

Abstract
Genome-wide association studies showed variation in insulin-like growth factor-2 binding protein 2 (IGF2BP2) to be associated with type 2 diabetes mellitus (T2DM). We examined a 20-kb region of IGF2BP2 for association with T2DM-related quantitative traits in Mexican American families of a proband with gestational diabetes mellitus (GDM) from the BetaGene study. We genotyped 14 single-nucleotide polymorphisms (SNPs) in 717 individuals from 146 families phenotyped by oral glucose tolerance test (OGTT), intravenous glucose tolerance tests (IVGTTs) with minimal model analysis, and dual-energy X-ray absorptiometry scan for percent body fat. Three SNPs and one SNP combination that captured the majority of the variation in the region were tested for association with T2DM-related quantitative traits using a variance components framework. After correction for multiple testing, rs11705701 showed association with percent body fat (PACT = 0.041) with body fat decreasing ∼1.5–2% per copy of the A allele. We next tested whether the interaction between rs11705701 and body fat was associated with T2DM-relative quantitative traits. rs11705701 was significantly associated with insulin sensitivity (Bonferroni P = 0.028) and marginally associated with OGTT 2-h insulin (Bonferroni P = 0.066) and disposition index (DI) (Bonferroni P = 0.072). We conclude that rs11705701 in IGF2BP2 is associated with body fat and this effect on body fat influences insulin resistance which may contribute to T2DM risk.
IntroductIon
Genome-wide association studies in samples of Northern European ancestry identified variation in insulin-like growth factor-2 binding protein 2 (IGF2BP2) as a susceptibility gene for type 2 diabetes mellitus (T2DM) (1–3). IGF2BP2 is part of a family of binding proteins (4,5). Insulin-like growth factor-2 binding protein 1 binds to the leader 3 mRNA in the 5′-UTR of the insulin-like growth factor 2 (IGF2) gene to regulate IGF2 translation (5). Mobility shift experiments show that both IGF2BP2 and insulin-like growth factor-2 binding protein 3 (IGF2BP3) bind to a specific RNA segment with similar affinity to that of insulin-like growth factor-2 binding protein 1, suggesting that all three members of this binding protein family may regulate IGF2 translation in a similar manner (5). IGF2, in turn, is a polypeptide growth factor that plays important roles in growth and development and stimulates insulin action. However, the exact functional role of IGF2BP2 is not known and even less is known how variation in IGF2BP2 may affect quantitative traits that contribute to diabetes risk.
We have shown that Mexican American women with a previous diagnosis of gestational diabetes mellitus (GDM) have a 5-year T2DM risk that exceeds 50% (6,7); an observation that has now been extended to other populations (8). We have also shown that the increased risk for T2DM in these women is due to a progressive loss of β-cell function in the face of chronic insulin resistance (9–11). Furthermore, risk for T2DM in these women can be significantly reduced by ameliorating insulin resistance, thereby reducing insulin secretory demand on the pancreatic β-cells and improving β-cell function (12,13). Therefore, the study of women with previous GDM provides an opportunity to examine potential genetic influences on T2DM-related quantitative traits.
The BetaGene study is a family-based study to identify such influences where we have hypothesized that if there exist genetic variation influencing T2DM-related quantitative traits, then such variants should be segregating within families. In BetaGene, we are performing detailed phenotyping of Mexican American probands with recent GDM and their family members to obtain quantitative estimates of body composition, insulin sensitivity (SI), acute insulin response (AIR), and β-cell compensation (disposition index, DI), which are traits associated with the pathogenesis of T2DM (cf. (14–16)) and that we have shown to be heritable in Mexican American families (17,18). This report focuses on the effects of genetic variation in a 20-kb region surrounding rs1470579 in IGF2BP2, based on unpublished preliminary results from the stage I analysis of the Finland United States Investigation of Non-insulin Dependent Diabetes Mellitus (FUSION) study genome-wide association (2), which suggested that variation in this region may be associated with SI derived from the intravenous glucose tolerance test (IVGTT). We determined that rs11705701, a variant that appears to be correlated with an alternative splice form of IGF2BP2 (19), is associated with percent body fat and that the interaction between this variant and adiposity is associated with SI.
Methods And Procedures
Subject recruitment
Subject recruitment for BetaGene is ongoing, and for the purpose of this report we describe only those subjects, clinical protocols, and assays related to the results presented herein. Subjects are Mexican American (both parents and ≥3 grandparents Mexican or of Mexican descent). Self-reported parental and grandparental birthplace information was obtained to assess ancestry. Subjects are either probands with GDM diagnosed within the previous 5 years and their family members, or non-GDM probands with normal glucose levels in pregnancy in the past 5 years. All probands are identified from the patient populations at Los Angeles County/USC Medical Center, the Kaiser Permanente of Southern California health plan membership, and OB/GYN clinics at local hospitals. GDM probands are recruited for phenotyping if they have (i) a confirmed diagnosis of GDM within the previous 5 years, (ii) glucose levels associated with poor pancreatic β-cell function and a high risk of diabetes when not pregnant (6), (iii) no evidence of β-cell autoimmunity by GAD-65 testing, and (iv) available for study at least two siblings. If at least two first cousins of the GDM proband are available from the same nuclear family, those cousins are recruited as well. Non-GDM probands are recruited if they had a 1-h 50-g glucose screening result <130 mg/dl (7.2 mmol/l) during their most recent pregnancy and have no family history of diabetes. They are frequency-matched to GDM probands by age, BMI, and parity. Subjects with chronic medical conditions or chronic medication use known to have important effects on glucose metabolism are excluded.
All protocols for BetaGene have been approved by the Institutional Review Boards of participating institutions and all participants provided written informed consent prior to participation.
Clinical protocols
Phenotyping is performed on two separate visits to the General Clinical Research Center. Visit 1 includes a study-directed medical history and physical examination, screening serum chemistries, complete blood count, DNA collection, and a 75-g oral glucose tolerance test (OGTT) with blood samples obtained before and 30, 60, 90, and 120 min after glucose ingestion. Participants from GDM families with fasting glucose <126 mg/dl on the OGTT and non-GDM probands with normal OGTT results are invited for a second visit, which consists of a dual-energy X-ray absorptiometry scan for determination of body composition and an IVGTT performed as described previously (11).
Assays
Plasma glucose is measured on an autoanalyzer using the glucose oxidase method (YSI Model 2300, Yellow Springs Instruments, Yellow Springs, OH). Insulin is measured by two-site immunoenzymometric assay (TOSOH Biosciences, San Francisco, CA) that has <0.1% crossreactivity with proinsulin and intermediate split products.
Molecular analysis
Given the unpublished preliminary results from the FUSION study, we focused our examination of IGF2BP2 to a 20-kb region surrounding rs1470579. Because it was equivocal whether tag SNP selection based on HapMap data would reflect the underlying linkage disequilibrium (LD) pattern in Mexican Americans, we selected SNPs at ∼2.5-kb intervals across this 20-kb region, preferentially selecting SNPs that were polymorphic in all four HapMap populations (HapMap Build 35). Fourteen SNPs, including rs4402960, the SNP originally shown to be associated with T2DM in samples of Northern European ancestry (1–3), were selected and genotyped using the Applied Biosystems TaqMan system (20,21). These SNPs were all located within introns 1 or 2 of IGF2BP2. Genotyping assays were either selected through Applied Biosystems’ “Assays on Demand” database (http://www3.appliedbiosystems.com/AB_Home/Products/Guides/AssaySelection/Index.html) or custom designed using Applied Biosystems’ “Assays by Design” service.
Data analysis
We calculated 2 measures of insulin response to glucose: the difference between the 30′ and fasting plasma insulin concentration from OGTT (30′ ΔInsulin) and the incremental area under the insulin curve for during the first 10 min of the IVGTT (AIR). IVGTT glucose and insulin data were analyzed using the minimal model (MINMOD Millennium V5.18) to derive measures of glucose effectiveness and SI (22,23). The DI was computed as the product of SI and AIR and used as a measure of β-cell compensation for insulin resistance (24).
Genotype data were tested for deviation from Hardy–Weinberg equilibrium and non-Mendelian inheritance using PEDCHECK V1.1 (http://watson.hgen.pitt.edu/register/) (25) and PEDSTATS V0.6.4 (http://www.sph.umich.edu/csg/abecasis/PedStats) (26). Allele frequencies were estimated by maximum likelihood using data from all available family members and SOLAR V2.1.4 (27,28). Haplotype frequencies were estimated using FUGUE (http://www.sph.umich.edu/csg/abecasis/FUGUE/). Tag SNPs were selected from among the genotyped SNPs using TAGGER with 2- and 3-maker haplotypes as implemented in Haploview V3.32 (29). In addition, LD and haplotype block structure were assessed using Haploview and the method of Gabriel et al. (30).
Quantitative trait data were statistically transformed to approximate univariate normality prior to analyses. Association between SNPs and quantitative traits was tested using likelihood ratio testing under a variance components framework as implemented in SOLAR (27,28). Because families were ascertained through probands based on previous GDM status, we corrected for ascertainment bias by conditioning each model on the proband's phenotype.
Each SNP was initially tested for association with quantitative traits assuming an additive genetic model. The minor allele was used as the reference allele, and the models were adjusted for age, sex, and where appropriate, percent body fat. The AG combination of rs13060777 and rs6444082 (cf. Results) was modeled as the number of copies of the AG haplotype. Any result under the additive model that showed a trend for association, defined as P = 0.1 Bonferroni-corrected for the number of markers examined (corrected P = 0.025), was subsequently tested for association under dominant and/or recessive genetic models. Because Bonferroni correction for multiple testing is overly conservative for correlated tests, we used PACT to better control for the number of tests performed and to account for the residual correlation among the tag SNPs and additional correlation among the quantitative traits tested for association (31).
The univariate association between rs11705701 and percent body fat (cf. Results) led us to examine whether the association between rs11705701 and T2DM-related quantitative traits was modified by percent body fat by testing the interaction between rs11705701 and body fat. We compared a model with age, sex, and percent body fat as main effects with a second model that included a main effect for rs11705701 and the multiplicative interaction between rs11705701 and percent body fat. We assumed an additive genetic model for rs11705701. We tested for the overall gene effect using a 2-df test and also tested for a significant interaction effect using a 1-df test. PACT is not applicable under the 2-df test (31), so for these analyses, we performed a Bonferroni correction to account for the multiple traits tested (n = 9). However, because many of the quantitative traits examined are correlated and Bonferroni assumes independence, the corrected P values should be overly conservative.
Linear modeling results are reported as age, sex, and percent body fat–adjusted means and standard deviations. For results from the interaction analysis that included percent body fat, trait values are reported as age- and sex-adjusted means and standard deviations. All other results were reported as unadjusted means and standard deviations.
Results
We report results from 717 individuals in 146 families for whom all phenotype and genotype data were available (Table 1). There was an average of 5.0 (range 1–11) members from each family including probands, siblings, and cousins from each family. Probands, siblings, and cousins were of similar age, although cousins were significantly younger compared to siblings and probands (P = 0.0067). Among probands, siblings, and cousins, median BMI exceeded the threshold for overweight (25 kg/m2). Median BMI and percent body fat were both significantly lower in siblings (P = 0.01 for both) and cousins (P < 0.0001 for both) compared to probands. Overall, quantitative trait values for glucose regulation were better in siblings and cousins compared to probands (cf. Table 1).
Table 1. Subject Characteristics.
| GDM probands | Siblings | Cousins | |
|---|---|---|---|
| Male/female | 0/118 | 150/229 | 95/125 |
| Age (years) | 35.8 (5.8) | 35.1 (8.7) | 33.0 (9.2)*,** |
| BMI (kg/m2) | 32.5 (7.0) | 29.9 (6.1)* | 28.6 (6.4)*,** |
| Body fat (%) | 39.2 (5.3) | 32.8 (8.5)* | 30.9 (8.6)*,** |
| Fasting glucose (mmol/l) | 5.4 (0.6) | 5.2 (0.5)* | 5.1 (0.6)* |
| Two-hour glucose (mmol/l) | 9.2 (2.3) | 7.7 (2.2)* | 6.9 (2.0)*,** |
| Fasting insulin (pmol/l) | 83 (57) | 62 (45)* | 59 (52)* |
| 30′ Insulin (pmol/l) | 500 (322) | 524 (346) | 549 (368) |
| Two-hour insulin (pmol/l) | 721 (500) | 526 (411)* | 451 (385)*,** |
| SG (×10-2 min-1) | 1.48 (0.48) | 1.71 (0.68)* | 1.83 (0.71)*,** |
| SI (×10-3 min-1 per pmol/l) | 2.76 (1.76) | 2.94 (1.63) | 3.16 (1.73)* |
| AIR (pmol/l × 10 min) | 4171 (4281) | 5678 (4521)* | 6245 (5285)* |
| Disposition index | 8694 (5829) | 14055 (9579)* | 15710 (9394)*,** |
Values are unadjusted median and interquartile range.
AIR, acute insulin response; GDM, gestational diabetes mellitus; SG, glucose effectiveness; SI, insulin sensitivity.
Uncorrected P < 0.05 vs. probands.
Uncorrected P < 0.05 vs. siblings.
Among the 14 SNPs genotyped, 2 SNPs (rs1374910 and rs9862583) failed the test for Hardy–Weinberg equilibrium, and 3 SNPs (rs9843367, rs9869293, and rs9867882) had minor allele frequencies <1% in our sample and were not further analyzed. The average intermaker distance among the nine remaining SNPs was 2.97 kb with smallest gap being 1.2 kb and the largest 8.1 kb. Estimated pairwise LD and haplotype blocks are shown in Figure 1. Eight of the nine SNPs formed a single 24-kb haplotype block comprised five haplotypes with frequency >1% (Supplementary Table S1 online). The observed LD and haplotype block patterns were similar, although not identical to the pattern derived from the HapMap CEU samples (Figure 1b), where there is a single 32-kb haplotype block encompassing all SNPs. A similar LD and haplotype block pattern was observed in the combined HapMap Japanese and Han Chinese samples (data not shown), whereas the Yoruban samples showed multiple smaller haplotype blocks across this region (data not shown). Tagger identified three SNPs (rs13060777, rs6444082, and rs11705701) and one SNP pair (AG combination of rs13060777 and rs6444082), as tags for this region of IGF2BP2 (Table 2).
Figure 1.
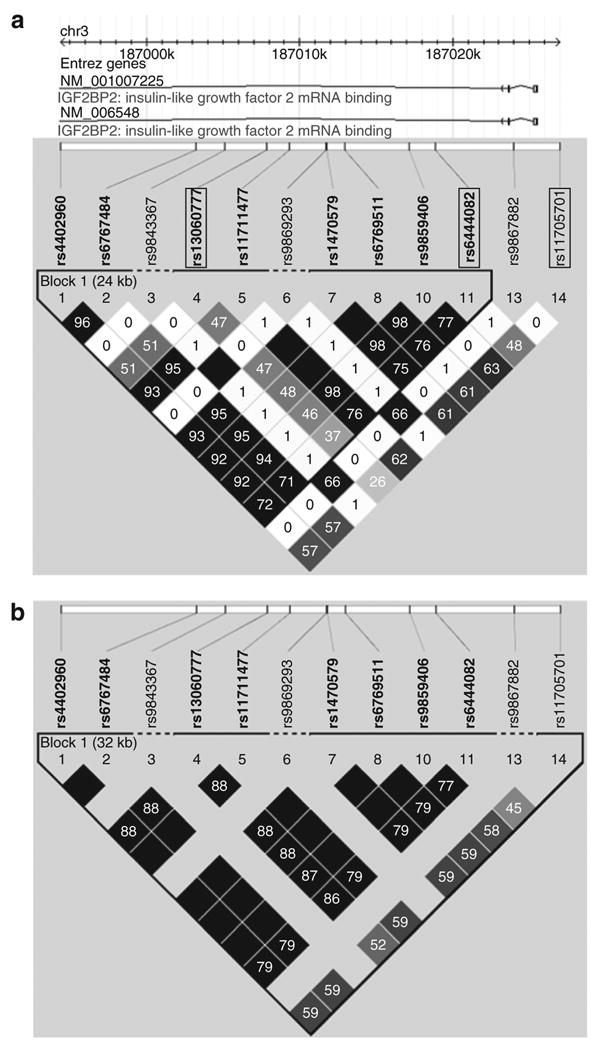
IGF2BP2 pairwise linkage disequilibrium and haplotype block structure. (a) Pairwise LD and haplotype block structure determined by the Gabriel et al. method as implemented in Haploview V4.0 for SNPs genotyped in the BetaGene study. Tagging SNPs are identified by a box. A single 24-kb haplotype block was identified, and rs11705701 only was not included in this block. (b) Pairwise LD and haplotype block structure for the identical SNPs from the HapMap CEU (Utah residents with ancestry from northern and western Europe) sample. A similar LD and haplotype block pattern was observed for the combined HapMap Japanese in Tokyo and Han Chinese in Beijing samples. LD is displayed as pairwise r 2 values (values within boxes), where varying shades of white indicate r 2 = 0, gray indicates 0 < r 2 < 1, and black indicates r 2 = 1. IGF2BP2, insulin-like growth factor-2 binding protein 2; LD, linkage disequilibrium; SNP, single-nucleotide polymorphism.
Table 2. Tag SNP characteristics.
| Tag SNP | Position (kb)a | Minor allele | MAF | SNPs tagged |
|---|---|---|---|---|
| rs13060777 | 187007901 | G | 0.15 | rs13060777 |
| rs6444082 | 187018925 | T | 0.23 | rs6444082 |
| rs11705701 | 187027011 | A | 0.33 | rs11705701 |
| rs1306077/rs6444082 | — | AG | — | rs4402960, rs11711477, rs6769511, rs1470579, rs9859406, rs6767484 |
MAF, minor allele frequency; NCBI, National Center for Biotechnology Information; SNP, single-nucleotide polymorphism.
NCBI build 35.
Two of the tag SNPs (rs6444082 and rs11705701) and the AG combination were nominally associated with percent body fat under an additive genetic model (Table 3, nominal P = 0.016, 0.001, and 0.011, respectively). These same SNPs also showed nominal association with 30′ ΔInsulin and 2-h insulin from the OGTT (Table 3). Despite the nominal association of rs64440872, rs11705701, and the AG combination with percent body fat, none showed evidence for association with BMI (P > 0.12, cf. Table 3). For results showing a trend for association under the additive genetic model, neither dominant nor recessive genetic models showed stronger associations, suggesting the association is best characterized by an additive model (data not shown).
Table 3. Results of univariate tests of association between SNPs in IGF2BP2 and T2DM-related quantitative traits.
| rs13060777 | rs6444082 | rs11705701 | AG combinationa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||||||
| Uncorrected P value |
Uncorrected P value |
Uncorrected P value |
Uncorrected P value |
|||||||||
|
|
|
|
|
|||||||||
| Effectb | Age, sex | Age, sex, % fat | Effectb | Age, sex | Age, sex, % fat | Effectb | Age, sex | Age, sex, % fat | Effectb | Age, sex | Age, sex, % fat | |
| BMI | - | 0.783 | N/A | - | 0.124 | N/A | - | 0.309 | N/A | + | 0.244 | N/A |
| % Body fat | - | 0.134 | N/A | - | 0.016 | N/A | - | 0.001 | N/A | + | 0.011 | N/A |
| Fasting glucose | - | 0.422 | 0.736 | - | 0.677 | 0.885 | - | 0.720 | 0.664 | + | 0.645 | 0.813 |
| Two-hour glucose | + | 0.963 | 0.850 | + | 0.386 | 0.942 | + | 0.225 | 0.834 | - | 0.418 | 0.862 |
| Fasting insulin | - | 0.530 | 0.940 | - | 0.186 | 0.774 | + | 0.352 | 0.479 | + | 0.386 | 0.483 |
| 30′ ΔInsulin | + | 0.125 | 0.311 | + | 0.040 | 0.137 | + | 0.018 | 0.109 | - | 0.035 | 0.141 |
| Two-hour insulin | - | 0.055 | 0.104 | - | 0.014 | 0.175 | - | 0.008 | 0.126 | + | 0.011 | 0.156 |
| SG | + | 0.660 | 0.850 | + | 0.926 | 0.790 | - | 0.256 | 0.100 | - | 0.922 | 0.722 |
| SI | + | 0.718 | 0.595 | + | 0.090 | 0.967 | + | 0.150 | 0.743 | - | 0.166 | 0.703 |
| AIR | - | 0.199 | 0.396 | - | 0.051 | 0.249 | - | 0.047 | 0.191 | + | 0.093 | 0.337 |
| DI | - | 0.154 | 0.135 | - | 0.340 | 0.182 | - | 0.200 | 0.065 | + | 0.361 | 0.169 |
Uncorrected P values are shown for test of association under an additive genetic model adjusted for age and sex or with the additional adjustment for percent body fat. AIR, acute insulin response; DI, disposition index; IGF2BP2, insulin-like growth factor-2 binding protein 2; N/A, not available; SI, insulin sensitivity; SNP, single-nucleotide polymorphism; T2DM, type 2 diabetes mellitus.
The AG combination of rs13060777 and rs6444082 was modeled as the number of copies of the AG haplotype (see Methods and Procedures for details).
The directional effect of the minor allele on the quantitative trait based on the regression coefficient.
The most significant association between rs11705701 and percent body fat remained significant even after PACT was applied to account for the correlation among SNPs and phenotypes and for multiple testing (PACT = 0.041). Percent body fat decreased ∼1.5–2% (corresponding to ∼2.5–4 pounds of fat) with each copy of the A allele for rs11705701 (GG = 33.1 ± 6.1%, GA = 31.5 ± 6.3%, AA = 29.5 ± 6.6%). Given the association with body fat, we next assessed whether adding percent body fat as an additional covariate altered our results for other T2DM-related quantitative traits. All nominal associations initially observed when only age and sex were included as covariates became nonsignificant when percent body fat was added as a covariate to the analysis (Table 3).
The modest association between rs11705701 and percent body fat and the strong effect of percent body fat to account for association between SNPs and T2DM-related quantitative traits suggested the interaction between percent body fat and rs11705701 may be associated with T2DM-related quantitative traits. To assess this possibility, we initially tested whether there was an overall association between T2DM-related quantitative traits and the joint effect of rs11705701 and its interaction with percent body fat using a 2-df test. The joint effect was marginally associated with OGTT 2-h insulin (Bonferroni P = 0.066) and DI (Bonferroni P = 0.072) and significantly associated with SI (Bonferroni P = 0.028). The interaction between rs11705701 and percent body fat alone was also significantly associated with these traits; OGTT 2-h insulin (Bonferroni interaction P = 0.019), SI (Bonferroni interaction P = 0.002), and DI (Bonferroni interaction P = 0.037).
Figure 2a illustrates the association between rs11705701 and SI stratified by percent body fat tertile. Figure 2b illustrates the same effect using the model-predicted trait values to better visualize the interaction between rs11705701 and percent body fat. At low body fat, SI was highest in AA homozygotes and lowest in GG homozygotes. SI decreased with increasing percent body fat across all three genotype groups, illustrating the presumed effect of adiposity to induce insulin resistance. However, the decline in SI across the range of percent body fat followed the opposite order of the pattern observed at low body fat. The fall in SI was greatest among AA homozygotes, less for heterozygotes, and least for GG homozygotes.
Figure 2.

Interaction between rs11705701 and percent body fat on SI. (a) Age- and gender-adjusted mean SI and standard deviation stratified by rs11705701 genotype assuming an additive genetic model and percent body fat tertiles. There was an overall effect of rs11705701 on SI (Bonferroni 2-df P = 0.028), and the interaction between rs11705701 and percent body fat was significantly associated with SI (Bonferroni P = 0.002). SI decreases with increasing body fat among all rs11705701 genotypes; however, the decrease is least among GG homozygotes and greatest among AA homozygotes. (b) Interaction based on the model parameter estimates and covering the range of percent body fat observed in the BetaGene study. The consequence of the differential effect of the variant across the range of body fat is clearly depicted where in the low body fat range SI is higher with each copy of the A allele, whereas within the high body fat range SI is lower with each additional copy of the A allele. SI, insulin sensitivity.
By comparison, we did not observe a significant interaction between rs11705701 and adiposity on AIR (Figure 3). By itself, rs11705701 was only marginally associated with AIR (uncorrected P = 0.047). AIR tended to increase with increasing body fat, but was reduced with each copy of the A allele across the range of percent body fat observed in our sample.
Figure 3.

Interaction between rs11705701 and percent body fat on AIR. (a) Age- and gender-adjusted mean AIR and standard deviation stratified by rs11705701 genotype assuming an additive genetic model and percent body fat tertiles. rs11705701 was marginally associated with AIR (uncorrected P = 0.047), and there was no evidence for percent body fat to modify this association. AIR is similar within each genotype group, indicating no effect of percent body fat to alter AIR. (b) Interaction based on the model parameter estimates and covering the range of percent body fat observed in the BetaGene study. AIR increases with increasing body fat similarly for all genotype groups across the range of body fat. AIR is lower with each copy of the A allele across the range of body fat. AIR, acute insulin response.
Simultaneous examination of Figures 2 and 3 reveals an apparent dissociation between SI and normal β-cell compensation. With increased adiposity, SI fell most in AA individuals, whereas manifesting the smallest rise in AIR across the range of body fat. This lack of compensatory insulin secretion hints at a possible β-cell defect, which is reflected in Figure 4. Figure 4 demonstrates an interaction between rs11705701 and percent body fat on DI, our measure of β-cell compensation from the IVGTT. DI decreased with increasing percent body fat in all genotype groups, but the decline was most precipitous among AA homozygous individuals. In the lowest body fat tertile, all three genotypes had similar DI, but in the middle and highest body fat tertile, DI among AA homozygotes was lowest among the three genotype groups. Thus, the deleterious effect of adiposity on β-cell function was greatest among individuals with the AA genotype.
Figure 4.

Interaction between rs11705701 and percent body fat on DI. (a) Age- and gender-adjusted mean DI and standard deviation stratified by rs11705701 genotype assuming an additive genetic model and percent body fat tertiles. There was a marginal effect of rs11705701 on DI (Bonferroni 2-df P = 0.072), and the interaction between rs11705701 and percent body fat was significantly associated with DI (Bonferroni P = 0.037). Within each genotype group, DI is lower as body fat increases, but the fall in DI is higher among AA homozygotes compared to GG homozygotes. (b) Interaction based on the model parameter estimates and covering the range of percent body fat observed in the BetaGene study. The consequence of the differential effect of the variant across the range of body fat is clearly depicted where in the low body fat range DI is higher with each copy of the A allele, whereas within the high body fat range DI is lower with each additional copy of the A allele. DI, disposition index.
Discussion
SNP rs4402960, located in intron 2 of IGF2BP2, was initially shown to be associated with T2DM in samples of Northern European ancestry (1–3), and this association has been replicated in several other populations (32–35). However, to our knowledge, evidence for association with T2DM-related quantitative traits is lacking. One group reported a modest association between rs4402960 and homeostasis model assessment-beta in a Japanese sample (34), whereas the Insulin Resistance Atherosclerosis Study Family study reported association between rs4402960 and DI in their Hispanic sample (36). Our approach differed from these prior studies in three ways. First, we used more sophisticated and direct methods for phenotyping body composition, insulin resistance, and β-cell function. Second, we applied those methods to a Mexican American population with a wide range of diabetes risk. Third, instead of focusing specifically on rs4402960 or SNPs in high LD with it, we screened a 20-kb region of IGF2BP2, including rs4402960, to test for association with our carefully ascertained traits. With this approach, we observed nominal association between the AG combination of rs13060777 and rs6444082, which tags rs4402960 and percent body fat, OGTT 30′ ΔInsulin, and OGTT 2-h insulin. However, these did not remain significant after correction for multiple testing. By contrast, we found significant associations between a separate SNP in the region, rs11705701, and several T2DM-related quantitative traits. rs11705701 and rs4402960 are ∼32.6-kb apart and in strong LD (D′ = 0.902), although they are only modestly correlated (r2 = 0.579) in our sample. rs11705701 is in the promoter region ∼1.48-kb upstream from exon 1 of IGF2BP2 and is not included in the haplotype block that encompasses all other SNPs we genotyped in our sample (cf. Figure 1). The fact that our associations are observed with rs11705701 and not rs4402960 suggests the former may be in LD with an SNP with functional consequences for IGF2BP2 in Mexican Americans.
The first association that we observed for rs11705701 was with percent body fat, which yielded a nominal P value of 0.001, and remained significant after correcting for multiple testing and the correlation among both SNPs and traits (PACT = 0.041). This finding led us to test whether associations between rs117050701 and other T2DM-related quantitative traits were modified by body fat. Our results show that body fat has very strong effects on the association between rs11705701 and SI and modest effects on OGTT 2-h insulin and DI. We observed an overall effect of rs11705701 on these traits, as reflected by the 2-df test of the SNP main effect and its interaction with percent body fat, while the significant interaction term points to heterogeneity with respect to percent body fat. Plots of the data revealed that both SI and DI decreased most with rising body fat in AA homozygotes and decreased least with rising body fat for GG homozygotes. Of note, there were no analogous associations with BMI, suggesting that IGF2BP2 has its main effects on body fat and demonstrating the potential limitation of BMI as a surrogate measure of body fat in the context of genetic studies.
Given our study population, it is possible that population stratification may confound our analyses. However, we cite two lines of evidence to suggest that this is not the case. First, our results remain similar when individual birthplace is included as an additional covariate in our analyses. Although birthplace is not a perfect proxy for ancestry, the fact that there is little effect of birthplace on our association results would suggest that ancestry is not a major contributor to our observations. Second, when we re-examine the association between rs11705701 and percent body fat using the family-based association test (37,38), which is protected from population stratification, we still see evidence for association (P = 0.018). The reduction in the magnitude of the association is expected, given the family-based association test is based on allelic transmissions, which reduces the efficiency of the test relative to the variance components approach used here.
Overall, the pattern of associations we observed suggests variation in IGF2BP2 may have direct effects on body fat, which may elicit secondary effects on insulin resistance and β-cell function. However, recent work by Prokunina-Olsson et al. (19) provides biological evidence for dual effects IGF2BP2 in human adipose tissue and pancreatic islets. They have shown that the A allele of rs11705701 was associated with increased expression of a functionally distinct alternative splice form of IGF2BP2 that encodes for five RNA-binding domains, compared to the six RNA-binding domains determined by sequence information. This isoform appears to be expressed in many human tissues, but is primarily expressed in human adipose and pancreatic islets and only in carriers of the A allele. Of note, rs4402960 did not correlate with expression of this alternative splice form. These authors also tested rs11705701 for association with T2DM-related quantitative traits in the FUSION sample of Finns. Unfortunately, the FUSION study does not have measures of body fat, and this SNP did not show association with BMI, which is consistent with our observations.
In summary, we tested variation in a limited 20-kb region of IGF2BP2 for association with T2DM-related quantitative traits in our sample of Mexican Americans in the BetaGene study. Our selection of the limited 20-kb region was motivated by preliminary results emanating from the FUSION study. We show that rs11705701 is associated with percent body fat in our subjects and may be associated with insulin secretion. We also show that SI is associated with the interaction between rs11705701 and percent body fat and that β-cell function is marginally associated with the same interaction. Our results are supported by in vitro studies from the FUSION study showing that rs11705701 correlates with an alternative splice form of IGF2BP2 that appears to be primarily expressed in adipose tissue and pancreatic islets. We conclude that variation in IGF2BP2 is associated with body fat. This effect on body fat influences insulin resistance in peripheral tissues and in combination with a modest effect of IGF2BP2 on insulin secretion results in poor β-cell function, which in turn contributes to risk for type 2 diabetes.
Supplementary Material
Supplemental Table I. Estimated haplotype frequencies.
Acknowledgments
We thank the families who participated in the BetaGene study and the support of the USC General Clinical Research Center (M01-RR-00043). We also acknowledge the efforts of our recruiting and technical staff. We also thank our colleagues in the FUSION study for sharing initial results from their genome-wide association scan and permitting us to pursue IGF2BP2, and in particular thank Ludmila Prokunina-Olsson and Francis S. Collins for sharing results from their functional studies on IGF2BP2. This work was supported by NIH grant DK-61628 and an American Diabetes Association Distinguished Clinical Scientist Award to T.A.B. A portion of this work was conducted in a facility constructed with support from Research Facilities Improvement Program grant number C06 (RR10600-01, CA62528-01, RR14514-01) from the National Center for Research Resources.
Footnotes
Supplementary Material: Supplementary material is linked to the online version of the paper at http://www.nature.com/oby
Disclosure: The authors declared no conflict of interest.
References
- 1.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Saxena R, Voight BF, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 2.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen J, Christiansen J, Lykke-Andersen J, et al. A family of insulinlike growth factor II mRNA-binding proteins represses translation in late development. Mol Cell Biol. 1999;19:1262–1270. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kjos SL, Peters RK, Xiang A, et al. Predicting future diabetes in Latino women with gestational diabetes. Diabetes. 1995;44:586–591. doi: 10.2337/diab.44.5.586. [DOI] [PubMed] [Google Scholar]
- 7.Peters RK, Kjos SL, Xiang A, Buchanan TA. Long-term diabetogenic effect of single pregnancy in women with previous gestational diabetes mellitus. Lancet. 1996;347:227–230. doi: 10.1016/s0140-6736(96)90405-5. [DOI] [PubMed] [Google Scholar]
- 8.Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes. Diabetes Care. 2002;25:1862–1868. doi: 10.2337/diacare.25.10.1862. [DOI] [PubMed] [Google Scholar]
- 9.Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol. 1990;162:1008–1014. doi: 10.1016/0002-9378(90)91306-w. [DOI] [PubMed] [Google Scholar]
- 10.Buchanan TA, Xiang A, Kjos SL, et al. Gestational diabetes: antepartum characteristics that predict postpartum glucose intolerance and type 2 diabetes in Latino women. Diabetes. 1998;47:1302–1310. doi: 10.2337/diab.47.8.1302. [DOI] [PubMed] [Google Scholar]
- 11.Buchanan TA, Xiang AH, Kjos SL, et al. Antepartum predictors of the development of type 2 diabetes in Latino women 11-26 months after pregnancies complicated by gestational diabetes. Diabetes. 1999;48:2430–2436. doi: 10.2337/diabetes.48.12.2430. [DOI] [PubMed] [Google Scholar]
- 12.Buchanan TA, Xiang AH, Peters RK, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes. 2002;51:2796–2803. doi: 10.2337/diabetes.51.9.2796. [DOI] [PubMed] [Google Scholar]
- 13.Xiang AH, Peters RK, Kjos SL, et al. Effect of pioglitazone on pancreatic b-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes. 2006;55:517–522. doi: 10.2337/diabetes.55.02.06.db05-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeFronzo RA. The triumvirate: b-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1987;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 15.Bergman RN. Toward physiological understanding of glucose tolerance.Minimal model approach. Diabetes. 1989;38:1512–1527. doi: 10.2337/diab.38.12.1512. [DOI] [PubMed] [Google Scholar]
- 16.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe RM, Xiang A, Trigo E, et al. Evidence of genetic predisposition for B-cell dysfunction in Mexican-American families of probands with gestational diabetes. Diabetes. 2003;52(Suppl 1):A257. [Google Scholar]
- 18.Bergman RN, Zaccaro DJ, Watanabe RM, et al. Minimal model-based insulin sensitivity has greater heritability and a different genetic basis than homeostasis model assessment or fasting insulin. Diabetes. 2003;52:2168–2174. doi: 10.2337/diabetes.52.8.2168. [DOI] [PubMed] [Google Scholar]
- 19.Prokunina-Olsson L, Hanisch JJ, Jackson AU, et al. A novel variant of IGF2BP2 is associated with type 2 diabetes and affects expression of a functional splicing isoform. Diabetes. 2008;57(Suppl 1):A61. [Google Scholar]
- 20.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ. SNP genotyping by the 5′-nuclease reaction. Methods Mol Biol. 2003;212:129–147. doi: 10.1385/1-59259-327-5:129. [DOI] [PubMed] [Google Scholar]
- 22.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;236:E667–E677. doi: 10.1152/ajpendo.1979.236.6.E667. [DOI] [PubMed] [Google Scholar]
- 23.Boston RC, Stefanovski D, Moate PJ, et al. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther. 2003;5:1003–1015. doi: 10.1089/152091503322641060. [DOI] [PubMed] [Google Scholar]
- 24.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68:1456–1467. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- 27.Blangero J, Almasy L. Multipoint oligogenic linkage analysis of quantitative traits. Genet Epidemiol. 1997;14:959–964. doi: 10.1002/(SICI)1098-2272(1997)14:6<959::AID-GEPI66>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 28.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 30.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 31.Conneely KN, Boehnke M. So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am J Hum Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascoe L, Tura A, Patel SK, et al. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes. 2007;56:3101–3104. doi: 10.2337/db07-0634. [DOI] [PubMed] [Google Scholar]
- 33.Grarup N, Rose CS, Andersson EA, et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes. 2007;56:3105–3111. doi: 10.2337/db07-0856. [DOI] [PubMed] [Google Scholar]
- 34.Horikoshi M, Hara K, Ito C, et al. Variations in the HHEX gene are associated with increased risk of type 2 diabetes in the Japanese population. Diabetologia. 2007;50:2461–2466. doi: 10.1007/s00125-007-0827-5. [DOI] [PubMed] [Google Scholar]
- 35.Omori S, Tanaka Y, Takahashi A, et al. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes. 2008;57:791–795. doi: 10.2337/db07-0979. [DOI] [PubMed] [Google Scholar]
- 36.Palmer ND, Goodarzi MO, Langefeld CD, et al. Quantitative trait analysis of type 2 diabetes susceptibility loci identified from whole genome association studies in the Insulin Resistance Atherosclerosis Family study. Diabetes. 2008;57:1093–1100. doi: 10.2337/db07-1169. [DOI] [PubMed] [Google Scholar]
- 37.Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- 38.Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table I. Estimated haplotype frequencies.