coenzyme A: back in action. Prog. Lipid Res. 2005, 44, 125–153. [DOI] [PubMed] [Google Scholar]
- Jackowski S.; Rock C. O. Regulation of coenzyme A biosynthesis. J. Bacteriol. 1981, 148, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Westaway S. K.; Levinson B.; Johnson M. A.; Gitschier J.; Hayflick S. J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden–Spatz syndrome. Nature Genet. 2001, 28, 345–349. [DOI] [PubMed] [Google Scholar]
- Zhang Y. M.; Rock C. O.; Jackowski S. Feedback regulation of murine pantothenate kinase 3 by coenzyme A and coenzyme A thioesters. J. Biol. Chem. 2005, 280, 32594–32601. [DOI] [PubMed] [Google Scholar]
- Rock C. O.; Karim M. A.; Zhang Y. M.; Jackowski S. The murine pantothenate kinase (Pank1) gene encodes two differentially regulated pantothenate kinase isozymes. Gene 2002, 291, 35–43. [DOI] [PubMed] [Google Scholar]
- Johnson M. A.; Kuo Y. M.; Westaway S. K.; Parker S. M.; Ching K. H.; Gitschier J.; Hayflick S. J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [DOI] [PubMed] [Google Scholar]
- Kotzbauer P. T.; Truax A. C.; Trojanowski J. Q.; Lee V. M. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J. Neurosci. 2005, 25, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. M.; Duncan J. L.; Westaway S. K.; Yang H.; Nune G.; Xu E. Y.; Hayflick S. J.; Gitschier J. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M.; Leonardi R.; Zhang Y. M.; Rehg J. E.; Jackowski S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PloS One 2012, 7, e40871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R.; Rehg J. E.; Rock C. O.; Jackowski S. Pantothenate kinase 1 is required to support the metabolic transition from the fed to the fasted state. PloS One 2010, 5, e11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R.; Rock C. O.; Jackowski S. Pank1 deletion in leptin-deficient mice reduces hyperglycaemia and hyperinsulinaemia and modifies global metabolism without affecting insulin resistance. Diabetologia 2014, 57, 1466–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatti C.; Service S. K.; Hartikainen A. L.; Pouta A.; Ripatti S.; Brodsky J.; Jones C. G.; Zaitlen N. A.; Varilo T.; Kaakinen M.; Sovio U.; Ruokonen A.; Laitinen J.; Jakkula E.; Coin L.; Hoggart C.; Collins A.; Turunen H.; Gabriel S.; Elliot P.; McCarthy M. I.; Daly M. J.; Jarvelin M. R.; Freimer N. B.; Peltonen L. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nature Genet. 2009, 41, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R.; Zhang Y. M.; Yun M. K.; Zhou R.; Zeng F. Y.; Lin W.; Cui J.; Chen T.; Rock C. O.; White S. W.; Jackowski S. Modulation of pantothenate kinase 3 activity by small molecules that interact with the substrate/allosteric regulatory domain. Chem. Biol. 2010, 17, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithson D. C.; Shelat A. A.; Baldwin J.; Phillips M. A.; Guy R. K. Optimization of a non-radioactive high-throughput assay for decarboxylase enzymes. Assay Drug Dev. Technol. 2010, 8, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B. S.; Senisterra G.; Rabeh W. M.; Vedadi M.; Leonardi R.; Zhang Y. M.; Rock C. O.; Jackowski S.; Park H. W. Crystal structures of human pantothenate kinases—insights into allosteric regulation and mutations linked to a neurodegeneration disorder. J. Biol. Chem. 2007, 282, 27984–27993. [DOI] [PubMed] [Google Scholar]
- Ye Q.; Shen Y.; Zhou Y.; Lv D.; Gao J.; Li J.; Hu Y. Design, synthesis and evaluation of 7-azaindazolyl-indolyl-maleimides as glycogen synthase kinase-3β (GSK-3beta) inhibitors. Eur. J. Med. Chem. 2013, 68, 361–371. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Jiang L. L.; Chen C. N.; Yang G. F. The first example of a regioselective Biginelli-like reaction based on 3-alkylthio-5-amino-1,2,4-triazole. J. Heterocycl. Chem. 2009, 46, 139–148. [Google Scholar]
- Shultz M. D. Setting expectations in molecular optimizations: strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [DOI] [PubMed] [Google Scholar]
- Jabeen I.; Pleban K.; Rinner U.; Chiba P.; Ecker G. F. Structure–activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J. Med. Chem. 2012, 55, 3261–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nature Rev. Drug. Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.