

Abstract
Because of increasingly widespread sedentary lifestyles and diets high in fat and sugar, the global diabetes and obesity epidemic continues to grow unabated. A substantial body of evidence has been accumulated which associates diabetes and obesity to dramatically higher risk of cancer development, particularly in the liver and gastrointestinal tract. Additionally, diabetic and obese individuals have been shown to suffer from dysregulation of bile acid (BA) homeostasis and dysbiosis of the intestinal microbiome. Abnormally elevated levels of cytotoxic secondary BAs and a pro-inflammatory shift in gut microbial profile have individually been linked to numerous enterohepatic diseases including cancer. However, recent findings have implicated a detrimental interplay between BA dysregulation and intestinal dysbiosis that promotes carcinogenesis along the gut–liver axis. This review seeks to examine the currently investigated interactions between the regulation of BA metabolism and activity of the intestinal microbiota and how these interactions can drive cancer formation in the context of diabesity. The precarcinogenic effects of BA dysregulation and gut dysbiosis including excessive inflammation, heightened oxidative DNA damage, and increased cell proliferation are discussed. Furthermore, by focusing on the mediatory roles of BA nuclear receptor farnesoid x receptor, ileal transporter apical sodium dependent BA transporter, and G-coupled protein receptor TGR5, this review attempts to connect BA dysregulation, gut dysbiosis, and enterohepatic carcinogenesis at a mechanistic level. A better understanding of the intricate interplay between BA homeostasis and gut microbiome can yield novel avenues to combat the impending rise in diabesity-related cancers.
Keywords: Gut–liver axis, gut dysbiosis, intestinal microbiota, bile acids, farnesoid X receptor, G-protein-coupled BA receptor 1 (TGR5), apical sodium dependent bile acid transporter, polymorphism, inflammation, bacterial translocation, diabetes, obesity, metabolic syndrome, gastrointestinal carcinogenesis
Introduction
As the worldwide obesity epidemic continues to grow, the prevalence of type II diabetes is also rising to a projected 439 million of individuals globally by 2030.1 Of all obesity-related chronic conditions, diabetes is most strongly associated because of their similar symptomatic manifestations. Type II diabetes and obesity are both characterized by insulin resistance, glucose intolerance, hypoadiponectinemia, endoplasmic reticulum (ER) stress, and low-grade inflammation. Over 11% of the 34% of U.S. obese adults are reported to be diabetic in 2011.2 Obesity is generally thought to stem from a combination of genetic and environmental factors. Increased dietary consumption of fat and refined carbohydrates along with decreased physical activity contributes to excessive weight gain while underlying genetic dispositions may lead to differential clinical progression. It has become clear that modest weight reduction can improve glycemic control and alleviate insulin resistance as obesity is considered a modifiable risk factor for diabetes.2 Furthermore, the comorbidity of diabetes and obesity has been linked to liver and colon cancer risk, although the precise mechanisms remain unresolved. Meta-analysis of cohort studies has identified increased risk in obese individuals (relative risk (RR)=1.89, 95% confidence interval (CI): 1.51–2.36) for developing liver cancer compared to normal weight individual while meta-analysis of prospective studies correlated a larger waist circumference and waist–hip ratio with increased risk for colon cancer.3,4 Similarly, diabetics are more than twice as likely to be diagnosed with cancer of the liver, pancreas, endometrium, and to a lesser extent, colon, breast, and bladder.5
Many possible underlying mechanisms including hyperinsulinemia, hyperglycemia, and inflammation have been proposed to explain the increased cancer incidence. Insulin, produced by b-cells in the pancreas, is released to promote cellular absorption of blood glucose and many factors including excess weight and increased plasma triglyceride (TG) levels can raise circulating levels of insulin. Chronic insulin elevation results in resistance, which then increases the biological activity of insulin-like growth factor (IGF-1), an endocrine and paracrine hormone regulating tissue growth and metabolism.6 Epidemiological studies have linked IGF-1 to several cancer types, including hepatocellular carcinoma (HCC) and colorectal cancer (CRC).6,7 Moreover, excess adiposity leads to the derangement of other peptide hormones such as resistin, leptin, adiponectin, and tumor necrosis factor α (TNFα), contributing to the metabolic abnormalities commonly observed in obese and diabetic individuals. Indeed, increased leptin and reduced adiponectin have been identified as risk factors for the progression of liver steatosis, fibrosis, and tumorigenesis as well as CRC formation.8–11
Both bile acids (BAs) and the intestinal microbiota have been extensively studied in the context of various health conditions, particularly obesity and type II diabetes-associated HCC and CRC.12–15 Although the exact mechanism of how gut microbes and BAs affect one another remains unclear, it is evident that the introduction of intestinal microbes increased liver cholesterol and altered BA profiles in germ-free mice.16,17 Conversely, dietary BA supplementation can modulate gut microbial profile in animal models.18,19 This review will focus on the current understanding of the complex interplay between BA homeostasis and gut microbial profiles in regards to obesity and diabetes-associated liver and colon carcinogenesis.
Method
This review article covers information obtained from peerreviewed papers published in the past 15 years. We researched various combinations of key words in PubMed that were accessible through University of California-Davis Institutional database subscriptions. The key words used in the search were as listed: intestinal microbiome, intestinal microbiota, gut–liver axis, GI health, bile acid, liver cancer, colon colorectal cancer, DNA damage, inflammation, gastric bypass, obesity, diabetes, FXR, TGR5, ASBT, polymorphism, genetic variation, prebiotics, probiotics, bile acid dysregulation, gut dysbiosis, oxidative stress.
BA homeostasis in the gut–liver axis
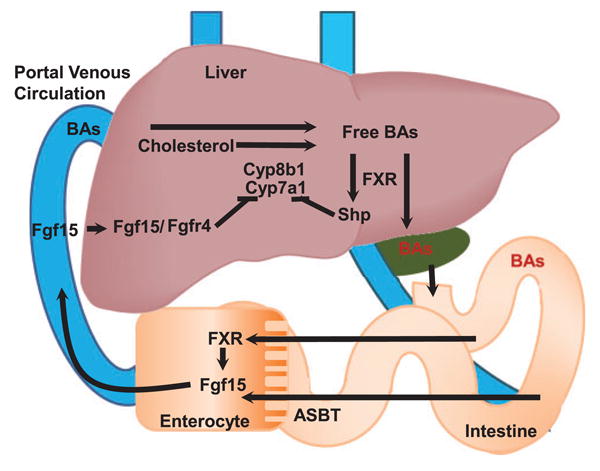
BAs are the primary facilitators of lipid absorption in the gastrointestinal (GI) tract. They are synthesized by cholesterol catabolism in the liver through both the classical (CYP7A1 and CYP8B1) and the acidic pathway (CYP27A1 and CYP8B1), which differ in the modification order of the sterol ring and side chain oxidation.20 BAs synthesized in the liver are subsequently conjugated for storage in the gallbladder. Upon ingestion of fat and protein, cholecystokinin, a peptide hormone in the small intestine will stimulate the release of bile containing digestive enzymes and primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), from the gall bladder. These BAs will then activate farnesoid x receptor (FXR) in the liver which induces the expression of small heterodimer partner (SHP) to inhibit the activity of liver receptor homolog-1 responsible for upregulating the rate-limiting BA synthesis enzyme CYP7A1.21,22 Intestinal FXR activity on the other hand, induces the expression of fibroblast growth factor 19 (human FGF19/rodent Fgf15) which binds hepatic fibroblast growth factor receptor 4 and activates c-Jun N-terminal kinase 1/2 (JNK1/2) and extracellular signal-regulated kinase 1/2 (ERK1/2) to inhibit BA synthesis. BAs are actively reabsorbed from the ileum by the ileal BA transporters and circulated back to the liver through the hepatic portal vein. This highly efficient process ensures that a majority of synthesized BAs are recycled with only 1-2% being converted into secondary BAs, deoxycholic acid (DCA), and lithocholic acid (LCA), by bacterial 7α-dehydroxylation in the terminal ileum and colon and excreted in feces. The regulation of BA circulation between the liver and intestines is summarized in Figure 1.
Figure 1. Overview of enterohepatic circulation of bile acids. (A color version of this figure is available in the online journal).
BA related molecular players
BA nuclear receptor FXR
In addition to modulating BA synthesis, FXR also regulates the expression of several transporters including apical sodium dependent BA transporter (ASBT), fatty acidbinding protein subclass 6, and organic solute transporter α and β to control the absorption of not only BAs but also lipids, vitamins, and xenobiotics.23 Interestingly, recent studies have implicated FXR in the interplay between obesity-associated BA dysregulation and gut dysbiosis to potentially promote carcinogenesis in the liver and colon. Within the context of obesity and diabetes, FXR can regulate glucose and lipid homeostasis through actions at various sites along the gut-liver axis. Agonist activation or hepatic overexpression of FXR significantly lowered blood glucose levels in both diabetic and wild type24 mice.24,25 FXR stimulation also decreased blood low density lipoprotein levels and inhibited fatty acid and TG synthesis in mice fed a high sugar and fat diet22. These combined effects of FXR protected mice against body weight gain, liver and muscle fat deposition, and reversed insulin resistance.26
At the metabolic level, FXR functions to repress hepatic gluconeogenesis, lipogenesis, and fatty acid synthesis genes. Consequently, FXR stimulation promotes glycogen synthesis and enhances insulin sensitivity in obese mice.25,26 FXR activity in pancreatic β-cell lines and human islets can regulate transcription factor Kruppellike factor 11 to increase insulin gene expression and protein kinase B-dependent phosphorylation and translocation of glucose transporter 2 at the plasma membrane of hepatocytes.27 By stimulating pancreatic insulin secretion and hepatic glucose uptake, FXR can effectively delay the pathological progression of insulin resistance, hyperglycemia, and glucosuria in diabetic mice. Consistently, FXR knockout (KO) mice exhibited glucose intolerance, insulin insensitivity, elevated serum TG, cholesterol, and BA levels resulting in greater hepatic fat accumulation compared to wild type (WT) mice.22 Hepatic FXR expression is conversely regulated by glucose levels in streptozotocininduced diabetic rats.28 Chromatin immunoprecipitation in mice also revealed that long-term high-glucose exposure increased histone acetylation and demethylation on the FXR-target Cyp7a1 gene promoter region leading to elevated basal expression and consequently, a larger BA pool with altered composition.29 These observations strongly support the existence of crosstalk between the cellular mechanisms regulating glucose, lipid, and BA homeostasis in the liver and intestines with FXR serving mediator.
A link between FXR and enterohepatic cancer was firmly established when FXR KO mice were found to have markedly elevated hepatic inflammatory and oxidative stress markers compared to WT mice and a striking 100% incidence rate of spontaneous liver tumors between 13 and 15 months of age.23,30 BA-containing diet further exacerbated inflammation and oxidative stress in FXR KO mouse liver supporting that BA dysregulation subjects hepatocytes to higher oxidative stress.31 Hepatocyte-specific overexpression of SHP failed to alter liver tumor incidence or size in FXR KO mice, but did result in lower neoplasia grade, decreased cell proliferation, and increased apoptosis.32 Moreover, FXR stimulation can down-regulate lipopolysaccharide (LPS)-induced, nuclear factor kappa-light-chain-enhancer of activated B cells (NFκβ)-mediated hepatic inflammation by suppressing the expression of proinflammatory mediators in human HCC cells and mouse primary hepatocytes.33 FXR KO mice displayed higher hepatic mRNA levels of inducible nitric oxide synthase, prostaglandin-endoperoxide synthase 2 (COX-2), chemokine ligand 10, and interferon type II (IFN-γ) which resulted in exaggerated inflammation and necrosis after LPS exposure at a dose that failed to elicit measurable liver injury or inflammation in WT mice.33 The HCC in FXR KO mice was associated with sustained oncogenic Wnt/B-catenin signaling through Wnt4 and disheveled induction, E-cadherin repression, and glycogen synthase kinase-3B inactivation as the mice aged.34
Furthermore, microarray analysis of FXR KO mouse liver revealed altered gene expression profiles related to metabolism, inflammation, and fibrosis compared to WT liver recapitulating human HCC progression.35 Liver tumor bearing FXR KO mice showed elevated levels of interleukin 6 (IL-6) and signal transducer and activator of transcription 3 (STAT3) due to diminished expression of suppressor of cytokine signaling 3, a direct FXR target gene.32 STAT3 activation in conjunction with elevated TNFα and IL-6 levels has been shown to potentiate HCC formation.36 Additionally, FXR can epigenetically silence the promoter of gankyrin, a proteasome subunit responsible for the degradation of retinoblastoma, p53, hepatic nuclear factor 4 alpha, and CCAAT (a sequence of DNA nucleotides)/enhancer-binding tumor suppressor proteins. The loss of FXR in mice increased gankyrin expression to promote tumorigenesis. Interestingly, long-lived little mice with high basal FXR expression do not develop liver cancer with age or carcinogen administration due to insufficient gankyrin induction.37 FXR activation in human hepatocytes and hepatoma cells protected against cytotoxicity induced by cisplatin and other DNA-damaging agents.38 These findings support that in addition to its metabolic regulation, FXR also functions to modulate oxidative stress, inflammation, and cell proliferation to inhibit cancer development.
Evidence also exists to suggest that FXR may act as a modulator of intestinal inflammation and a link between BA homeostasis and the intestinal microbiome. In the small intestine, FXR negatively regulates the expression of transporters involved in BA reabsorption while inducing the production and secretion of FGF19/Fgf15 to inhibit hepatic BA synthesis.23 Colon inflammation in Crohn's disease patients and rodent colitis models is correlated with reduced FXR mRNA levels. The progression of colon inflammation is exacerbated in FXR KO mice while treatment with FXR agonist attenuated colonic tissue damage and immune cell activation.39 Conversely, FXR stimulation protected WT mice from chemical-induced colitis by reducing epithelial permeability, ulceration, and inflammatory cell infiltration. Moreover, FXR agonist-treated WT mice and differentiated enterocyte-like cells displayed lower pro-inflammatory cytokines and better preserved epithelial barrier function.40
In addition to its beneficial effects on intestinal function and inflammation, a connection between FXR and intestinal microbes was observed when ampicillin-treated mice had inhibited ileal expression of FXR, SHP, and FGF19/Fgf15. Expression of FXR and its target genes levels were rescued by combination treatment with CA, but not taurocholic acid, in ampicillin-treated mice suggesting that enterobacteria can enhance BA-mediated FXR activity via taurocholic acid deconjugation.41 Furthermore, intestinal inflammation in mice down-regulated FXR expression in a toll-like receptor 9 (TLR9)-dependent manner since the FXR promoter contains a response element to interferon regulatory factor 7, a TLR9-regulated factor.42 These preliminary findings suggest a possible role of intestinal FXR as a mediator between BA homeostasis, the gut microbiome, and host immunity to prevent excessive inflammation and maintain GI health.
Examination of FXR in human HCC samples and cell lines has yielded further evidence to support its protective role against cancer formation. Marked reduction in FXR levels and activity were observed in human HCC samples compared to normal liver tissue. This reduction resulted from inhibition of hepatic nuclear factor 1 alpha activity on the FXR gene promoter by elevated pro-inflammatory mediators.34,35 The 3′ untranslated region of FXR mRNA was found to be a target of miR-421 and FXR downregulation by miR-421 promoted proliferation, migration, and invasion in human HCC cells.43 Decreased FXR levels in HCC cells also correlated with overexpression of active Ras resulting in strong activation of ERK1/2, a common characteristic of malignant cells.44,45 However, additional studies are required to determine whether dysregulated FXR activity increases the risk of HCC and CRC.
One possible underlying cause of FXR insufficiency in humans is genetic variation in the gene itself resulting in diminished expression or function. Indeed, sequencing analysis of FXR in intrahepatic cholestasis of pregnancy patients revealed four functional heterozygous variants, three of which demonstrated functional defects in either translation efficiency or signaling activity.46 Additionally, FXR polymorphism identification analysis of European-, African-, Chinese-, and Hispanic-Americans identified a common, hypomorphic single nucleotide polymorphism (SNP) with population allelic frequencies ranging from 2.5% (European Americans) to 12.1% (Chinese Americans). The in vitro transactivation activity of this hypomorphic SNP was lower relative to that of WT allele and human carriers of this allele showed significantly reduced hepatic SHP levels.47 Furthermore, the global FXR haplotype distribution between inflammatory bowel disease and healthy individuals was significantly different which emphasizes the link between FXR-mediated BA signaling and intestinal inflammation.48 Since chronic inflammation is widely considered a predisposition to cancer development, enhancement of FXR signaling appears to be a promising clinical target to not only normalize the BA dysregulation seen in obese and diabetic individuals but also combat chronic hepatic and intestinal inflammation.
BA transporter ASBT
The appropriate circulation of BAs between the liver and small intestine is crucial to the maintenance of BA homeostasis and consequently, normal GI physiology. The ileum is where approximately 90% of secreted BAs are actively reabsorbed into the bloodstream by ASBT for transport back to the liver through the hepatic portal vein.49,50 Because of its predominantly ileal expression and central role in enterohepatic cycling of BAs, ASBT is another potential participant in the interplay between BA dysregulation and gut dysbiosis. In Caco-2 cells, 25-hydroxycholesterol and CDCA treatments greatly reduced ASBT promoter activity and mRNA levels through the actions of FXR, SHP, retinoic acid receptor, and retinoid x receptor (RXR).51,52 Mice fed a cholesterol-enriched diet exhibited down-regulation of ASBT at both the mRNA and protein levels, decreased ileal BA uptake, and elevated fecal BA excretion.53 Interestingly, exposure of Caco-2 cells to pro-inflammatory factor IL-1B also caused a 65% reduction in ASBT mRNA level.54 Elevated levels of cholesterol in the intestinal lumen and pro-inflammatory mediators in the intestinal epithelium appear to down-regulate ASBT activity, thereby disrupting enterohepatic BA circulation. Consequently, a greater amount of unabsorbed BAs remain in the intestines where they can be transformed by intestinal microbes into toxic, hydrophobic BAs.55 Indeed, ASBT KO mice had a 10 - to 20-fold increase in fecal BA excretion and an 80% reduction in BA pool size compared to WT mice despite up-regulated BA synthesis.56
Paralleling its upstream regulator FXR, ASBTactivity can also be modulated by the gut microbiome. Pharmacological inhibition of ASBT in diabetic fatty rats significantly raised fecal BA concentrations and non-fasting plasma total glucagon-like peptide 1 (GLP-1) while decreasing hemoglobin A1c and blood glucose. However, ASBT inhibition also reduced FXR mRNA levels in both the liver and small intestine, likely as compensation for the disrupted BA circulation.57 Interestingly, ASBT deficiency or inhibition in mice lowered serum glucose, insulin, and TG as a result of diminished sterol regulatory element-binding protein 1 c expression.58 Based on these results, ASBT inhibition appears as a possible clinical intervention for the management of obesity and diabetes. However, it remains to be determined whether such beneficial effects also occur in humans and whether they outweigh the adverse effects of disrupted BA cycling.
An association between ileal ASBT activity and intestinal microbes was established when ampicillin-treated mice showed markedly decreased BA fecal excretion and elevated BA concentrations in hepatic portal blood. The ampicillin-treated mice displayed significantly higher ileal ASBT mRNA and brush-border membrane protein levels, elevated total BAs, and reduced intestinal enterobacteria-biotransformed BAs. These observations indicate negative regulation of ileal ASBT expression by the gut microbiome.59 This modulation of ASBT activity by intestinal microbes was shown to occur in part through a proteasomal degradation pathway since proteasome inhibition attenuated the CA-induced reduction of ileal ASBT protein level in ampicillin-treated mice.60 Consistent murine findings showed Caco-2 cells infected with E. coli had drastically diminished ASBT activity due to decreased transport Vmax and protein level at the plasma membrane.61 This suggests that certain species of intestinal microbiome can alter the enterohepatic circulation of BAs by influencing ileal ASBT.
Furthermore, hypomorphic genetic variants in human ASBT have also been linked to increased cancer risk of the lower GI tract. One particular human ASBT SNP was associated with a twofold higher risk of colorectal adenomas potentially due to malabsorption of BAs leading to elevated colonic levels of toxic BAs.55,62 Another sequencing study revealed that carriers of several minor ASBT variants had significantly reduced ileal ASBT expression at both the mRNA and protein levels.63 Lastly, three ASBT nonsynonymous SNPs exhibited partially impaired to near complete loss of BA transport compared to WT allele in vitro.64 Being required for normal enterohepatic BA circulation, ASBT appears to be not only influenced by ileal FXR, cholesterol levels, and microbiota activity but also genetic variation.
G-coupled protein receptor TGR5
Although investigation into the mechanisms through which BAs function in the liver and intestines have been predominantly focused on nuclear receptors such as FXR, there is accumulating evidence for the participation of G-protein coupled BA receptor 1 (GPBAR1/TGR5) in mediating the systemic actions of BAs. Highly expressed in the liver, intestine, and brown adipose tissue, TGR5 complements FXR in regulating BA homeostasis, glucose metabolism, and enterohepatic inflammation.65 Therefore, examination of the regulatory roles of TGR5 and its potential contribution to BA dysregulation, gut dysbiosis and ultimately, liver and colon carcinogenesis is pertinent. TGR5 activation at the plasma membrane initiates the canonical G-protein signal transduction resulting in initiation of downstream pathways that remain poorly defined.66
Nevertheless, TGR5 was implicated in modulating BA and energy metabolism when TGR5 KO mice on a highfat diet experienced a 21–25% reduction in BA pool size and greater body fat accumulation accompanied by body weight gain relative to WT mice.50 Intriguingly, TGR5 can stimulate GLP-1 release and suppress hepatic glycogenolysis resulting in improved glucose tolerance, hepatic and pancreatic functions in obese mice.67,68 Treatment of enteroendocrine cells with a TGR5 agonist also enhanced GLP-1 secretion.68 Administration of BA sequestrant to dietinduced obese mice caused markedly higher energy expenditure, body weight reduction, and improved glycemic control.69,70 These findings in mice were replicated in monkeys with TGR5 agonist triggering co-secretion of peptide YY (PYY) and GLP-1 from distal GI L-cells to produce sustained improvements in glucose tolerance.71 From these studies, TGR5 appears to control BA and glucose metabolism through its up-regulation of PYY and GLP-1. However, it is unknown whether if TGR5 expression or function is inhibited in obese and diabetic individuals.
Complementary to FXR, TGR5 can prevent excessive inflammation in the liver and intestines to preserve normal physiology. In mouse macrophages and Kupffer cells, TGR5 activation suppressed IκBα phosphorylation, p65 translocation, NFκβ DNA binding, and transcriptional regulation activity by stabilizing the interaction between IkBa and β-arrestin 2. Consistently, TGR5 KO mice exhibited more severe LPS-induced liver necrosis and inflammation relative to WT mice whereas TGR5 agonist inhibited LPS-induced NFκβ-mediated expression of pro-inflammatory mediators in WT mouse liver.72 Loss of TGR5 in mice also increased susceptibility to diethylnitrosamine-induced acute liver injury and cancer due to increased hepatocyte death, compensatory proliferation, and expression of inflammatory cytokines.73 Moreover, TGR5 KO mice displayed abnormal hydrophobic BA composition, severe hepatocyte necrosis, prolonged cholestasis, exacerbated inflammatory response, and delayed regeneration compared to WT mice post partial hepatectomy.74 Treatment of non-alcoholic fatty liver disease (NAFLD) mice with dual FXR/TGR5 agonist decreased intrahepatic inflammation and improved histological features.75 Furthermore, TGR5 KO mice developed abnormal colonic mucous cell morphology with an altered molecular architecture of epithelial tight junctions resulting in increased intestinal permeability and microbial translocation.76 TGR5 stimulation reduces TNFα production by Crohn's disease-associated macrophages after LPS exposure by inhibiting c-FOS phosphorylation and NFκβ activation.77 Taken together, these findings implicate TGR5 as another important player in protecting the liver and intestines against excessive inflammation.
In addition to regulating energy homeostasis and inflammation, altered TGR5 expression and activity can also influence signaling pathways associated with cancer formation in cells of the liver, intestines, and beyond. Treatment of human gastric carcinoma cells with BAs activated epidermal growth factor receptor (EGFR) and ERK1/2 proliferative signaling in a TGR5-dependent manner.78,79 DCA exposure redistributed TGR5 to plasma membrane microdomains to transactivate EGFR resulting in downstream ERK1/2 activation.80 Low concentrations of CDCA can stimulate human endometrial cancer cell growth by upregulating cyclin D1 expression through TGR5-mediated recruitment of cAMP response element-binding protein to the cyclinD1 gene promoter.81 In human HCC cells, TGR5 inhibition decreased BA-induced caspase 8 activation by interfering with the JNK-mediated recruitment of caspase 8 to the death-inducing signaling complex.30 Furthermore, TGR5 stimulation inhibited cell proliferation and migration by suppressing STAT3 phosphorylation, transcription, and DNA binding activity.73 These results implicate TGR5 as a regulator of survival and growth pathways in liver and GI cells in response to BAs. Thus, within the context of BA dysregulation, overstimulation of TGR5 may encourage carcinogenesis.
Indeed, a significant positive correlation was found between BA concentrations and the grades of intestinal atrophy and metaplasia with GI cancer developing more frequently in individuals with high BA concentrations.82 Strong TGR5 staining was present in 12% of human intestinal metaplasia cases but none in normal gastric epithelium cases and moderate to strong membranous and cytoplasmic TGR5 staining was present in 52% of intestinal but only 25% of diffuse subtype adenocarcinomas.79 Sequencing of primary sclerosing cholangitis patients identified six TGR5 variants, five of which were found to exhibit reduced or abolished function because of altered localization or activity.83 Akin to FXR and ASBT, TGR5 appears to be another intersection between BA and glucose homeostasis, inflammation, and cell proliferation in the liver and intestines.
Relationship between diabesity, BA dysregulation, and gut dysbiosis
Crosstalk between BA homeostasis and the intestinal microbiota
Beyond the previously discussed molecular players, BAs also interact with other receptors such as constitutive androstane receptor, pregnane x receptor, and vitamin D receptor to regulate glucose and lipid homeostasis as well as innate immunity.84 Although BAs are critical for regulating lipid absorption and glucose homeostasis, they can exert harmful effects when their levels become dysregulated. In abnormally high concentrations, hydrophobic secondary BAs are cytotoxic, leading to DNA damage and cell death.85 BAs also have antimicrobial and amphipathic properties that are regarded as important regulators for the gut microbe environment with DCA being the most potent of all BAs at physiological concentrations in the human colon.14 DCA at a concentration of 0.5mM can effectively inhibit intestinal bacteria growth in cell culture, indicating that BA can regulate intestinal microbe composition through environmental stress.86 The human intestines house 10–100 trillion microbes, providing efficient metabolic capability to process indigestible dietary sources.87 Since the gut microbiome profile is largely dependent on the host diet and surrounding intestinal environment, there is a degree of variability among individuals, even in twins. However, shared microbial genes are identifiable to construct a “core microbiome” at the genetic level.88,89 Disturbances to the gut microbiome with phylum-level changes can result in altered nutrient acquisition, energy extraction, and metabolism.90
The crosstalk between BAs and the gut microbiome was established in the mid-1960s and continues to be a major research focus. A direct relationship was observed between the intestinal microbiota and BA levels using nuclear magnetic resonance spectroscopy (NMR) after microbial colonization in germ-free mice.16 These intestinal microbes stimulated multiple pathways in xenobiotic metabolism and pharmacokinetics in the liver, kidney, plasma, urine, and colon.16 CYP8B1, required for the production of CA, helps to control the hydrophobicity of the BA pool by regulating the CA/CDCA ratio in conjunction with CYP7A1 and CYP27A1. Together, these enzymes are regulated by FXR, in a negative feedback manner. However, overexpression of FXR in germ-free mice did not lead to down-regulation of Cyp8b1, Cyp7a1, or Cyp27a1.16 Therefore, the regulation of these genes by FXR is more complex than previously thought with possible participation by the gut microbiome. In addition to regulating intestinal Fgf15, gut microbes also regulated hepatic Cyp7a1 through intestinal FXR as well as endogenous FXR antagonists, tauro-conjugated alpha, and beta-muricholic acid as demonstrated in a mice feeding study.17 Conventionally raised mice had reduced expression level of FXR antagonists and Cyp7a1 and increased expression of Cyp8b1 allowing for greater intestinal FXR activity compared to germ-free mice.16,17 Since gut microbe colonization did not affect liver FXR, it is postulated that the intestinal microbiome acts mainly in the ileum to create a more hydrophobic BA profile by suppressing Cyp7a1 expression through increased induction of intestinal Fgf15.17
Diabesity associated shifts in BA and gut microbe profiles
Previous studies have shown that consumption of a high-fat diet increased BA secretion and altered the gut microbial profiles in obese animal models and in patients with type 2 diabetes.86,91 The human gut contains a variety of species with different segments of the GI tract varying in bacterial density and diversity.89 For instance, the ileum contains around 107 colony-forming units per gram of bacteria with a dominance of gram-negative aerobes and obligate anaerobes while the colon houses 1012 colony-forming units per gram of bacteria with a dominance of anaerobes.89,92 Gram-positive firmicutes and actinobacteria and gram-negative bacteroidetes are the most prominent phyla with each taxon differing in their ability to extract and utilize dietary and host-derived resources, leading to variations in host lipid, glucose, and amino acid metabolism. It was found that differences in body composition between obese (ob) twins and lean (ln) twins were attributable in part to variations in bacterial fermentation of short-chain fatty acids, metabolism of branched-chain amino acids, and transformation of BAs.93 Co-housing ln mice with ob mice prevented diet-induced obesity in the latter by facilitating an ingression of bacteroidetes from the ln to ob mouse gut and altered several BA concentrations to resemble those in ln cage mates and ln control. Carbohydrate fermentation and BA metabolism in co-housed ob mice were phenotypically rescued. Notably, a high fat diet with minimal fruits and vegetables selected against microbial profiles associated with leanness and prevented successful ingression by ln-microbes.93
The association between the intestinal microbiota and diet-induced weight gain in both mice and human models mentioned earlier suggests that BA may be interacting with the intestinal microbes in the pathogenesis of obesity and diabetes. Studies have found that increasing levels of the primary BA cause a shift toward firmicutes, particularly genus Clostridium cluster XIVa, which raises production of hydrophobic secondary BAs.18,19 Firmicutes increased significantly from 54.1% in the control group to 93.4% in the high CA group at the expense of bacteroidetes and other minority bacterial populations. Potentially pathogenic proteobacteria, specifically gammaproteobacteria E. coli, also expanded in the gut of rats fed the high CA diet, decreasing bacterial densities and diversity.18 Metagenome sequencing of the fecal microbiome of obese individuals was consistent with these murine study findings.94,95 Weight loss studies further showed that a decrease in firmicutes and an increase of bacteroides were correlated with percent body weight loss, but not caloric intake.96,97 Even though most studies showed a shift toward firmicutes in obese and diabetic individuals, others have shown opposite results: a shift toward bacteroidetes or no difference between these two phyla.98,99 Nevertheless, several bacterial species and families have been associated with biological markers of obesity and diabetes. E. coli and Lactobacillus, for example, are linked to leptin variation independent of weight changes while the Coriobacteriaceae family is correlated with changes in hepatic TG, glucose, and glycogen levels.16,100
Because various signaling pathways may be altered in the diseased state, crosstalk between the gut microbiome and host may also be disrupted.101 The most studied human gut bacteria responsible for 7α-dehydroxylation of BAs belong to the genus Clostridium, which are grampositive anaerobic members of the Firmicutes family. They are capable of regulating BA synthesis in the liver by removing FXR-antagonist tauro-beta-muricholic acid in the ileum and converting primary BAs into secondary BAs.102 Rats fed with CA displayed increased BA levels in the colon which selected against bacteroidetes and actinobacteria in favor of firmicutes, leading to greater generation of DCA and LCA, which can be cytotoxic at higher concentrations than the physiological range of 0.046–0.210 and 0–0.03 mM, respectively.14,19,103 Conversely, cirrhotic patients were found to have a collapse of Clostridium XIVa family because of a smaller BA pool.19 This direct relationship between the size of the BA pool and colonization by the Clostridium genus supports an intimate interaction between the intestinal microbiome and BA metabolism. The harmful effects of BA dysregulation and gut dysbiosis in the gut–liver axis are summarized in Figure 2.
Figure 2. The proposed mechanisms by which dysregulated bile acid and gut dysbiosis induce carcinogenesis. (A color version of this figure is available in the online journal.).

Gastric bypass alters BA synthesis and intestinal microbe composition
Rou-En-Y-Gastric Bypass (RYGB) is an increasingly popular bariatric surgical procedure for the treatment of obese and diabetic patients because of its resulting weight loss, improved insulin resistance and glucose intolerance, as well as reduction in other comorbidities. A systematic review and meta-analysis conducted for reviews from January 1990 to April 2006 showed that a striking 86.6% of type II diabetes mellitus patients who underwent gastric bypass surgery experienced resolution or dramatically alleviated symptoms of improved glycemic index and insulin levels.104 The mechanisms underlying these beneficial outcomes remain subjects of intense investigation. Leptin circulation, pH changes, GLP-1 secretion, as well as BA synthesis and gut microbiome shifts are all postulated as potential mechanisms.105–109 Due to the nature of the procedure, gastric bypass patients were thought to have poorer reabsorption rate, thus increasing BA synthesis and improving their glucose tolerance. Several studies reported a positive correlation between the increased postprandial GLP-1 and insulin secretion and the significant increase in serum BA in obese individuals compared to lean controls.110,111 In an attempt to elucidate the effect of RYGB on BA synthesis, a similar technique was used in rats to study the molecular changes during intestinal adaptation. A decrease in mRNA expression of Cyp7a1 and Cyp27a1 and major BA exporters such as bile salt export pump and organic anion-transporting polypeptide was observed in rats subjected to bile diversion surgery.112 Ileal FXR and Fgf15 were also reduced at the mRNA level, but no changes were observed for hepatic FXR, which is consistent with previous results from ileal interposition.112 Causing increased serum BA levels and reduced in ER stress markers, bile diversion improved glucose tolerance and liver steatosis in diet-induced obese rats.112 RYGB on obese patients, similarly, had restored blunted BA mobilization, suggesting a possible weight loss mechanism postsurgery.113
An altered intestinal lumen microbiota can also contribute to the weight loss seen after RYGB. Several studies reported that intestinal microbes quickly adapt to the starvation-like, less acidic GI environment. The transfer of the gut microbial community from RYGB mice to germ-free mice resulted in similar weight loss and adiposity observed in RYGB patients.114 Microbiota analysis via next generation sequencing revealed that a notable increase in the relative abundance of Escherichia and Faecalibacterium prausnitzii across the fecal content of mice, rats, and humans after receiving RYGB independent of calorie restriction.100,114 F. prausnitzii abundance was also associated with reduced low-grade inflammation and reduction in colitis.115 Furthermore, plasma leptin levels negatively correlated with E. coli levels and positively correlated with Lactobacillus levels. A marked decrease in blood bacterial DNA was also noted suggesting that RYGB restricted bacterial translocation in the intestines. Meanwhile, the role of Bifidobacteria in regulating GI health remains unclear. After RYGB, Bifidobacterium count increases and negatively correlated with adiponectin, a marker for insulin sensitivity and inflammation in the liver.100,101 Additional investigation at the genetic and molecular levels is needed to elucidate specific microbial and metabolomic mechanisms from which the beneficial results of RYGB are derived. Nevertheless, the aforementioned findings further associate BA homeostasis and the gut microbiome in the pathogenesis of obesity and diabetes.
BA dysregulation and gut dysbiosis in the gut–liver axis
Effects on inflammation, immunity, and metabolism
A growing body of evidence has linked various inFammatory signaling pathways including TLR/MyD88, NFκB, and COX2 as bridging factors between pathogen-triggered inflammation and carcinogenesis. More recently, BA dysregulation and gut dysbiosis were also implicated in modulating the inflammatory process. Serum BA levels have increasingly served as biomarkers for liver diseases, obesity, and diabetes.116 BAs not only regulate hepatic de novo lipogenesis, TG export, and plasma turnover, but also hepatic gluconeogenesis and insulin sensitivity through the actions of FXR and TGR5.15 For instance, ursodeoxycholic acid (UDCA), a weak FXR and TGR5 binding ligand, improved hepatic ER stress and insulin sensitivity in diabetic mice, while norUDCA, a short chained homologue of UDCA, lowered hepatic TG levels.15 BA-activated FXR inhibited bacterial overgrowth and mucosal injury in the distal small intestine through promotion of innate defense mechanisms to prevent epithelial barrier deterioration and bacterial translocation, thus reducing the risk for HCC and CRC.117,118
Conversely, BA dysregulation causes increased bacterial translocation by disrupting barrier function in the small intestine leading to systemic infection.117,119,120 High concentration of DCA and CDCA can damage tissue by solubilizing the cell membrane and acting as immunosuppressive agents.120 Indeed, BAs, especially DCA over 500 μM, can increase chloride secretion and intestinal permeability, inhibit mucosal healing, and eventually elicit intestinal inflammation in the colon often seen in irritable bowel disease (IBD) patients.119,120 Similar dose-dependent findings were also found in mice feeding studies. The physiological DCA concentration under a high-fat diet (1–3 mM) can impair gut barrier function whereas DCA concentration under a low-fat diet (0–1 mM) did not disrupt gut permeability.103,121 Secondary BA UDCA and DCA exhibited opposing roles, although higher than physiological concentrations of UDCA were necessary to produce significant protective effects against DCA-induced gut permeability, especially in the colon. A high-fat diet was reported to decrease the ratio of UDCA and DCA, even though the overall fecal BA pool is larger.121 The enterohepatic circulation of toxic BAs such as DCA increased the senescence-associated secretory phenotype in mouse hepatic stellate cells, induced greater levels of inflammatory and tumor-promoting factors, and promoted non-alcoholic steatohepatitis and HCC development.122
Furthermore, the integrity of the intestinal mucosal barrier and immunological activities are governed by a network of nuclear receptors including FXR. Increases in intestinal FXR and TNFα expression were observed in mice fed a high fat diet.121 Elevated BA not only up-regulated numerous pro-inflammatory mediators and NFκβ, but also shifted intestinal microbe composition toward endotoxin-producing species. Endotoxic microbes are capable of aggravating glucose tolerance and insulin resistance to elicit increased metabolic inflammation and gut permeability.123,124 Low grade inflammation and harmful metabolites generated by endotoxic microbes in the gut permitted microbial translocation into the enterohepatic circulation. Bacterial translocation through compromised tight junctions in the small intestine was found to precede intestinal bacterial overgrowth in acute liver injury caused by cholestasis, alcohol toxicity, and obesity in mice.117 Given that plasma endotoxin levels correlate with the severity of liver disease and that KO mice lacking toll-like receptor 2 and TNFα signaling pathways are resistant to liver injury and fibrosis, bacterial translocation appears to play a role in the progression of chronic liver disease.117,125,126 Although bacterial LPS may not be involved in the onset of gut barrier dysfunction, it translocates through the intestinal mucosa in the presence of a leaky gut.121 Consistent with murine findings, elevated bacterial and antigen uptake in the intestines of obese and diabetic individuals resulted from impaired tight junctions.119
It is uncertain whether microbial profile shift is the cause or consequence of increased intestinal permeability, but it is clear that the intestinal microbiome significantly impacts intestinal and colonic health. Individuals suffering from gut dysbiosis, characterized by a reduction in beneficial, anti-inflammatory Bifidobacterium and Lactobacillus and elevation in aerobic, pro-inflammatory Enterobacter and Clostridium species, are at higher risk for liver diseases.127 Hydrophobic, taurine-conjugated BAs enhanced the growth of sulfate-reducing gut bacteria, antigen and bacterial translocation resulting in ulcerative colitis and CRC.128–130 One study positively correlated fecal levels of CDCA with members of the Enterobacteriaceae family while another found a positive linkage between Enterobacteriaceae, endotoxemia, and hepatic inflammation.19,131 On the other hand, F. prausnitzii has been consistently associated with anti-inflammatory properties and a marked decrease in F. prausnitzii is seen in IBD patients compared to healthy individuals.91,100,115
Intestinal microbes not only play a role in gut permeability regulation and innate immunity, but also influence the expression of genes involved in regulating inflammation and energy homeostasis. A significant increase in gramnegative proteobacteria and the disappearance of Bifidobacteria were observed in the gut of mice under a high-fat diet.132 Inflammasome, a multiprotein oligomer that activates pro-inflammatory cytokines, has recently been proposed to mediate liver injury progression. In the gut, inflammasome protects against gram-negative bacterial infection whereas in the liver, it aggravates hepatic steatosis and inflammation through activation of TLR4 and TLR9, driving tumorigenesis.133 LPS can induce inflammasome through activation of caspase-11 and specific toll-like receptors.134 Interestingly, mice infused with LPS for 4 weeks displayed a similar phenotype to those under a high-fat diet exhibiting increased weight gain, insulin resistance, hepatic TG content, and adipose tissue inflammation.135
Furthermore, circulating LPS correlated positively with insulin levels, adipose macrophage infiltration, and fasting blood glucose in diabetic patients.136 Impaired tight junctions as previously discussed and chylomicron-facilitated transport are two mechanisms proposed for how LPS promotes inflammation in the liver and intestines. FXR KO mice with dysregulated BAs and increased inflammation also exhibit impaired glucose tolerance and decreased insulin sensitivity in a manner similar to diabetic mice and patients with type 2 diabetes mellitus.25,35,137 The role of FXR in the regulating glucose homeostasis was demonstrated from multiple murine studies and a Phase 2 clinical study for type 2 diabetes mellitus and NAFLD in which FXR stimulation improves glucose tolerance, hepatic steatosis, and insulin sensitivity.23,26,137 The lack of FXR up-regulation by elevated BA levels in mice fed a high-fat diet may suggest a disrupted feedback system in BA synthesis.
Intriguingly, the intestinal microbiota may be a potential contributor to the development of NAFLD by modulating lipid and glucose metabolism through FXR and TGR5 signaling. NAFLD is characterized by an accumulation of TG in hepatocytes, which increase inflammation and progresses into fibrosis, cirrhosis, and ultimately HCC.111 The condition is associated with low-grade chronic inflammation, a common feature in obese individuals with high visceral adiposity.6 Hepatocyte apoptosis resulting from increased fat in the liver and insulin resistance are common pathological signs of NAFLD.138,139 Elevated serum free fatty acid can impair insulin signaling, increase hepatic glucose production while hepatic fatty acid overload induces inflammatory cytokines, fibrogenic cytokines, and oxidative stress. Hepatic BA accumulation due to hepatic steatosis can up-regulate NFκb signaling along with major inflammatory markers IL-6, IL-8, and TNFα in the progression of liver carcinogenesis.140 Similar to the liver and the intestines, adipose tissue in obese and diabetic patients also exhibit increased expression of proinflammatory cytokines. Hypertrophic adipocytes and macrophage infiltration exacerbate this chronic inflammatory microenvironment and enhance the survival and proliferation of neoplastic liver and intestinal cells.141
Effects on genome integrity
As previously mentioned, obese and diabetic individuals suffer from abnormally elevated BA levels, particularly hydrophobic secondary BA such as DCA and LCA. A growing body of evidence exists to support that hydrophobic secondary BA can increase intracellular production of reactive oxygen and nitrogen species resulting in elevated oxidative stress and DNA damage. Insights into the mechanism of BA induced-genotoxicity were obtained when treatment of human Barrett's esophagus tissues with a combination of BAs led to significant elevation in oxidative stress biomarker 8-hydroxy-deoxyguanosine and reactive oxygen species (ROS).142,143 Moreover, esophageal cells treated with DCA showed increased generation of reactive nitrogen species and DNA damage which were inhibited by co-treatment with nitric oxide scavenger or pretreatment with nitric oxide synthase inhibitor.143,144 DCA-treated colon cancer cells exhibited higher ROS generation, membrane blebbing, activation of mitochondrial apoptotic pathway, and formation of apoptotic bodies compared to untreated cells.145 DCA exposure also resulted in micronuclei formation accompanied by a dose-dependent response in NFκb activation that was attenuated by cotreatment.146
The above studies clearly demonstrate that hydrophobic BAs induce cellular oxidative stress through elevated reactive oxygen and nitrogen species production with similar results observed between human liver and colon cell lines. Interestingly, human HCC cells treated with hydrophobic primary CDCA also displayed cell cycle arrest, caspase-9-like activity, poly ADP-ribose polymerase cleavage, dose-dependent sites of DNA lesions, and extensive nuclear fragmentation.147,148 These cells also showed activation of ERK1/2 leading to the phosphorylation and stabilization of myeloid cell leukemia sequence 1 (MCL-1) in a mitogenactivated protein kinase-dependent (MEK-dependent) manner which conferred greater resistance to chemotherapeutic drugs.147 These findings strongly support that excessive levels of hydrophobic BA can exert a carcinogenic effect on enterohepatic tissues by promoting genomic instability through oxidative injury. Figure 3 provides an overview of the implicated carcinogenic consequences of BA dysregulation, gut dysbiosis, and insulin resistance.
Figure 3. Overview of the interplay between bile acid dysregulation and gut dysbiosis in diabesity. (A color version of this figure is available in the online journal.).

Potential preventative measures
Synthetic compounds
Several synthetic drugs such as colesevelam and colestimide, designed to sequester BAs, have demonstrated efficacy in improving insulin resistance and glucose tolerance.149–151 BA receptor agonists also appeared to be promising in the management of obesity and diabetes related symptoms.152,153 Specifically, synthetic retinoid such as acyclic retinoid (ACR) was investigated for its beneficial effect on obesity-related liver tumorigenesis, since RXRα, a heterodimer partner of FXR, was found to be repressed in early stages of HCC due to phosphorylation by the Ras/MAPK signaling pathway. Results indicate that ACR inhibited the Ras/MAPK pathway, ameliorated liver steatosis, improved insulin sensitivity, and decreased inflammation. Ultimately, ACR appears promising in restoring RXRα function in the liver, making it an effective chemoprevention drug against HCC progression.36
Prebiotics, probiotics, and synbiotics
With the intestinal microbiota implicated as one of the key players in the progression of liver and colon carcinogenesis in obese and diabetic patients, selective modification of the gut microbial composition has been extensively researched as a viable alternative or additive to current treatment plans. Prebiotics promote the growth of beneficial bacteria while probiotics are live microorganisms, administered exogenously, which provide a benefit beyond nutrition. Most prebiotics exist in the form of non-digestible carbohydrates and exert protective effects against liver and colon cancer.154–157 Dietary fibers shortened GI transit time to reduce the length of exposure to toxic metabolites such as hydrophobic BAs and bacterial toxins while aiding in their incorporation in feces for excretion.158 In addition, these fibers enhanced the growth of beneficial bacteria such as Bifidobacteria, lowered intestinal pH, and inhibited the growth of harmful bacteria.158
An increase in bifidobacteria is the signature of prebiotic treatment using insulintype fructans or galactooligosaccharides.159 Other resistant dietary fibers such as pomegranate peel extract and natural phytoalexin resveratrol can alter the gut microbiome in favor of bifidobacteria while lowering inflammatory markers in the colon and visceral adipose tissue.160,161 Short chain fatty acids (SCFAs), the fermented end products of dietary fibers, are recognized for their ability to inhibit growth and promote apoptosis in colon and liver cancer cells.162 SCFAs can also activate various drug metabolizing enzymes to decrease DNA mutation and reduce cancer risk.162 Complementing SCFAs with fish oil selectively reduced unsaturated fatty acid accumulation in the liver, improving hepatic fat oxidation and inflammation.163
Furthermore, probiotics have been suggested as a tool to manage inflammatory bowel disease.164 Although no general consensus exists for the beneficial effects of probiotics in obese, diabetic, and NAFLD patients, reduced hepatic total fatty acid content and serum alanine aminotransferase levels were noted in rodent models treated with probiotics.165,166 A synergistic effect of probiotics and blueberry husks in preventing colon carcinomas and subsequent liver tumors has been observed in rats.167 Blueberry husks alone significantly decrease the number of colonic ulcers and dysplastic lesions while probiotics alone improved liver function by decreasing parenchymal infiltration and bacterial translocation. Thus, the combination treatment of probiotics and blueberry husks delayed colonic carcinogenesis and prevented hepatic injuries. The combined treatment also reduced endotoxin-producing enterobacteriaceae and increased beneficial, anti-inflammatory lactobacilli.167 Synbiotics, mixtures of probiotics and prebiotics, were noted to lower colon cancer risk through improved insulin resistance, reduced inflammation, and preservation of gut barrier integrity in rats fed a high-fat diet.168 In terms of gut microbial ecology, synbiotic treatment significantly increased fecal Lactobacillus species at the expense of potentially pathogenic E. coli and Staphylococcus, thus lowering the endotoxin level in cirrhotic patients.169
Dietary changes and natural derivatives
Simple dietary changes can also aid in reversing BA dysregulation and intestinal microbiome derangement. Vitamin B6 can improve colon health by significantly reducing hydrophobic LCA levels in the colon, creating a reduced LCA to DCA ratio. LCA was 20-fold more toxic than DCA toward liver and colon cancer cells, and vitamin B6 helped with detoxifying enzymes and decreased DNA damage.170 Moreover, various naturally derived products are also under investigation including the antioxidant tempol which reduced obesity and improved insulin resistance in mice fed a high-fat diet by activating bile salt hydrolase to increase intestinal tauro-beta-muricholic acid, an FXR antagonist.102 Tempol appears to exert its antiobesity effects through FXR since inhibition of intestinal FXR promoted diet-induced obesity and insulin resistance. Tempol was also able to shift the gut microbial profile from firmicutes toward bacteroidetes dominance.102 Plant-based products such as burdock powder and genistein also showed moderate efficacy in normalizing BA homeostasis and the gut microbiome in animal models.102,171 The proposed beneficial effects on BA dysregulation and gut dysbiosis by pharmacological and dietary intervention are described in Figure 4.
Figure 4. Beneficial effects of normalized bile acid homeostasis and gut microbiota. (A color version of this figure is available in the online journal.).

Conclusion
Great strides have been made in our understanding of the intricate relationship between BAs and the intestinal microbiome. There is growing interest in the roles of BAs and gut microbes in the pathophysiology of chronic liver and GI diseases. However, the precise mechanisms of how BA dysregulation and gut dysbiosis exacerbate the chronic symptoms of obesity and diabetes to promote liver and colon cancer remain poorly defined. Substantial experimental data suggest that in addition to chronic hyperinsulinemia and derangement of peptide hormones, gut dysbiosis and BA dysregulation, frequently observed in obese and diabetic patients, are also contributors to inflammation and injury of the liver and colon. The NIH Common Fund Human Microbiome Project should help provide some definitive answers to many of these partially explained findings on the role of the gut microbes in metabolic disorders. Even though our current knowledge on how BAs and gut microbial affect liver and intestinal health has substantially expanded, the molecular pathways of their interplay in enterohepatic diseases require further elucidation.
Acknowledgments
This review is supported by grants funded by National Institutes of Health CA53596 (YW), DK092100 (YW), and R01AT007079 (DM).
Footnotes
Author contributions: JT: researched relevant papers, synthesized information, and prepared manuscript. TC: researched relevant papers, synthesized information, and prepared manuscript. DM: provided guidance for synthesizing information, and edited manuscript. YYW: generated ideas, provided guidance for research topics and manuscript writing.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ, Smith SR Endocrine Society, American Diabetes Association. European Association for the Study of Diabetes, Obesity and type 2 diabetes: What can be unified and what needs to be individualized? Diabetes Care. 2011;34:1424–30. doi: 10.2337/dc11-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86:556–65. doi: 10.1093/ajcn/86.3.556. [DOI] [PubMed] [Google Scholar]
- 4.Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005–8. doi: 10.1038/sj.bjc.6603932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: A consensus report. Diabetes Care. 2010;33:1674–85. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calle EE, Kaaks R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 7.Tsugane S, Inoue M. Insulin resistance and cancer: epidemiological evidence. Cancer Sci. 2010;101:1073–9. doi: 10.1111/j.1349-7006.2010.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baranova A, Gowder SJ, Schlauch K, Elariny H, Collantes R, Afendy A, Ong JP, Goodman Z, Chandhoke V, Younossi ZM. Gene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistance. Obes Surg. 2006;16:1118–25. doi: 10.1381/096089206778392149. [DOI] [PubMed] [Google Scholar]
- 9.Fenton JI, Birmingham JM, Hursting SD, Hord NG. Adiponectin blocks multiple signaling cascades associated with leptin-induced cell proliferation in Apc Min/+ colon epithelial cells. Int J Cancer. 2008;122:2437–45. doi: 10.1002/ijc.23436. [DOI] [PubMed] [Google Scholar]
- 10.Kamada Y, Matsumoto H, Tamura S, Fukushima J, Kiso S, Fukui K, Igura T, Maeda N, Kihara S, Funahashi T, Matsuzawa Y, Shimomura I, Hayashi N. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J Hepatol. 2007;47:556–64. doi: 10.1016/j.jhep.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 11.Kumor A, Daniel P, Pietruczuk M, Malecka-Panas E. Serum leptin, adiponectin, and resistin concentration in colorectal adenoma and carcinoma (CC) patients. Int J Colorectal Dis. 2009;24:275–81. doi: 10.1007/s00384-008-0605-y. [DOI] [PubMed] [Google Scholar]
- 12.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011;19:349–59. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Shackel NA, Vadas MA, Gamble JR, McCaughan GW. Beyond liver fibrosis: Hepatic stellate cell senescence links obesity to liver cancer via the microbiome. Hepatology. 2013;59:2413–5. doi: 10.1002/hep.26932. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28:220–4. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- 16.Claus SP, Ellero SL, Berger B, Krause L, Bruttin A, Molina J, Paris A, Want EJ, de Waziers I, Cloarec O, Richards SE, Wang Y, Dumas ME, Ross A, Rezzi S, Kochhar S, Van Bladeren P, Lindon JC, Holmes E, Nicholson JK. Colonization-induced host-gut microbial metabolic interaction. MBio. 2011;2:e00271–10. doi: 10.1128/mBio.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–35. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141:1773–81. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 19.Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids, and gut microbiota: Unraveling a complex relationship. Gut Microbes. 2013;4:382–7. doi: 10.4161/gmic.25723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang JY. Regulation of bile acid synthesis. Front Biosci. 1998;3:d176–93. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 21.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–26. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 22.Wei J, Qiu de K, Ma X. Bile acids and insulin resistance: Implications for treating nonalcoholic fatty liver disease. J Dig Dis. 2009;10:85–90. doi: 10.1111/j.1751-2980.2009.00369.x. [DOI] [PubMed] [Google Scholar]
- 23.Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368:17–29. doi: 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weljie AM, Newton J, Mercier P, Carlson E, Slupsky CM. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal Chem. 2006;78:4430–4442. doi: 10.1021/ac060209g. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. Activation of the nuclear receptor FXR improves hyper-glycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. 2006;103:1006–11. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–84. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim Biophys Acta. 2010;1802:363–72. doi: 10.1016/j.bbadis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–8. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 29.Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ. Nuclear receptors HNF4alpha and LRH-1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem. 2012;287:41334–41. doi: 10.1074/jbc.M112.421834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JI, Yoon JH, Myung SJ, Gwak GY, Kim W, Chung GE, Lee SH, Lee SM, Kim CY, Lee HS. Bile acid-induced TGR5-dependent c-Jun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007;361:156–61. doi: 10.1016/j.bbrc.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Nomoto M, Miyata M, Yin S, Kurata Y, Shimada M, Yoshinari K, Gonzalez FJ, Suzuki K, Shibasaki S, Kurosawa T, Yamazoe Y. Bile acid-induced elevated oxidative stress in the absence of farnesoid X receptor. Biol Pharm Bull. 2009;32:172–8. doi: 10.1248/bpb.32.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G, Zhu Y, Tawfik O, Kong B, Williams JA, Zhan L, Kassel KM, Luyendyk JP, Wang L, Guo GL. Mechanisms of STAT3 activation in the liver of FXR knockout mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G829–37. doi: 10.1152/ajpgi.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–43. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338:12–21. doi: 10.1124/jpet.111.179390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, Zhang L, Liu X, Yen Y, Lai L, Forman BM, Xu Z, Xu R, Huang W. Hepatocarcinogenesis in FXR-/- mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol. 2012;26:775–85. doi: 10.1210/me.2011-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimizu M, Tanaka T, Moriwaki H. Obesity and hepatocellular carcinoma: Targeting obesity-related inflammation for chemoprevention of liver carcinogenesis. Semin Immunopathol. 2013;35:191–202. doi: 10.1007/s00281-012-0336-6. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, Anakk S, Mayor A, Darlington G, Finegold M, Moore D, Timchenko NA. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology. 2013;57:1098–106. doi: 10.1002/hep.26146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaquero J, Briz O, Herraez E, Muntane J, Marin JJ. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim Biophys Acta. 2013;1833:2212–9. doi: 10.1016/j.bbamcr.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–61. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 40.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S, Penna G, Laverny G, Adorini L, Moschetta A, van Mil SW. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–72. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 41.Kuribayashi H, Miyata M, Yamakawa H, Yoshinari K, Yamazoe Y. Enterobacteria-mediated deconjugation of taurocholic acid enhances ileal farnesoid X receptor signaling. Eur J Pharmacol. 2012;697:132–8. doi: 10.1016/j.ejphar.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 42.Renga B, Mencarelli A, Cipriani S, D'Amore C, Carino A, Bruno A, Francisci D, Zampella A, Distrutti E, Fiorucci S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS One. 2013;8:e54472. doi: 10.1371/journal.pone.0054472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Gong W, Dai S, Huang G, Shen X, Gao M, Xu Z, Zeng Y, He F. Downregulation of human farnesoid X receptor by miR-421 promotes proliferation and migration of hepatocellular carcinoma cells. Mol Cancer Res. 2012;10:516–22. doi: 10.1158/1541-7786.MCR-11-0473. [DOI] [PubMed] [Google Scholar]
- 44.Fujino T, Takeuchi A, Maruko-Ohtake A, Ohtake Y, Satoh J, Kobayashi T, Tanaka T, Ito H, Sakamaki R, Kashimura R, Ando K, Nishimaki-Mogami T, Ohkubo Y, Kitamura N, Sato R, Kikugawa K, Hayakawa M. Critical role of farnesoid X receptor for hepatocellular carcinoma cell proliferation. J Biochem. 2012;152:577–86. doi: 10.1093/jb/mvs101. [DOI] [PubMed] [Google Scholar]
- 45.Lax S, Schauer G, Prein K, Kapitan M, Silbert D, Berghold A, Berger A, Trauner M. Expression of the nuclear bile acid receptor/farnesoid X receptor is reduced in human colon carcinoma compared to nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int J Cancer. 2012;130:2232–9. doi: 10.1002/ijc.26293. [DOI] [PubMed] [Google Scholar]
- 46.Van Mil SW, Milona A, Dixon PH, Mullenbach R, Geenes VL, Chambers J, Shevchuk V, Moore GE, Lammert F, Glantz AG, Mattsson LA, Whittaker J, Parker MG, White R, Williamson C. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133:507–16. doi: 10.1053/j.gastro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 47.Marzolini C, Tirona RG, Gervasini G, Poonkuzhali B, Assem M, Lee W, Leake BF, Schuetz JD, Schuetz EG, Kim RB. A common polymorphism in the bile acid receptor farnesoid X receptor is associated with decreased hepatic target gene expression. Mol Endocrinol. 2007;21:1769–80. doi: 10.1210/me.2007-0025. [DOI] [PubMed] [Google Scholar]
- 48.Attinkara R, Mwinyi J, Truninger K, Regula J, Gaj P, Rogler G, Kullak-Ublick GA, Eloranta JJ Swiss I.B.D. C.S.G. Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res Notes. 2012;5:461. doi: 10.1186/1756-0500-5-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claro da Silva T, Polli JE, Swaan PW. The solute carrier family 10 (SLC10): Beyond bile acid transport. Mol Aspects Med. 2013;34:252–69. doi: 10.1016/j.mam.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosters A, Karpen SJ. Bile acid transporters in health and disease. Xenobiotica. 2008;38:1043–71. doi: 10.1080/00498250802040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alrefai WA, Sarwar Z, Tyagi S, Saksena S, Dudeja PK, Gill RK. Cholesterol modulates human intestinal sodium-dependent bile acid transporter. Am J Physiol Gastrointest Liver Physiol. 2005;288:G978–85. doi: 10.1152/ajpgi.00379.2004. [DOI] [PubMed] [Google Scholar]
- 52.Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–56. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- 53.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 54.Thomas C, Landrier JF, Gaillard D, Grober J, Monnot MC, Athias A, Besnard P. Cholesterol dependent downregulation of mouse and human apical sodium dependent bile acid transporter (ASBT) gene expression: Molecular mechanism and physiological consequences. Gut. 2006;55:1321–31. doi: 10.1136/gut.2005.085555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alrefai WA, Gill RK. Bile acid transporters: Structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803–23. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 56.Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278:33920–7. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, Nystrom C, Croom D, Ross S, Collins J, Rajpal D, Hamlet K, Smith C, Gedulin B. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab. 2012;302:E68–76. doi: 10.1152/ajpendo.00323.2011. [DOI] [PubMed] [Google Scholar]
- 58.Lundasen T, Andersson EM, Snaith M, Lindmark H, Lundberg J, Ostlund-Lindqvist AM, Angelin B, Rudling M. Inhibition of intestinal bile acid transporter Slc10a2 improves triglyceride metabolism and normalizes elevated plasma glucose levels in mice. PLoS One. 2012;7:e37787. doi: 10.1371/journal.pone.0037787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyata M, Yamakawa H, Hamatsu M, Kuribayashi H, Takamatsu Y, Yamazoe Y. Enterobacteria modulate intestinal bile acid transport and homeostasis through apical sodium-dependent bile acid transporter (SLC10A2) expression. J Pharmacol Exp Ther. 2011;336:188–96. doi: 10.1124/jpet.110.171736. [DOI] [PubMed] [Google Scholar]
- 60.Miyata M, Yamakawa H, Hayashi K, Kuribayashi H, Yamazoe Y, Yoshinari K. Ileal apical sodium-dependent bile acid transporter protein levels are down-regulated through ubiquitin-dependent protein degradation induced by bile acids. Eur J Pharmacol. 2013;714:507–14. doi: 10.1016/j.ejphar.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 61.Annaba F, Sarwar Z, Gill RK, Ghosh A, Saksena S, Borthakur A, Hecht GA, Dudeja PK, Alrefai WA. Enteropathogenic Escherichia coli inhibits ileal sodium-dependent bile acid transporter ASBT. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1216–22. doi: 10.1152/ajpgi.00017.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang W, Xue S, Ingles SA, Chen Q, Diep AT, Frankl HD, Stolz A, Haile RW. An association between genetic polymorphisms in the ileal sodium-dependent bile acid transporter gene and the risk of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 2001;10:931–6. [PubMed] [Google Scholar]
- 63.Renner O, Harsch S, Schaeffeler E, Winter S, Schwab M, Krawczyk M, Rosendahl J, Wittenburg H, Lammert F, Stange EF. A variant of the SLC10A2 gene encoding the apical sodium-dependent bile acid transporter is a risk factor for gallstone disease. PLoS One. 2009;4:e7321. doi: 10.1371/journal.pone.0007321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ho RH, Leake BF, Urquhart BL, Gregor JC, Dawson PA, Kim RB. Functional characterization of genetic variants in the apical sodium-dependent bile acid transporter (ASBT; SLC10A2) J Gastroenterol Hepatol. 2011;26:1740–8. doi: 10.1111/j.1440-1746.2011.06805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, Lou G, Meng Z, Huang W. TGR5: A novel target for weight maintenance and glucose metabolism. Exp Diabetes Res. 2011;2011:853501. doi: 10.1155/2011/853501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54:1263–72. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G371–80. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–77. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, Schoonjans K. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep. 2012;2:430. doi: 10.1038/srep00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Watanabe M, Morimoto K, Houten SM, Kaneko-Iwasaki N, Sugizaki T, Horai Y, Mataki C, Sato H, Murahashi K, Arita E, Schoonjans K, Suzuki T, Itoh H, Auwerx J. Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS One. 2012;7:e38286. doi: 10.1371/journal.pone.0038286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ullmer C, Alvarez Sanchez R, Sprecher U, Raab S, Mattei P, Dehmlow H, Sewing S, Iglesias A, Beauchamp J, Conde-Knape K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br J Pharmacol. 2013;169:671–84. doi: 10.1111/bph.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54:1421–32. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen WD, Yu D, Forman BM, Huang W, Wang YD. Deficiency of G-protein-coupled bile acid receptor Gpbar1 (TGR5) enhances chemically induced liver carcinogenesis. Hepatology. 2013;57:656–66. doi: 10.1002/hep.26019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, Spraul A, Guettier C, Humbert L, Schoonjans K, Rainteau D, Tordjmann T. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–60. doi: 10.1002/hep.26463. [DOI] [PubMed] [Google Scholar]
- 75.McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi M, Rosen HR. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–70. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, Baldelli F, Donini A, Fiorucci S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoneno K, Hisamatsu T, Shimamura K, Kamada N, Ichikawa R, Kitazume MT, Mori M, Uo M, Namikawa Y, Matsuoka K, Sato T, Koganei K, Sugita A, Kanai T, Hibi T. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn's disease. Immunology. 2013;139:19–29. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007;354:154–9. doi: 10.1016/j.bbrc.2006.12.168. [DOI] [PubMed] [Google Scholar]
- 79.Cao W, Tian W, Hong J, Li D, Tavares R, Noble L, Moss SF, Resnick MB. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol Gastrointest Liver Physiol. 2013;304:G322–7. doi: 10.1152/ajpgi.00263.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jensen DD, Godfrey CB, Niklas C, Canals M, Kocan M, Poole DP, Murphy JE, Alemi F, Cottrell GS, Korbmacher C, Lambert NA, Bunnett NW, Corvera CU. The bile acid receptor TGR5 does not interact with beta-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem. 2013;288:22942–60. doi: 10.1074/jbc.M113.455774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Casaburi I, Avena P, Lanzino M, Sisci D, Giordano F, Maris P, Catalano S, Morelli C, Ando S. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle. 2012;11:2699–710. doi: 10.4161/cc.21029. [DOI] [PubMed] [Google Scholar]
- 82.Tatsugami M, Ito M, Tanaka S, Yoshihara M, Matsui H, Haruma K, Chayama K. Bile acid promotes intestinal metaplasia and gastric -carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2012;21:2101–7. doi: 10.1158/1055-9965.EPI-12-0730. [DOI] [PubMed] [Google Scholar]
- 83.Hov JR, Keitel V, Laerdahl JK, Spomer L, Ellinghaus E, ElSharawy A, Melum E, Boberg KM, Manke T, Balschun T, Schramm C, Bergquist A, Weismuller T, Gotthardt D, Rust C, Henckaerts L, Onnie CM, Weersma RK, Sterneck M, Teufel A, Runz H, Stiehl A, Ponsioen CY, Wijmenga C, Vatn MH, Group IS, Stokkers PC, Vermeire S, Mathew CG, Lie BA, Beuers U, Manns MP, Schreiber S, Schrumpf E, Haussinger D, Franke A, Karlsen TH. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS One. 2010;5:e12403. doi: 10.1371/journal.pone.0012403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fiorucci S, Cipriani S, Mencarelli A, Renga B, Distrutti E, Baldelli F. Counter-regulatory role of bile acid activated receptors in immunity and inflammation. Curr Mol Med. 2010;10:579–95. doi: 10.2174/1566524011009060579. [DOI] [PubMed] [Google Scholar]
- 85.Baptissart M, Vega A, Maqdasy S, Caira F, Baron S, Lobaccaro JM, Volle DH. Bile acids: From digestion to cancers. Biochimie. 2013;95:504–17. doi: 10.1016/j.biochi.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 86.Yokota A, Fukiya S, Islam KB, Ooka T, Ogura Y, Hayashi T, Hagio M, Ishizuka S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes. 2012;3:455–9. doi: 10.4161/gmic.21216. [DOI] [PubMed] [Google Scholar]
- 87.Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26:5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 88.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–10. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Graessler J, Qin Y, Zhong H, Zhang J, Licinio J, Wong ML, Xu A, Chavakis T, Bornstein AB, Ehrhart-Bornstein M, Lamounier-Zepter V, Lohmann T, Wolf T, Bornstein SR. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: Correlation with inflammatory and metabolic parameters. Pharmacogenomics J. 2012;13:514–22. doi: 10.1038/tpj.2012.43. [DOI] [PubMed] [Google Scholar]
- 92.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care. 2010;33:2277–84. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F, Wu GD. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137:1716–24e1. 2. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–5. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 98.Angelakis E, Armougom F, Million M, Raoult D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012;7:91–109. doi: 10.2217/fmb.11.142. [DOI] [PubMed] [Google Scholar]
- 99.Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, Flint HJ. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–4. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 100.Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, Mariat D, Corthier G, Dore J, Henegar C, Rizkalla S, Clement K. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–57. doi: 10.2337/db10-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kong LC, Tap J, Aron-Wisnewsky J, Pelloux V, Basdevant A, Bouillot JL, Zucker JD, Dore J, Clement K. Gut microbiota after gastric bypass in human obesity: Increased richness and associations of bacterial genera with adipose tissue genes. Am J Clin Nutr. 2013;98:16–24. doi: 10.3945/ajcn.113.058743. [DOI] [PubMed] [Google Scholar]
- 102.Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. doi: 10.1038/ncomms3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song P, Zhang Y, Klaassen CD. Dose-response of five bile acids on serum and liver bile Acid concentrations and hepatotoxicty in mice. Toxicol Sci. 2011;123:359–67. doi: 10.1093/toxsci/kfr177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, Bantle JP, Sledge I. Weight and type 2 diabetes after bariatric surgery: Systematic review and meta-analysis. Am J Med. 2009;122:248–256 e5. doi: 10.1016/j.amjmed.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 105.Beckman LM, Beckman TR, Sibley SD, Thomas W, Ikramuddin S, Kellogg TA, Ghatei MA, Bloom SR, le Roux CW, Earthman CP. Changes in gastrointestinal hormones and leptin after Roux-en-Y gastric bypass surgery. J Parenter Enteral Nutr. 2011;35:169–80. doi: 10.1177/0148607110381403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glu-cagon-like peptide-1 and blunted glucose-dependent insulinotropic peptide secretion are associated with Roux-en-Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis. 2007;3:597–601. doi: 10.1016/j.soard.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, Auwerx J, Goldfine AB. Serum bile acids are higher in humans with prior gastric bypass: Potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671–7. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Werling M, Vincent RP, Cross GF, Marschall HU, Fandriks L, Lonroth H, Taylor DR, Alaghband-Zadeh J, Olbers T, Le Roux CW. Enhanced fasting and post-prandial plasma bile acid responses after Roux-en-Y gastric bypass surgery. Scand J Gastroenterol. 2013;48:1257–64. doi: 10.3109/00365521.2013.833647. [DOI] [PubMed] [Google Scholar]
- 109.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, Krajmalnik-Brown R. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106:2365–70. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pournaras DJ, Glicksman C, Vincent RP, Kuganolipava S, Alaghband-Zadeh J, Mahon D, Bekker JH, Ghatei MA, Bloom SR, Walters JR, Welbourn R, le Roux CW. The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology. 2012;153:3613–9. doi: 10.1210/en.2011-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zarrinpar A, Loomba R. Review article: The emerging interplay among the gastrointestinal tract, bile acids and incretins in the pathogenesis of diabetes and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;36:909–21. doi: 10.1111/apt.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kohli R, Kirby M, Setchell KD, Jha P, Klustaitis K, Woollett LA, Pfluger PT, Balistreri WF, Tso P, Jandacek RJ, Woods SC, Heubi JE, Tschoep MH, D'Alessio DA, Shroyer NF, Seeley RJ. Intestinal adaptation after ileal interposition surgery increases bile acid recycling and protects against obesity-related comorbidities. Am J Physiol Gastrointest Liver Physiol. 2010;299:G652–60. doi: 10.1152/ajpgi.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ahmad NN, Pfalzer A, Kaplan LM. Roux-en-Y gastric bypass normalizes the blunted postprandial bile acid excursion associated with obesity. Int J Obes (Lond) 2013;37:1553–9. doi: 10.1038/ijo.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liou AP, Paziuk M, Luevano JM, Jr, Machineni S, Turnbaugh PJ, Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med. 2013;5:178ra41. doi: 10.1126/scitranslmed.3005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P, Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–6. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, Erickson SK, Chiang JY. Glucose and insulin induction of bile acid synthesis: Mechanisms and implication in diabetes and obesity. J Biol Chem. 2012;287:1861–73. doi: 10.1074/jbc.M111.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283–92. doi: 10.1016/j.jhep.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–5. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Munch A, Strom M, Soderholm JD. Dihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsies. Scand J Gastroenterol. 2007;42:1167–74. doi: 10.1080/00365520701320463. [DOI] [PubMed] [Google Scholar]
- 120.Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, Apicella C, Capasso L, Paludetto R. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–13. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 121.Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: Epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G227–34. doi: 10.1152/ajpgi.00267.2012. [DOI] [PubMed] [Google Scholar]
- 122.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 123.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–86. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ravussin Y, Koren O, Spor A, LeDuc C, Gutman R, Stombaugh J, Knight R, Ley RE, Leibel RL. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity (Silver Spring) 2012;20:738–47. doi: 10.1038/oby.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gabele E, Froh M, Arteel GE, Uesugi T, Hellerbrand C, Scholmerich J, Brenner DA, Thurman RG, Rippe RA. TNFalpha is required for cholestasis-induced liver fibrosis in the mouse. Biochem Biophys Res Commun. 2009;378:348–53. doi: 10.1016/j.bbrc.2008.10.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hartmann P, Haimerl M, Mazagova M, Brenner DA, Schnabl B. Tolllike receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology. 2012;143:1330–40 e1. doi: 10.1053/j.gastro.2012.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao HY, Wang HJ, Lu Z, Xu SZ. Intestinal microflora in patients with liver cirrhosis. Chin J Dig Dis. 2004;5:64–7. doi: 10.1111/j.1443-9573.2004.00157.x. [DOI] [PubMed] [Google Scholar]
- 128.Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–72. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- 129.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–8. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Szabo C, Hellmich MR. Endogenously produced hydrogen sulfide supports tumor cell growth and proliferation. Cell Cycle. 2013;12:2915–6. doi: 10.4161/cc.26064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, White MB, Noble NA, Monteith P, Fuchs M, Thacker LR, Sikaroodi M, Bajaj JS. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58:949–55. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, Marzioni M, Benedetti A, Svegliati-Baroni G. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury. Hepatology. 2013;59:1738–49. doi: 10.1002/hep.26695. [DOI] [PubMed] [Google Scholar]
- 133.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–40. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 135.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 136.Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. 2011;34:392–7. doi: 10.2337/dc10-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–9. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li Z, Xue J, Chen P, Chen L, Yan SP, Liu L. Prevalence of nonalcoholic fatty liver disease in mainland of China: A meta-analysis of published studies. J Gastroenterol Hepatol. 2013;29:42–51. doi: 10.1111/jgh.12428. [DOI] [PubMed] [Google Scholar]
- 139.Park JS, Seo JH, Youn HS. Gut microbiota and clinical disease: Obesity and nonalcoholic fatty liver disease. Pediatr Gastroenterol Hepatol Nutr. 2013;16:22–27. doi: 10.5223/pghn.2013.16.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jonsson JR, Barrie HD, O'Rourke P, Clouston AD, Powell EE. Obesity and steatosis influence serum and hepatic inflammatory markers in chronic hepatitis C. Hepatology. 2008;48:80–7. doi: 10.1002/hep.22311. [DOI] [PubMed] [Google Scholar]
- 141.Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 2013;25:403–16. doi: 10.1016/j.cellsig.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 142.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A, Bernstein C, Green SB, Prasad A, Garewal HS. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett's oesophagus. Gut. 2007;56:763–71. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, Wang JY, Cheng E, Meyer F, Wang DH, Zhang Q, Spechler SJ, Souza RF. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-kappaB activation in benign Barrett's epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G278–86. doi: 10.1152/ajpgi.00092.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jolly AJ, Wild CP, Hardie LJ. Sodium deoxycholate causes nitric oxide mediated DNA damage in oesophageal cells. Free Radic Res. 2009;43:234–40. doi: 10.1080/10715760802684211. [DOI] [PubMed] [Google Scholar]
- 145.Smith AF, Longpre J, Loo G. Inhibition by zinc of deoxycholate-induced apoptosis in HCT-116 cells. J Cell Biochem. 2012;113:650–7. doi: 10.1002/jcb.23394. [DOI] [PubMed] [Google Scholar]
- 146.Jenkins GJ, Cronin J, Alhamdani A, Rawat N, D'Souza F, Thomas T, Eltahir Z, Griffiths AP, Baxter JN. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis. 2008;23:399–405. doi: 10.1093/mutage/gen029. [DOI] [PubMed] [Google Scholar]
- 147.Liao M, Zhao J, Wang T, Duan J, Zhang Y, Deng X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol Cancer. 2011;10:44. doi: 10.1186/1476-4598-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rolo AP, Palmeira CM, Holy JM, Wallace KB. Role of mitochondrial dysfunction in combined bile acid-induced cytotoxicity: The switch between apoptosis and necrosis. Toxicol Sci. 2004;79:196–204. doi: 10.1093/toxsci/kfh078. [DOI] [PubMed] [Google Scholar]
- 149.Bays HE. Long-term (52–78 weeks) treatment with colesevelam HCl added to metformin therapy in type 2 diabetes mellitus patients. Diabetes Metab Syndr Obes. 2012;5:125–34. doi: 10.2147/DMSO.S32018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Beysen C, Murphy EJ, Deines K, Chan M, Tsang E, Glass A, Turner SM, Protasio J, Riiff T, Hellerstein MK. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: A randomised controlled study. Diabetologia. 2012;55:432–42. doi: 10.1007/s00125-011-2382-3. [DOI] [PubMed] [Google Scholar]
- 151.Goldfine AB, Fonseca VA, Jones MR, Wang AC, Ford DM, Truitt KE. Long-term safety and tolerability of colesevelam HCl in subjects with type 2 diabetes. Horm Metab Res. 2010;42:23–30. doi: 10.1055/s-0029-1241195. [DOI] [PubMed] [Google Scholar]
- 152.Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol. 2012;165:414–23. doi: 10.1111/j.1476-5381.2011.01561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sato H, Macchiarulo A, Thomas C, Gioiello A, Une M, Hofmann AF, Saladin R, Schoonjans K, Pellicciari R, Auwerx J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. 2008;51:1831–41. doi: 10.1021/jm7015864. [DOI] [PubMed] [Google Scholar]
- 154.Wu WT, Cheng HC, Chen HL. Ameliorative effects of konjac glucomannan on human faecal beta-glucuronidase activity, secondary bile acid levels and faecal water toxicity towards Caco-2 cells. Br J Nutr. 2011;105:593–600. doi: 10.1017/S0007114510004009. [DOI] [PubMed] [Google Scholar]
- 155.Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC, Scott KP, Buc Calderon P, Feron O, Muccioli GG, Sonveaux P, Cani PD, Delzenne NM. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer. 2012;107:1337–44. doi: 10.1038/bjc.2012.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Serban DE. Gastrointestinal cancers: Influence of gut microbiota, pro-biotics and prebiotics. Cancer Lett. 2013;345:258–70. doi: 10.1016/j.canlet.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 157.Wollowski I, Rechkemmer G, Pool-Zobel BL. Protective role of probiotics and prebiotics in colon cancer. Am J Clin Nutr. 2001;73:451S–5S. doi: 10.1093/ajcn/73.2.451s. [DOI] [PubMed] [Google Scholar]
- 158.Slavin J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients. 2013;5:1417–35. doi: 10.3390/nu5041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Davis LM, Martinez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One. 2011;6:e25200. doi: 10.1371/journal.pone.0025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Larrosa M, Yanez-Gascon MJ, Selma MV, Gonzalez-Sarrias A, Toti S, Ceron JJ, Tomas-Barberan F, Dolara P, Espin JC. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J Agric Food Chem. 2009;57:2211–20. doi: 10.1021/jf803638d. [DOI] [PubMed] [Google Scholar]
- 161.Neyrinck AM, Van Hee VF, Bindels LB, De Backer F, Cani PD, Delzenne NM. Polyphenol-rich extract of pomegranate peel alleviates tissue inflammation and hypercholesterolaemia in high-fat diet-induced obese mice: Potential implication of the gut microbiota. Br J Nutr. 2013;109:802–9. doi: 10.1017/S0007114512002206. [DOI] [PubMed] [Google Scholar]
- 162.Zampa A, Silvi S, Fabiani R, Morozzi G, Orpianesi C, Cresci A. Effects of different digestible carbohydrates on bile acid metabolism and SCFA production by human gut micro-flora grown in an in vitro semi-continuous culture. Anaerobe. 2004;10:19–26. doi: 10.1016/j.anaerobe.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 163.Pedersen MH, Lauritzen L, Hellgren LI. Fish oil combined with SCFA synergistically prevent tissue accumulation of NEFA during weight loss in obese mice. Br J Nutr. 2011;106:1449–56. doi: 10.1017/S0007114511001917. [DOI] [PubMed] [Google Scholar]
- 164.Whelan K, Quigley EM. Probiotics in the management of irritable bowel syndrome and inflammatory bowel disease. Curr Opin Gastroenterol. 2013;29:184–9. doi: 10.1097/MOG.0b013e32835d7bba. [DOI] [PubMed] [Google Scholar]
- 165.Kelishadi R, Farajian S, Mirlohi M. Probiotics as a novel treatment for non-alcoholic fatty liver disease; a systematic review on the current evidences. Hepat Mon. 2013;13:e7233. doi: 10.5812/hepatmon.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J Gastroenterol. 2013;19:6911–8. doi: 10.3748/wjg.v19.i40.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hakansson A, Branning C, Molin G, Adawi D, Hagslatt ML, Jeppsson B, Nyman M, Ahrne S. Blueberry husks and probiotics attenuate colorectal inflammation and oncogenesis, and liver injuries in rats exposed to cycling DSS-treatment. PLoS One. 2012;7:e33510. doi: 10.1371/journal.pone.0033510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Raso GM, Simeoli R, Iaconoa A, Santoroa A, Ameroa P, Paciellob O, Russoa R, D'Agostinoa G, Di Costanzoc M, Cananic RB, Calignanoa A, Melia R. Effects of a Lactobacillus paracasei B21060 based synbiotic on steatosis, insulin signaling and toll like receptor expression in rats fed a high fat diet. J Nutr Biochem. 2013;25:81–90. doi: 10.1016/j.jnutbio.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 169.Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology. 2004;39:1441–9. doi: 10.1002/hep.20194. [DOI] [PubMed] [Google Scholar]
- 170.Okazaki Y, Utama Z, Suidasari S, Zhang P, Yanaka N, Tomotake H, Sakaguchi E, Kato N. Consumption of vitamin B(6) reduces fecal ratio of lithocholic acid to deoxycholic acid, a risk factor for colon cancer, in rats fed a high-fat diet. J Nutr Sci Vitaminol (Tokyo) 2012;58:366–70. doi: 10.3177/jnsv.58.366. [DOI] [PubMed] [Google Scholar]
- 171.Okazaki Y, Sitanggang NV, Sato S, Ohnishi N, Inoue J, Iguchi T, Watanabe T, Tomotake H, Harada K, Kato N. Burdock fermented by Aspergillus awamori elevates cecal Bifidobacterium, and reduces fecal deoxycholic acid and adipose tissue weight in rats fed a high-fat diet. Biosci Biotechnol Biochem. 2013;77:53–7. doi: 10.1271/bbb.120551. [DOI] [PubMed] [Google Scholar]