

Abstract
The RadA/Sms protein is a RecA-related protein found universally in eubacteria and plants, implicated in processing of recombination intermediates. Here we show that the putative Zn finger, Walker A motif, KNRXG motif and Lon protease homology domain of the Escherichia coli RadA protein are required for DNA damage survival. RadA is unlikely to possess protease activity as the putative active site serine is not required. Mutants in RadA have strong synergistic phenotypes with those in the branch migration protein RecG. Sensitivity of radA recG mutants to azidothymidine (AZT) can be rescued by blocking recombination with recA or recF mutations or by overexpression of RuvAB, suggesting that lethal recombination intermediates accumulate in the absence of RadA and RecG. Synthetic genetic interactions for survival to AZT or ciprofloxacin exposure were observed between RadA and known or putative helicases including DinG, Lhr, PriA, Rep, RuvAB, UvrD, YejH and YoaA. These represent the first affected phenotypes reported for Lhr, YejH and YoaA. The specificity of these effects sheds new light on the role of these proteins in DNA damage avoidance and repair and implicates a role in replication gap processing for DinG and YoaA and a role in double-strand break repair for YejH.
Introduction
The process of homologous recombination ensures genomic stability by promoting the error-free repair of broken or incompletely replicated chromosomes. All free-living organisms encode an strand-exchange protein that is essential for homologous recombination: RecA in eubacteria, RadA in archaea and Rad51 in eukaryotes [reviewed in (Holthausen et al., 2010)]. These proteins facilitate homologous strand-pairing and strand transfer by initial formation of an extended nucleoprotein filament on single-strand DNA (ssDNA). In Escherichia coli, the RecA filament not only plays an essential role in the early steps of homologous recombination but also acts as a signaling structure to initiate a transcriptional response to DNA damage, known as the ‘SOS response’ (Simmons et al., 2008; Lovett, 2010). The RecA filament is competent to promote strand exchange by pairing with homologous duplex DNA, where the complementary strand is extracted from the duplex and paired with the ssDNA strand of the protein:DNA filament.
Strand exchange between homologous DNA molecules produces branched intermediate DNA structures, including the four-strand structure known as the ‘Holliday junctions’. These branched intermediates require resolution, via nuclease, helicase and/or topoisomerase functions to yield viable dsDNA products. Three such resolution systems have been identified in E. coli: the RuvABC DNA branch migration/resolvase complex, the RecG DNA branch migration protein and the topoisomerase III/RecQ helicase complex [reviewed in (Persky and Lovett, 2008)].
Bacteria encode both RecA and a paralogous protein known as RadA (‘radiation-sensitive’) or Sms (‘sensitivity to MMS’) (Song and Sargentini, 1996). Despite its name, RadA/Sms proteins of eubacteria are not orthologs to the strand exchange protein RadA of archaea and are arguably more similar to the archaeal RadB paralog proteins. Genetic analysis of E. coli RadA implicates the protein as a late recombination function because of synergistic genetic effects in combination with mutations affecting known Holliday-junction processing functions, such as the RecG helicase and the RuvABC helicase/nuclease complex (Beam et al., 2002). Although these proteins have been denoted as helicases, they function in recombination, strictly speaking, as DNA pumps. Mutations in RadA eliminate the residual levels of recombination, as measured by conjugation, of recG ruvABC mutants, further reducing recombination about 100-fold. Both RecG and the Ruv complex process recombination intermediates in vitro: RecG and RuvAB catalyze Holliday junction branch migration and RuvC cleaves such junctions (reviewed in (Persky and Lovett, 2008). Effects of mutations in RadA also are synergistic with those in ruvABC and, especially strongly, with recG for sensitivity to DNA damaging agents. Other types of recombination appear to require RadA even when RecG and RuvAB are present: RadA is required for the recovery of genetic rearrangements induced by defects in the replication fork helicase, DnaB (Lovett, 2006).
RadA has been implicated in DNA repair and recombination reactions in eubacteria other than E. coli. The extreme resistance to ionizing radiation of Deinococcus radiodurans, which occurs by an extensive DNA synthesis-dependent strand-annealing mechanism, requires RadA (Slade et al., 2009). RadA is required for gene conversion in Rhizobium etli, in a pathway genetically distinct from those requiring RecG or RuvABC (Castellanos and Romero, 2009). RadA is required for genetic transformation in Streptococcus pneumoniae (Burghout et al., 2007) and Bacillus subtilis (Krüger et al., 1997). Genetic epistasis analysis in the latter organism supports a role for RadA in stabilization or processing of recombination intermediates, independent of RuvAB and RecG (Carrasco et al., 2004). Eubacterial-related RadA is also found in higher plants (Ishibashi et al., 2006) and plays a role in tolerance of UV exposure; it likely entered the plant genome via the chloroplast endosymbiont (Chintapalli et al., 2013).
Alignment of RadA protein sequences derived from diverged bacterial genomes suggests a three-domain structure for the protein. At its N-terminus are four invariant cysteine residues, reminiscent of a C4 Zinc-finger structure. Internal in the protein is an extended region with homology to RecA, including Walker A and B motifs characteristic of AAA+ ATPases. At its C-terminus, RadA aligns with a region from the Lon protease protein in bacteria. This region in E. coli RadA includes the Lon active site serine (but not other histidine and aspartate residues of the catalytic triad); however, this residue is not conserved among eubacterial RadA, and the function of this domain remains unclear. In the region between the RecA and Lon homologies lies a characteristic, conserved eubacterial RadA motif, KNRXG, found in eubacterial and plant RadA but not in other archaeal or eukaryotic RecA-related proteins (Beam et al., 2002; Giri et al., 2009).
In this work, we examine mutants affecting these features, including the putative Zn finger, ATPase Walker A box, KNRFG and Lon protein homology domain and its active site serine (S372), on DNA damage sensitivity phenotypes. We show that radA mutants are particularly sensitive to ciprofloxacin (CFX), a fluoroquinilone antibiotic that traps covalent DNA cleavage complexes with type II topoisomerases, gyrase and topoisomerase IV (Drlica and Zhou, 1997). In addition, radA mutants are sensitive to azidothymidine (AZT), a chain-terminating nucleoside that produces replication gaps and double-strand breaks (DSBs) in vivo (Cooper and Lovett, 2011). Both of these treatments elicit recombinational repair reactions (diagrammed in Fig. 1). We examine radA mutations in combination with a number of helicases/DNA pumps for survival to UV irradiation, ciprofloxacin and AZT and find it acts synergistically with many of these. These helicases can be grouped into classes based on their effects when combined with radA. The combination of deficiency in RadA and RecG is especially synergistic: although neither single mutant is very sensitive, the double mutant shows severe reduction in survival to DNA damage and replication fork inhibition. [This synergy had been observed previously for different genotoxins (Beam et al., 2002)]. For AZT, much of the sensitivity exhibited by radA recG mutants can be suppressed by an additional mutation in recA or recF but less well by lexA3, an allele that prevents induction of the SOS response. Expression of RuvAB in radA recG cells leads to strong suppression of UV and azidothymidine sensitivity and modest suppression of CFX sensitivity. Our observations suggest that recombination necessary for survival to these agents is blocked at a late step by loss of RadA and RecG and that lesions can be channeled into other pathways by an additional earlier block in RecA filament assembly or strand exchange. Our results are consistent with a role of RadA in branch-migration or resolution of branched intermediate structures, as has been proposed for RecG (Rudolph et al., 2010).
Figure 1.
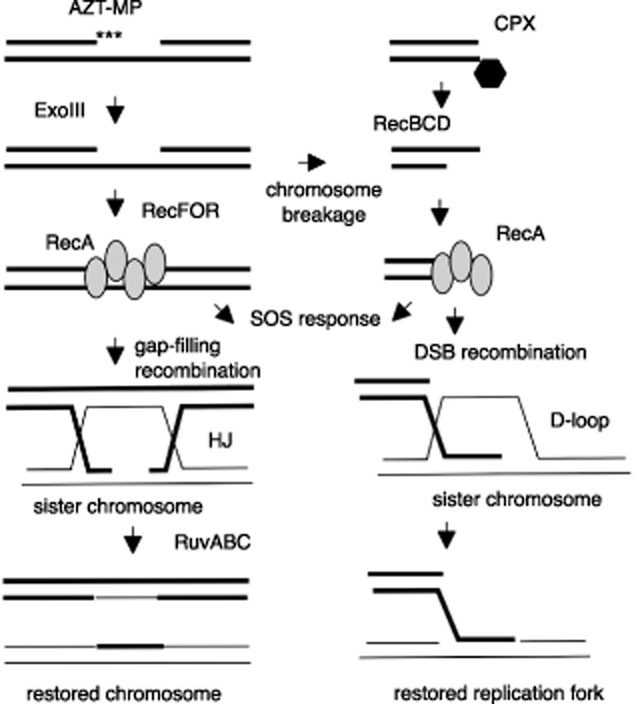
Possible recombination mechanisms for repair of AZT or ciprofloxacin damage. Left side: incorporation of AZT monophosphate into DNA causes chain termination during replication but can be removed by exonuclease III, leaving a replication gap. (Gaps can be converted to double-strand breaks, which are processed as shown on the right.) RecFOR promote RecA binding, which leads to induction of the SOS response. RecA also promotes recombination with the sister chromosome, producing intermediates such as Holliday junctions (HJs). After resolution, the chromosomes are healed. Right side: Ciprofloxacin traps topoisomerase II (gyrase or topo IV) covalent complexes, leading to a double-strand break. RecBCD resects the break and loads RecA, which signals the SOS response and promotes recombination with an intact sister chromosome to produce a displacement loop (D-loop) structure. Resolution of the D-loops restores a replication fork.
Results
Mutations were introduced into the identified motifs of RadA: the putative zinc finger [C28Y, previously characterized as radA100 (Song and Sargentini, 1996)], the Walker A ATPase motif (K108R), the KNRFG RadA motif (K258R) and the putative Lon-protease active site serine (S372A). In addition, two deletions, Δ228–460 and Δ275–460, were made to remove the Lon homology domain. Low-copy plasmids carrying these alleles were introduced into wild-type and radA mutant strain backgrounds, and survival to CFX was assayed for the resulting transformants (Table 1). The radA mutant is sensitive to CFX at 8 ng ml–1. This sensitivity phenotype was complemented by the plasmid-borne RadA+ and by the S372A mutant of RadA. In contrast, the remaining mutant radA plasmids failed to complement this defect, indicating that these affected motifs are required for RadA function. In the wild-type background, plasmids bearing K108R showed a significant reduction in CPX survival, indicating that the allele produces dominant-negative phenotypic effects and must interfere with the wild type protein function. Alleles C28Y, K258R and Δ228–460 showed mild reduction in survival and may likewise be semi-dominant.
Table 1.
Mutational analysis of RadA motifs
| % Survival (8 ng/ml CFX) |
||
|---|---|---|
| Plasmid | rad+ | radAΔ |
| Vector | 14 (± 5.6) | 0.70 (± 0.36) |
| RadA wt | 13 (± 1.9) | 47 (± 8.9) |
| RadA C28Y | 4.3 (± 1.6) | 0.69 (± 0.27) |
| RadA K108R | 1.5 (± 0.27) | 0.25 (± 0.099) |
| RadA K258R | 5.3 (± 1.1) | 0.32 (± 0.094) |
| RadA S372A | 11 (± 2.1) | 26 (± 4.5) |
| RadA Δ(228–460) | 6.6 (± 1.1) | 0.91 (± 0.35) |
| RadA Δ(275–460) | 9.9 (± 2.1) | 0.69 (± 0.27) |
Cultures of AB1157 or the isogenic radA deletion strain carrying the pWSK29 vector or plasmids containing the indicated RadA alleles were grown, diluted and plated on LB ampicillin plates with 8 ng/ml ciprofloxacin. Survival was determined after incubation at 37° overnight. At least eight isolates of each strain were tested over 2 or more days. Standard errors of the mean are shown in parentheses. Survival of strains lacking plasmids was 49% for wild-type and 1.5% for the radAΔ strain.
Our previous work had suggested strong synergy between RadA and RecG for survival after UV irradiation (Beam et al., 2002). Here we show that this synergy is also evident for survival in the presence of chain-terminating nucleoside, AZT as well as for CFX (Fig. 2). Neither the radA nor the recG single mutants strains are strongly sensitive to AZT or CFX; the double radA recG mutant exhibited extreme sensitivity at low doses of AZT (20 ng ml−1) or CFX (2 ng ml−1).
Figure 2.
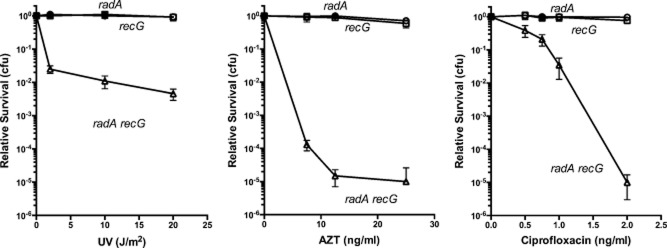
Fractional survival of radA and recG strains challenged with UV irradiation, AZT or ciprofloxacin. Cultures in early exponential phase were diluted and plated on LB medium followed by UV irradiation or on LB medium containing the indicated concentrations of AZT or ciprofloxacin. Fractional survival was determined after incubation for 24–36 hours at 37°. Each point represents the mean survival of 12–16 cultures plated on at least three different days. Error bars indicate standard deviation of the mean survival. (radA: open circles, recG: open squares, radA recG: open triangles).
For AZT, we noted that the sensitivity of the radA recG double mutant exceeded the sensitivity of a recA mutant, in which homologous recombination and SOS induction is blocked. Addition of a recA mutation to the radA recG strain suppressed the AZT sensitivity to the level of the recA mutant only (Fig. 3). Addition of a mutation in recF, in which loading of RecA to single-strand gaps is diminished [reviewed in (Cox, 2007)] also suppressed AZT sensitivity substantially (Fig. 3). This is consistent with the notion that AZT-induced replication gaps are processed into lethal intermediates in the radA recG mutant strain. Production of these lethal intermediates is inferred to depend on RecA filament formation, primarily mediated by the RecF pathway.
Figure 3.
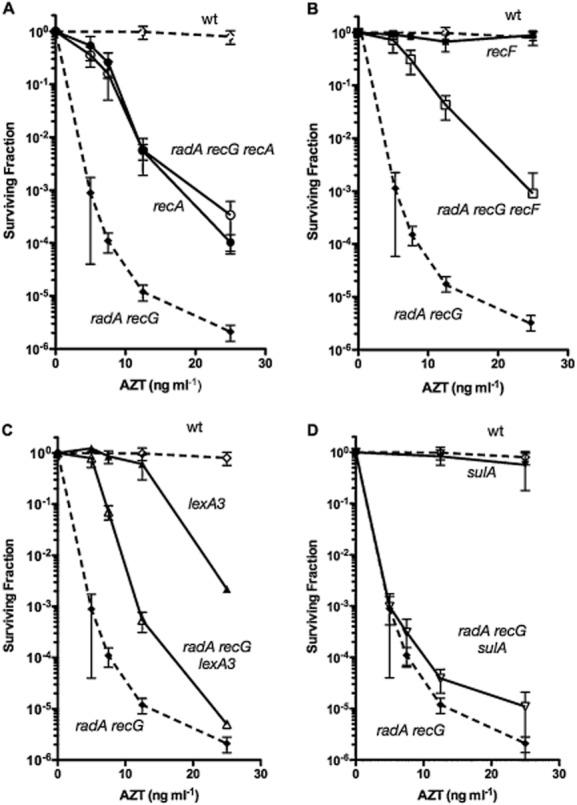
Suppression of sensitivity of the radA recG strain to AZT by defects in recombination and SOS genes. Early log phase cultures were diluted and plated on LB medium containing AZT at the indicated concentrations. Survival was determined after incubation at 37° for 24–36 hours. Each point represents mean survival of at least eight cultures plated on at least three different days. The standard deviation of the mean survival is indicated by error bars. Data for wild type (open diamonds) and recG radA (closed diamonds) strains are reproduced in each panel for reference (dashed lines).A. recA (closed circles) and recA radA recG (open circles).B. recF (closed squares) and recF radA recG (open squares).C. lexA3 (closed triangles) and lexA3 radA recG (open triangles).D. sulA (closed inverted triangles) and sulA radA recG (open inverted triangles).
Such lethal intermediates could represent branched recombination intermediates that require RecG or RadA for resolution. Alternatively or additionally, RecA filaments could persist longer in the radA recG mutant, which signal the SOS response. Inhibition of cell division by SulA is induced as part of the SOS response and radA recG mutants might fail to proliferate under AZT chronic exposure because of SulA-mediated division arrest.
Using a luciferase operon fusion to the RecA promoter, we measured constitutive expression of the SOS response in wild-type, radA, recG and radA recG strains (Fig. 4). The radA recG strain did show constitutive induction of the RecA promoter, to a level comparable with that observed with AZT treatment (Cooper and Lovett, 2011), indicating that spontaneous damage is accumulating in this strain background and signaling the SOS response. The single mutants in radA or recG did not exhibit any change in SOS induction, as judged by recA promoter activity in this assay.
Figure 4.
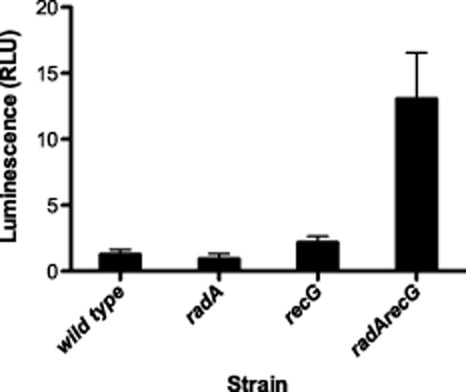
RecA promoter activity as measured by luciferase production. Luciferase assays were performed as described in the materials and methods. Luminescence was normalized to the OD590 of each culture to give relative luciferase units (RLUs). The mean of at least three different assays containing at least three different cultures for each point is shown. Error bars indicated the standard deviation of the mean. (Wild type, AB1157; radA, STL10550; recG, STL10067; radA recG, STL11258).
We introduced a lexA3-Ind- mutation into the radA recG strain, which prevents induction of the SOS response and measured the resulting survival to chronic AZT exposure during growth. Unlike recA mutations that restored AZT resistance of radA recG strains to the level of a single recA mutant, lexA3 had a more modest effect on AZT tolerance in the radA recG background and radA recG lexA3 were considerably more sensitive than lexA3 mutants (Fig. 3). Although both recA and lexA3 mutations negate induction of the SOS response, only recA blocks homologous recombination. Therefore, although there is some constitutive induction of the SOS response in radA recG strains that is deleterious for AZT survival, the inviability of radA recG strains in the presence of AZT is, at least in part, due to the accumulation of recombination intermediates. Although SOS induction may play some role in the inviability of radA recG mutants in the presence of AZT, this is not due to induction of SulA: a mutation in sulA, which blocks SOS-mediated cell division inhibition, does not appreciably suppress AZT sensitivity in the radA recG strain (Fig. 3).
To explore what additional SOS-induced genes might be deleterious to radA recG mutants, we introduced mutations in several SOS-induced functions including genes encoding recombination proteins recN, RecA filament regulators recX and dinI (data not shown), translesion DNA polymerases polB, dinB and umuC (Table S1). None of these had any effect on UV, CPX or AZT survival of the radA recG mutant. Introduction of a recA operator-constitutive mutants, recAo281 or recAo1403, likewise did not affect the survival of radA, recG or radA recG double mutants to UV, CPX or AZT (Table S1). This would suggest that high RecA concentrations do not contribute to the toxicity of the SOS response in the absence of RadA or RecG.
What could be toxic about recombination? Intermolecular recombination intermediates such as four-strand Holliday junctions may be toxic if not resolved. Loss of Holliday junction processing functions, RuvABC and RecG, are not normally lethal to the cell. However, the addition of a mutation in UvrD helicase, which elevates levels of homologous recombination (Zieg et al., 1978; Bierne et al., 1997) and inhibits RecA-mediated strand exchange (Veaute et al., 2005), causes lethality in combination with mutations with either RuvABC or RecG (Magner et al., 2007; Fonville et al., 2010). This lethality can be blocked by mutations that abolish early steps of recombination, such as RecA, RecF or RecQ. This suggests that elevated recombination leads to accumulation of lethal recombination intermediates that require both RecG and RuvABC for removal. Elevated recombination can also be elicited by exposure to genotoxins. We have demonstrated previously that AZT treatment induces the RecFOR pathway of recombination and likely presents a higher demand for resolution of recombination intermediates (Cooper and Lovett, 2011).
Mutations in uvrD were viable in combination with those in radA, although strains grew poorly and accumulated suppressors very quickly (data not shown), requiring that they be assayed immediately after construction. Mutants in uvrD were strongly sensitive to UV and AZT, modestly sensitive to CPX and sensitivities were enhanced by addition of a mutation in radA (Fig. 5). Introduction of a mutation in recF almost entirely suppressed the AZT sensitivity and partially suppressed the UV and CPX sensitivity of uvrD and uvrD radA mutants. These data are consistent with the idea that, that like RecG and RuvAB, RadA processes a class of recombination intermediates that accumulate in uvrD mutants; these intermediates are induced by exposure to AZT, and to a lesser extent, to UV and CPX.
Figure 5.
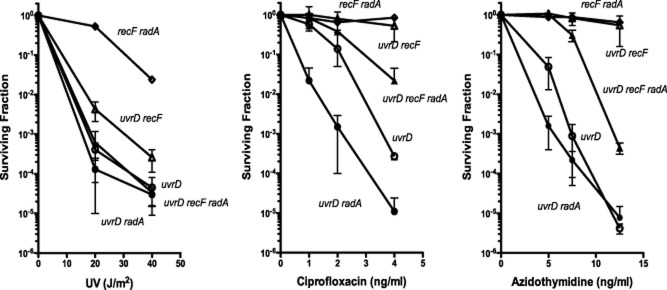
Suppression of sensitivity of the uvrD radA strain by recF to DNA damaging agents. Early log phase cultures were diluted and plated on LB medium containing AZT or ciprofloxacin at the indicted concentrations or UV-irradiated as indicated. Survival was determined after incubation at 37° for 24–36 hours. Each point represents mean survival of four to six cultures plated on at least two different days except for the radA recF strain that was included as control. Error bars show the standard deviation of the mean. (uvrD-open circles, uvrD radA-closed circles, uvrD recF-open triangles, uvrD recF radA-closed traingles, recF radA-open diamonds).
The RuvAB protein catalyzes the branch migration of three- and four-strand DNA junctions, which are potential intermediates of homologous recombination (reviewed in Persky and Lovett, 2008). The ruvA and ruvB genes are expressed constitutively and induced further during the SOS response, suggesting that there is an advantage in having elevated levels of RuvAB after DNA damage. We introduced a plasmid expressing RuvAB into wild-type, radA, recG and radA recG and determined whether RuvAB expression had any influence on survival of AZT, CPX or UV. Expression of RuvAB almost completely suppressed radA recG sensitivity to UV, strongly suppressed sensitivity to AZT at low doses and partially suppressed that to CPX at low doses (Fig. 6). RuvAB expression also suppressed the CPX sensitivity of recG mutants. These results are consistent with the notion that resolution of recombination intermediates, such as three- and four-stranded branched molecules, becomes limiting in radA recG mutants. These intermediates must be induced by UV and AZT and, to a lesser extent, CPX.
Figure 6.
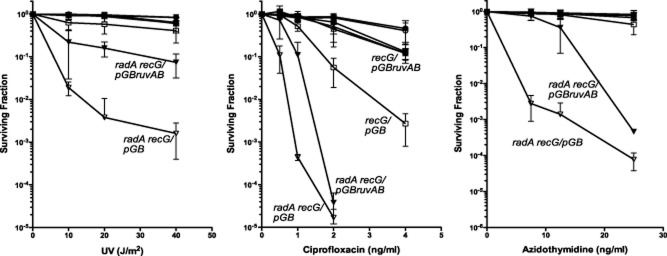
Suppression of sensitivity of the radA recG strain to DNA damaging agents by overexpression of ruvAB. Wild-type (circles), radA (triangles), recG (squares) or radA recG (inverted triangles) strains were transformed with pGB2 vector (open symbols) or pGB-ruvAB (closed symbols). The resulting strains were grown to early log phase in LB medium containing 100 g ml−1 streptomycin and plated on LB medium followed by UV irradiation or on LB medium containing ciprofloxacin or AZT. Plates were incubated at 37° and surviving colonies were counted after 18–36 hours. Mean fractional survival of at least eight different cultures plated on two different days is shown with the error bars representing the standard deviation of the mean.
To explore potential genetic interactions with other DNA helicases/DNA pumps, we combined RadA deficiency with DinG, HelD, PriA, RarA, RecQ, Rep, RuvAB, Lhr, YejH and YoaA and compared them to effects of UvrD and RecG described above. Survival to AZT, CFX and UV irradiation was assayed for the single and double mutants at several doses (Fig. 7). The results of these assays allowed the helicases to be grouped into five categories. The first category includes DNA pump proteins RuvAB and DNA helicases UvrD and PriA. PriA, in addition to its helicase activity, plays an important role in primosome assembly and replication restart after DNA damage repair (Gabbai and Marians, 2010). Mutants in ruvAB, uvrD or priA alone exhibited substantial sensitivity to AZT, CFX or UV and, when combined with an additional mutation in radA, showed enhanced AZT, CFX and UV sensitivity, compared with the single mutants. The uvrD effect is weaker than ruvAB or priA. Because of the extreme sensitivity of ruvAB mutants to AZT, potential synergy of ruvAB and radA for AZT survival could not be ascertained.
Figure 7.
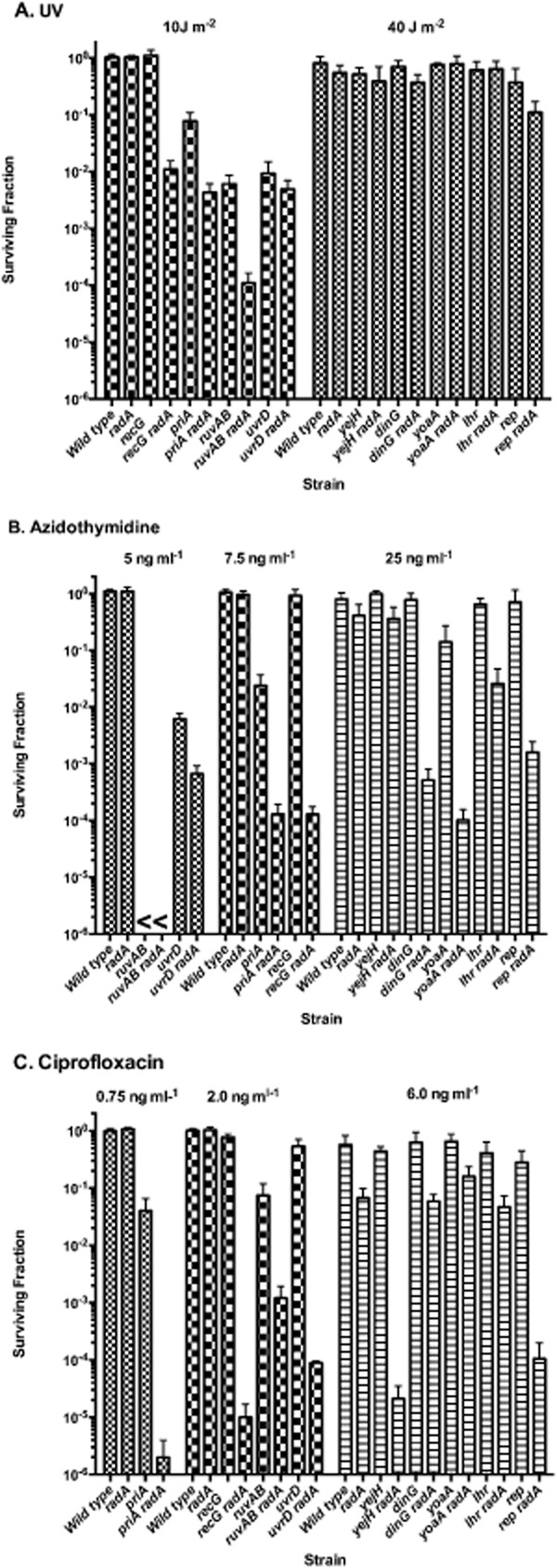
Synergistic response of radA and DNA helicase/DNA pump mutants to DNA damage. Log phase cultures were diluted and plated on LB agar and then UV irradiated (A) or plated on LB medium containing the indicated concentration of AZT (B) or ciprofloxacin (C). Plates were incubated at 37°, and surviving colonies were counted after 18–36 hours. Experiments were performed on at least two different days. The mean fractional survival and standard deviation of at least eight independent cultures is shown. Dark gray bars show survival in wild type or radA strains, gray bars show survival in single DNA helicase mutant strains and light gray bars show survival in radA –DNA helicase double mutants. Fractional survival in the ruvAB strains on AZT could not be determined and was assigned a < 1 × 10−6 value as indicated.
The second category of helicases includes known DNA helicase Rep and DNA pump RecG, discussed above. Rep, when mutated, did not by itself produce appreciable sensitivity to UV, AZT or CFX but when mutated in combination with radA produced synergistic sensitivity to both AZT and CFX and, more modestly, to UV. The third group was typified by mutants of putative helicase YejH, which produced sensitivity to CFX but not AZT when combined with radA. Our data did not show strong synergy between radA and yejH for effects on UV; recent work from the Cox laboratory demonstrates a synergistic effect on survival to UV at higher doses (Chen, Byrne and Cox, personal communication). The fourth group included the Fe-S helicase DinG and its paralog, putative helicase YoaA, which when absent in combination with radA caused sensitivity to AZT but not to ciprofloxacin or UV. Lhr, a putative helicase, may also be a member of this group: the lhr mutation had significant synergy with radA for effects on AZT survival but no significant effect on UV. The final fifth group, HelD, RarA and RecQ, showed no genetic interaction with radA for sensitivity to all agents tested (data not shown).
Discussion
This work confirms that several motifs found in RadA are required for its function, as assayed by resistance to ciprofloxacin, including the Walker A ATPase motif, the putative Zn finger and the conserved RadA KNRXG motif. The latter motif is required for structure-specific nuclease activity of the bacteriophage D29 gene 65 protein (Giri et al., 2009) and may therefore specify structure-specific binding or cleavage activity. Although its C-terminal domain with homology to Lon protease was required for function, RadA is unlikely to be a serine protease, because mutations to the putative active site serine had no phenotypic effects and it lacks the other two conserved residues of the catalytic triad.
We had previously implicated RadA in late steps of recombination because of synergistic genetic effects on recombination and DNA damage tolerance by RadA with RecG and/or RuvABC (Beam et al., 2002). These latter functions process Holliday junctions, displacement loops (D-loops) and other joint molecules in vitro, presumed intermediates in homologous recombination (West, 1997; Rudolph et al., 2010). Our unpublished biochemical analysis of RadA shows that it stimulates branch migration during in vitro RecA-mediated strand exchange (Cooper and Lovett, unpublished data), which readily explains the RadA's genetic properties reported here.
We show here that the genetic synergy of RadA with late recombination functions RecG and RuvAB extends to tolerance of AZT and CPX treatment. In addition, three other findings reported here provide additional support for a role for RadA in the late steps of recombination. Mutants in radA recG are extremely sensitive to the replication chain terminator, AZT, which leads to formation of gaps in DNA and stimulates recombination via the RecFOR pathway (Cooper and Lovett, 2011). We report here that much of this sensitivity can be suppressed by blocking RecA or its mediator protein, RecF, suggesting that in the absence of RadA and RecG formation of recombination intermediates is lethal to the cell. Expression of RuvAB from a plasmid is sufficient to restore considerable viability of radA recG mutants when exposed chronically to AZT and restores partial viability during exposure to CPX. An additional observation that supports a late recombination role for RadA is the poor viability and genotoxin-sensitivity of radA mutants when combined with loss of uvrD. UvrD is a helicase that dismantles RecA filaments in vivo and in vitro (Veaute et al., 2005), thereby suppressing recombination. The RecF dependence of the uvrD and uvrD radA strains’ sensitivity to AZT and CPX suggests that RecFOR-dependent recombination leads to death when these strains are exposed to these agents.
The genetic effects of mutants believed to affect the late steps of recombination, such as RuvAB, RecG and now RadA, have been puzzling to interpret. Because mutants affecting each individual function have phenotypes, these proteins must have some specificity in the types of DNA intermediates they process – that is, they cannot substitute for each other. On the other hand, mutants in these functions have additive or synergistic phenotypes when combined – that is, they may provide some common, redundant function. One possible solution to this conundrum is that these proteins do have some specificity in the types of intermediates that they process but that these intermediates are somewhat interconvertible. In addition, the kinetics and efficiency by which intermediates are processed may be critical. Incomplete recombination might become lethal after a second event, like the convergence of a replication fork onto the site, for example. In this way, loss of multiple processing pathways would exacerbate the race against time toward a lethal outcome and lead to the observed genetic synergy. Loss of either RecG, RadA or RuvAB could increase the demand, in this way, for the remaining functions.
Although genetic synergy can been used to argue for overlapping functions, we note that it can also be explained by changes in how intermediates are channeled into different pathways. For example, loss of RadA may channel more intermediates through a pathway more dependent on RecG. Effects on channeling likely explain the synergy between RadA and PriA. In absence of RadA, there does appears to be a higher demand for PriA-dependent replication restart after exposure to UV, AZT or CPX, judging from the strong sensitivity of radA priA mutants to these agents.
Because of the amino acid sequence similarity between RadA and RecA, an interesting question is whether RadA modulates the stability of the RecA filament, thereby altering induction of the SOS response. Double radA recG mutants are constitutively induced for the SOS DNA damage response, as measured by recA promoter expression, indicating that RecA filament formation is either elevated or longer lived in these mutants. Sandler et al. have also shown that loss of RadA leads to stronger SOS induction in vivo when combined with an SOS-constitutive recA4142 mutation and have hypothesized that RadA processes RecA filaments to maintain a level below the critical level for SOS induction (Massoni et al., 2012). Using microscopic analyses of sulA::GFP fluorescence, these authors observed a modest, 1.5-fold elevation of expression caused by loss of RadA in an otherwise wild-type strain. Using luciferase measurements of bulk cultures in early log phase and a recA promoter/luciferase fusion, we did not observe any difference in constitutive expression of this SOS-regulated gene. Cox et al. observed constitutive induction of sulA::GFP by several fold in radA mutants, as cultures entered the late log phase of growth and through stationary phase (Chen, Byrne, Wood and Cox, personal communication). Because we did not test radA effects under conditions where SOS would be induced or in later growth phases, our results are not inconsistent with these other observations. Elevated levels of RecA filaments or induction of the SOS response are not, however, the sole cause of AZT lethality in radA recG strains, because blocking the transcriptional response by a lexA3 mutation had only a partial effect on AZT tolerance. In addition, increased levels of RecA afforded by recA operator mutations did not increase AZT or CPX sensitivity in radA mutants.
We show here that the survival of strains to chronic doses of CPX and AZT can be useful to distinguish genetic function. The primary lesion of AZT is believed to be the formation of ssDNA replication gaps and that of CPX are broken chromosomes with trapped topoisomerase II complexes. We note, however, that subsequent processing can produce a constellation of DNA intermediates, any of which might lead to cell death. At the doses used here, we expect multiple lesions, and the functions required at low doses may be distinct from those at high doses.
Our experiments suggest that at low doses of AZT, lesions are processed by RecF-dependent, RecA-dependent recombination and that this pathway requires RuvAB and either RecG or RadA for completion. Increased levels of RuvAB can relieve the requirement for RadA or RecG. In contrast, enhancing the RecAF pathway by mutations in uvrD enhances its dependence on RadA. Blocking the early stages of recombination by mutations in recF or recA allows an alternative, nonrecombinational pathway (that also does not require RadA or RecG) to contribute to survival. The inability to induce the SOS response, afforded by a lexA3 mutation, either allows AZT-induced lesions to be channeled into the nonrecombinational pathway or, in some way, ameliorates the toxicity of the RecA RecF-dependent pathway when RecG and RadA are absent. At high doses of AZT, RecA is required for survival; RuvAB overexpression, loss of RecF or lexA3 cannot compensate for loss of RadA and RecG. This may be because, at high AZT concentrations, chromosomes become broken and require RecA-dependent DSB repair.
Survival to chronic doses of the fluoroquinolone CPX also requires RadA or RecG but does not have as strong a dependence on RuvAB as does survival to AZT. In addition, CPX sensitivity caused by the loss of RadA and RecG cannot be strongly suppressed by over-expression of RuvAB. This may be because CPX-induced DSBs involve formation of DNA intermediates that can be processed by either RecG or RadA but less so by RuvAB. Interestingly, uvrD mutants were found to be sensitive to high doses of CPX, and this sensitivity can be entirely suppressed by blocking RecF. This is surprising as it indicates that the RecF recombination pathway, believed to be inefficient on DSBs (reviewed in Persky and Lovett, 2008), can be activated in uvrD mutants by CPX.
RadA shows synthetic interaction with a number of helicases that may participate in DNA repair or avoidance of lesions, in addition to PriA, RecG, RuvAB and UvrD discussed above. These fall into distinct categories based on their effects on survival to two types of genotoxins, CFX or AZT, and the presumed primary types of DNA damage produced by them, DSBs vs. replication gaps respectively. For sensitivity to both CFX and AZT, mutations in RadA cause synergistic effects with those in the Rep and Lhr helicases. Because single mutants have little effect and double mutants have strong synthetic phenotypes, this may indicate that the Rep and Lhr proteins act redundantly to RadA in repair or are required to remove potential lesions that require RadA for repair. Rep promotes replication fork progression, presumably by removal of protein or DNA structures that impede replication (Lane and Denhardt, 1975; Seigneur et al., 1998; Petit and Ehrlich, 2002; Veaute et al., 2005). Lhr (‘long helicase-related’) protein is a putative helicase of unknown function; this is the first reported phenotype for an lhr mutant (Reuven et al., 1995). Mutants in RadA also showed synthetic sensitivity with those in putative helicase YejH; this effect was specific for CFX, suggesting both proteins play a role in DSB processing. Consistent with this, mutants YejH (recently renamed ‘RadD’) are sensitive to ionizing radiation and induced DSBs (Chen, Byrne, Wood and Cox, personal communication.) In contrast, RadA exhibited synthetic sensitivity to the gap-producing agent, AZT, but not DSB-producing CFX, with the DNA damage-inducible, Fe-S helicase DinG (Voloshin et al., 2003; Voloshin and Camerini-Otero, 2007; Ren et al., 2009; Cheng et al., 2012) and its paralog YoaA. This suggests a novel role for these proteins in replication-gap processing. DinG, in concert with Rep and UvrD helicases, is essential for replication after head-on collision with highly transcribed genes (Boubakri et al., 2010). DinG and YoaA helicases, like RadA and RecG proteins, may act either to prevent lethal lesions after AZT treatment or to repair them.
Experimental procedures
Bacterial strains, growth conditions and survival assays
Escherichia coli K-12 strains (Table S2) were grown at 37°C as previously described on Luria–Bertani (LB) medium, consisting of 1% Bacto-tryptone, 0.5% yeast extract, 0.5% sodium chloride and, for plates, 1.5% agar. Isogenic mutant strains in the AB1157 genetic background (Bachmann, 1972) were constructed by P1 virA transduction. Deletion mutations in various genes were initially constructed by published methods (Datsenko and Wanner, 2000) using pKD3 carrying the cat gene for chloramphenicol resistance (lhr, radA, rep, yejH, yoaA alleles) or pKD4 carrying the kan gene for kanamycin resistance (sulA allele). Alleles of uvrD and priA are derived from Tn5-EZ kan transposon (Epicentre) insertion screen described previously (Foti et al., 2005). Plasmids were introduced to strains using electroporation. For genetic selections, antibiotics were used at the following concentrations: ampicillin (Ap), 100 μg ml−1; kanamycin (Km), 60 μg ml−1; tetracycline (Tc), 30 μg ml−1, spectinomycin (Sp), 100 μg ml−1 and chloramphenicol (Cm), 15 μg ml−1. LB medium supplemented with 1% glucose, 2 mM calcium chloride and 1% agar for plates, was used to make P1 lysates and for transductions.
For survival assays, cells were grown to an OD590 of between 0.3 and 0.5, serially diluted in 56/2 salts and plated on LB plates with no drug or with AZT or CFX at the indicated concentrations. For UV survival assays, cells were diluted and plated on LB and subject to irradiation at 254 nm at the indicated fluence. Colony formation was determined after incubation at 37° for 24–36 hours. For strains carrying RadA plasmids derived from pWSK29, growth medium contained Ap and expression of the radA gene was induced by 1 mM IPTG for 30 minutes prior to plating on CFX. For strains carrying pGB2 (Churchward et al., 1984) or pGB2-RuvAB (Seigneur et al., 1998), growth medium contained Sp.
RadA mutant alleles
Mutations in the specific motifs of RadA were constructed by site-directed mutagenesis (Quikchange, Stratagene) of radA on a high-copy pBS SK-derived plasmid, pSTL307 (Beam et al., 2002). Primers used to generate the site-directed mutants included: K108R: 5′ AGG GTT ACC GCC AAT CAG AAT GG 3′; K258A: 5′ CAA GGT GCG AAA ACG GGA GTC G 3′; S372A: 5′ TTA CCT TCA CGC CGC CGA CCA CGT TCA 3′. RadA-C28Y was generated by Polymerase Chain Reaction (PCR) amplification of the radA gene derived from the radA100 strain SR776 (Diver et al., 1982) as described previously (Beam et al., 2002). The sequence of the radA alleles were confirmed by DNA sequence analysis of the entire gene and alleles were transferred to the low-copy plasmid pWSK29 (Wang and Kushner, 1991) by ApaI SacII digestion and ligation with T4 ligase (New England Biolabs). Mutant alleles with truncation of the C-terminal Lon domain were constructed by PCR amplification with Pfu polymerase (Stratagene) and the appropriate primers, followed by HindIII digestion and ligation. Primers included 5′ GGG GGG GAA GCT TTT ATT ACT GTT CGG TCA TCG CGA AGA C 3′ and 5′ GGG GGG GAA GCT TTG AGA TAT ACG GAG GGA GAT ATG TCG TCA T 3′ for RadAΔ275–460 or 5′ GGG GGG AAG CTT TTA AGC CAG CGA ACC ATC TTT GGT TAC G 3′ and 5′ GGG GGG AAG CTT CGA TAC CGT CGA CCT CGA GGG GGG GCC CGG TA 3′ for RadAΔ228–460.
SOS induction assay
Plasmids carrying the Photorhabdus luminescens luxCDABE luciferase operon were used for reporters for induction of the SOS response (Van Dyk et al., 2001). Promoter-less vector (pDEW201) or fusion to the E. coli recA promoter (pDEW238), were transformed into the appropriate strains and selected by Ap-resistance. Strains were grown and assayed as previously described (Goldfless et al., 2006). Arbitrary luciferase expression (Lux) values were calculated by normalizing the amount of bioluminescence (cpm) to the OD590 of the cultures.
Acknowledgments
This work was supported by NIH R01 grant GM51753. D. C. B. was partially supported by NSF REU grant 1062136. We thank Cynthia Beam, Bethany Dutra and Daniel Resnicow for strain and plasmid construction and S. Kushner, R. Lloyd, B. Michel, H. Ohmori, S. Sandler and T. Van Dyk for provision of strains and plasmids.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Reference
- Bachmann BJ. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev. 1972;36:525–557. doi: 10.1128/br.36.4.525-557.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam CE, Saveson CJ. Lovett ST. Role for radA/sms in recombination intermediate processing in Escherichia coli. J Bacteriol. 2002;184:6836–6844. doi: 10.1128/JB.184.24.6836-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Seigneur M, Ehrlich SD. Michel B. uvrD mutations enhance tandem repeat deletion in the Escherichia coli chromosome via SOS induction of the RecF recombination pathway. Mol Microbiol. 1997;26:557–567. doi: 10.1046/j.1365-2958.1997.6011973.x. [DOI] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E. Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghout P, Bootsma HJ, Kloosterman TG, Bijlsma JJE, de Jongh CE, Kuipers OP. Hermans PWM. Search for genes essential for pneumococcal transformation: the RADA DNA repair protein plays a role in genomic recombination of donor DNA. J Bacteriol. 2007;189:6540–6550. doi: 10.1128/JB.00573-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco B, Cozar MC, Lurz R, Alonso JC. Ayora S. Genetic recombination in Bacillus subtilis 168: contribution of Holliday junction processing functions in chromosome segregation. J Bacteriol. 2004;186:5557–5566. doi: 10.1128/JB.186.17.5557-5566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos M. Romero D. The extent of migration of the Holliday junction is a crucial factor for gene conversion in Rhizobium etli. J Bacteriol. 2009;191:4987–4995. doi: 10.1128/JB.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Caillet A, Ren B. Ding H. Stimulation of Escherichia coli DNA damage inducible DNA helicase DinG by the single-stranded DNA binding protein SSB. FEBS Lett. 2012;586:3825–3830. doi: 10.1016/j.febslet.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli SV, Bhardwaj G, Babu J, Hadjiyianni L, Hong Y, Todd GK, et al. Reevaluation of the evolutionary events within recA/RAD51 phylogeny. BMC Genomics. 2013;14:240. doi: 10.1186/1471-2164-14-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchward G, Belin D. Nagamine Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene. 1984;31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- Cooper DL. Lovett ST. Toxicity and tolerance mechanisms for azidothymidine, a replication gap-promoting agent, in Escherichia coli. DNA Repair (Amst) 2011;10:260–270. doi: 10.1016/j.dnarep.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- Datsenko KA. Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diver WP, Sargentini NJ. Smith KC. A mutation (radA100) in Escherichia coli that selectively sensitizes cells grown in rich medium to x- or u.v.-radiation, or methyl methanesulphonate. Int J Radiat Biol Relat Stud Phys Chem Med. 1982;42:339–346. doi: 10.1080/09553008214551251. [DOI] [PubMed] [Google Scholar]
- Drlica K. Zhou X. DNA gyrase, toposiomerase IV and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377–392. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonville NC, Blankschien MD, Magner DB. Rosenberg SM. RecQ-dependent death-by-recombination in cells lacking RecG and UvrD. DNA Repair (Amst) 2010;9:403–413. doi: 10.1016/j.dnarep.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Schienda J, Sutera VA. Lovett ST. A bacterial G protein-mediated response to replication arrest. Mol Cell. 2005;17:549–560. doi: 10.1016/j.molcel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Gabbai CB. Marians KJ. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri N, Bhowmik P, Bhattacharya B, Mitra M. Das Gupta SK. The mycobacteriophage D29 gene 65 encodes an early-expressed protein that functions as a structure-specific nuclease. J Bacteriol. 2009;191:959–967. doi: 10.1128/JB.00960-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfless SJ, Morag AS, Belisle KA, Sutera VA. Lovett ST. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol Cell. 2006;21:595–604. doi: 10.1016/j.molcel.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Holthausen JT, Wyman C. Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair (Amst) 2010;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Isogai M, Kiyohara H, Hosaka M, Chiku H, Koga A, et al. Higher plant RecA-like protein is homologous to RadA. DNA Repair (Amst) 2006;5:80–88. doi: 10.1016/j.dnarep.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Krüger E, Msadek T, Ohlmeier S. Hecker M. The Bacillus subtilis clpC operon encodes DNA repair and competence proteins. Microbiology. 1997;143:1309–1316. doi: 10.1099/00221287-143-4-1309. [DOI] [PubMed] [Google Scholar]
- Lane HE. Denhardt DT. The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J Mol Biol. 1975;97:99–112. doi: 10.1016/s0022-2836(75)80025-8. [DOI] [PubMed] [Google Scholar]
- Lovett ST. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair (Amst) 2006;5:1421–1427. doi: 10.1016/j.dnarep.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Lovett ST. The DNA damage response. In: Storz GT, editor. Bacterial Stress Responses. 2nd edn. Washington, DC: American Society of Microbiology Press; 2010. pp. 201–228. (ed.). [Google Scholar]
- Magner DB, Blanschien MD, Lee JA, Pennington JM, Lupski JR. Rosenberg SM. RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol Cell. 2007;26:273–286. doi: 10.1016/j.molcel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoni SC, Leeson MC, Long JE, Gemme K, Mui A. Sandler SJ. Factors limiting SOS expression in log-phase cells of Escherichia coli. J Bacteriol. 2012;194:5325–5333. doi: 10.1128/JB.00674-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persky NS. Lovett ST. Mechanisms of recombination: lessons from E. coli. Crit Rev Biochem Mol Biol. 2008;43:347–370. doi: 10.1080/10409230802485358. [DOI] [PubMed] [Google Scholar]
- Petit M-A. Ehrlich D. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 2002;21:3137–3147. doi: 10.1093/emboj/cdf317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Duan X. Ding H. Redox control of the DNA damage-inducible protein DinG helicase activity via its iron-sulfur cluster. J Biol Chem. 2009;284:4829–4835. doi: 10.1074/jbc.M807943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven NB, Koonin EV, Rudd KE. Deutscher MP. The gene for the longest known Escherichia coli protein is a member of helicase superfamily II. J Bacteriol. 1995;177:5393–5400. doi: 10.1128/jb.177.19.5393-5400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Briggs GS. Lloyd RG. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 2010;9:210–223. doi: 10.1016/j.dnarep.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD. Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Simmons L, Foti J, Cohen S. Walker G. 2008. , and The SOS Regulatory Network, EcoSal Plus. 2008; doi: 10.1128/ecosalplus.5.4.3.
- Slade D, Lindner AB, Paul G. Radman M. Recombination and replication in DNA repair of heavily irradiated Deinococcus radiodurans. Cell. 2009;136:1044–1055. doi: 10.1016/j.cell.2009.01.018. [DOI] [PubMed] [Google Scholar]
- Song Y. Sargentini NJ. Escherichia coli DNA repair genes radA and sms are the same gene. J Bacteriol. 1996;178:5045–5048. doi: 10.1128/jb.178.16.5045-5048.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyk T, Wei Y, Hanafey M, Dolan M, Reeve M, Rafalski J, et al. A genomic approach to gene fusion technology. Proc Natl Acad Sci USA. 2001;98:2555–2560. doi: 10.1073/pnas.041620498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, et al. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloshin ON. Camerini-Otero RD. The DinG protein from Escherichia coli is a structure-specific helicase. J Biol Chem. 2007;282:18437–18447. doi: 10.1074/jbc.M700376200. [DOI] [PubMed] [Google Scholar]
- Voloshin ON, Vanevski F, Khil PP. Camerini-Otero RD. Characterization of the DNA damage-inducible helicase DinG from Escherichia coli. J Biol Chem. 2003;278:28284–28293. doi: 10.1074/jbc.M301188200. [DOI] [PubMed] [Google Scholar]
- Wang RF. Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- West SC. Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet. 1997;31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- Zieg J, Maples VF. Kushner SR. Recombinant levels of Escherichia coli K-12 mutants deficient in various replication, recombination, or repair genes. J Bacteriol. 1978;134:958–966. doi: 10.1128/jb.134.3.958-966.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.