

Abstract
The Bcl-2 family of proteins plays a critical role regulating apoptosis, and pro-survival Bcl-2 family members are important therapeutic targets due to their overexpression in different cancers. Pro-apoptotic BH3-only proteins antagonize pro-survival Bcl-2 protein functions by binding directly to them, and a sub-class of BH3-only proteins termed sensitizers can initiate apoptosis via this mechanism in response to diverse signals. The five pro-survival proteins Bcl-xL, Mcl-1, Bcl-2, Bcl-w and Bfl-1 differ in their binding preferences, with Bcl-xL, Bcl-2 and Bcl-w sharing similar interaction profiles for many natural sensitizers and small molecules. Peptides that bind selectively to just one or a subset of family members have shown utility in assays that diagnose apoptotic blockades in cancer cells and as reagents for dissecting apoptotic mechanism. Combining computational design, combinatorial library screening and rational mutagenesis, we designed a series of BH3 sensitizer peptides that bind Bcl-xL with sub-nanomolar affinity and selectivity up to 1000-fold over each of the four competing pro-survival proteins. We demonstrate the efficacy of our designed BH3 peptides in assays that differentiate between cancer cells that are dependent on different pro-survival proteins.
Keywords: Bcl-2 proteins, BH3 peptides, yeast surface display, BH3 profiling, peptide design
Introduction
The Bcl-2 family of proteins plays a critical role controlling apoptosis in healthy tissues and inappropriately promoting or blocking apoptosis in disease1; 2. Pro-survival members of the Bcl-2 family are important therapeutic targets due to their over-expression in cancer cells, where they contribute to oncogenesis and resistance to chemotherapy2; 3; 4; 5. The pro-survival proteins Bcl-xL, Bcl-2, Bcl-w, Mcl-1 and Bfl-1 can bind to and prevent the pro-apoptotic activator and effector molecules Bim, Bid, Bax and Bak from initiating mitochondrial outer membrane permeabilization (MOMP), which leads to cell death6. A separate class of pro-apoptotic Bcl-2 proteins, BH3-only sensitizers, can antagonize this function by binding competitively to the pro-survival proteins7. At a structural level, pro-apoptotic Bcl-2 family effectors, activators and sensitizers are characterized by the presence of a short Bcl-2 homology 3 (BH3) sequence that forms an alpha helix and engages a conserved hydrophobic groove on the surface of the pro-survival proteins8. Small molecules and peptides that can bind to this groove to restore apoptotic function, acting as synthetic sensitizers, are being developed as candidate therapeutics for a variety of cancers9; 10; 11; 12; 13.
Individual BH3 motifs have varying affinities for the five pro-survival proteins Bcl-xL, Bcl-2, Bcl-w, Mcl-1 and Bfl-114. Activators Bid and Bim interact strongly with all five, whereas sensitizers exhibit different specificity profiles.15 Thus, the pro-survival proteins are non-equivalent in terms of molecular function, and a cancer cell that blocks pro-death signaling using up-regulation of Bcl-xL is physiologically distinct from one that up-regulates Mcl-1 or another protein. The dependence of a cancer cell on specific pro-survival proteins to bypass apoptosis has been described as “addiction” to those proteins16. Interestingly, because a delicate balance between numerous pro-survival and pro-apoptotic family members determines cell fate, the levels of various pro-survival proteins may not be reliable indicators of which proteins are critical for cell survival. Simple quantitation of protein levels has thus far generally proven insufficient for diagnosing addiction and predicting the response of a cell to treatment with natural or engineered sensitizers17; 18. Functional assays that can measure the level of sequestration of pro-apoptotic proteins by different pro-survival proteins have been shown to be a better probe of cellular state and a good predictor of apoptotic responses17; 18; 19. In such instances, pro-apoptotic activator proteins are often described as “priming” the pro-survival proteins and such a cellular condition is referred to as “primed”. In BH3 profiling assays, the addiction profiles of different cancer cells are diagnosed by treating mitochondria with synthetic sensitizer BH3 molecules that have different selectivity profiles for binding to the pro-survival proteins20; 21. High potency and specificity of sensitizer BH3 molecules for different pro-survival members is important for successful profiling.
Pro-survival proteins Bcl-xL, Bcl-2 and Bcl-w share 30-40% sequence identity and are discernible from Mcl-1 and Bfl-1 in terms of their binding preferences for naturally occurring BH3 motifs14. For example, Mcl-1 and Bfl-1, but not Bcl-xL, Bcl-2 or Bcl-w, bind to the BH3 region of Noxa, and the converse is true for binding to the BH3 region of Bad14. In previous studies, peptide variants of the BH3 region of natural pro-apoptotic proteins were designed to specifically bind Bcl-xL vs. Mcl-1 or vice versa22; 23; 24; 25. Although engineered peptides that bound Bcl-xL exhibited specificity over Mcl-1 and Bfl-1, they bound with varying affinities to Bcl-2 and Bcl-w, which is not surprising given the closer relationship of Bcl-xL to these proteins24. In this work, we engineered Bcl-xL-binding peptides with enhanced specificity for Bcl-xL over Bcl-2 and Bcl-w. By screening combinatorial libraries that were computationally designed to be enriched in Bcl-xL selective binders, we engineered BH3 peptides with >1000-fold specificity for Bcl-xL over Bcl-2 and Bcl-w. Furthermore, we demonstrated the utility of several Bcl-xL selective peptides in BH3 profiling.
Results and Discussion
Computational library design
Our goal was to design a combinatorial peptide library enriched with sequences that would preferentially bind Bcl-xL over Mcl-1 and Bfl-1, and that would also include specificity elements that could potentially discriminate Bcl-xL, Bcl-2 or Bcl-w individually. The design procedure was based on previously published SPOT arrays, which were used to measure 180 single point mutants of Bim-BH3 (a peptide corresponding to the BH3 region of Bim) binding to each of the 5 Bcl-2 family proteins of interest24; 26. The SPOT data were used to identify residue substitutions in Bim-BH3 that could be classified as “non-disruptive” or “specific” for Bcl-xL binding (Supplemental Table 1). Then, a computational optimization protocol was used to choose degenerate codons that balanced the inclusion of specific and non-disruptive residues in the library, within a total library size of ~107 DNA sequences. The library design procedure is described in detail under Materials and Methods. Sites that were varied in the final library are labeled in Fig. 1A, and the full library is described in Supplemental Table 1.
Figure 1.

Screening for Bcl-xL selective peptides using cell sorting. (A) Structure of Bim-BH3 bound to Bcl-xL (PDB ID: 3FDL). Residues of Bim-BH3 that were randomized in the combinatorial library are shown as sticks and labeled. Tyrosine at position 4e, which was not included in the library design, is also shown (see Results and Discussion). (B) Schematic of the yeast-surface display system. (C-H) Plots illustrating enrichment of a Bcl-xL selective population of peptide-displaying cells. FACS profiles of Bcl-xL binding to Bim-BH3 (C and D), the library population after four rounds of positive selection (E and F) and the final Bcl-xL specific population (G and H). Analysis was performed in the presence of 10 nM Bcl-xL only (left panels) or 10 nM Bcl-xL plus 1 μM of each of Mcl-1, Bfl-1, Bcl-2 and Bcl-w (right panels).
Library screening using yeast-surface display
We screened the designed library for peptides that bound Bcl-xL using a previously described yeast-surface display system (Fig. 1B)24; 27. After four rounds of cell sorting, the library demonstrated enhanced binding to Bcl-xL (Fig. 1E). Although binding of Bim-BH3 to Bcl-xL (Fig. 1C) could be blocked using 1 μM concentrations of competitor pro-survival proteins (Fig. 1D), a fraction of the Bcl-xL binding population after four rounds of cell sorting (Fig. 1E) was much more resistant towards binding to competitors (Fig. 1F). To isolate Bcl-xL specific clones, we included a pool of unlabeled proteins (Mcl-1, Bcl-2 and Bcl-w) as competitors in subsequent rounds of screening. A significant increase in specificity was observed for binding to Bcl-xL (at 10 nM) in the presence of unlabeled Bcl-2, Bcl-w and Mcl-1 (at 1 μM) after only two rounds of competition screening (data not shown). We performed four more rounds of competitive screening to further enrich Bcl-xL-specific peptides (Fig. 1G and H). Sequencing the BH3 insert in 96 clones from the final pool led to the identification of 10 unique sequences, of which two were predominant: XXA1 and XXA4 (>50% and ~15% of sequenced clones, respectively, Supplemental Table 2). Using dual color fluorescence to measure binding to two different partners, we observed that XXA1 exhibited robust binding to Bcl-xL in the presence of 100-fold excess of each of the competitor pro-survival proteins (Supplemental Fig. 1).
Fluorescence polarization binding assays
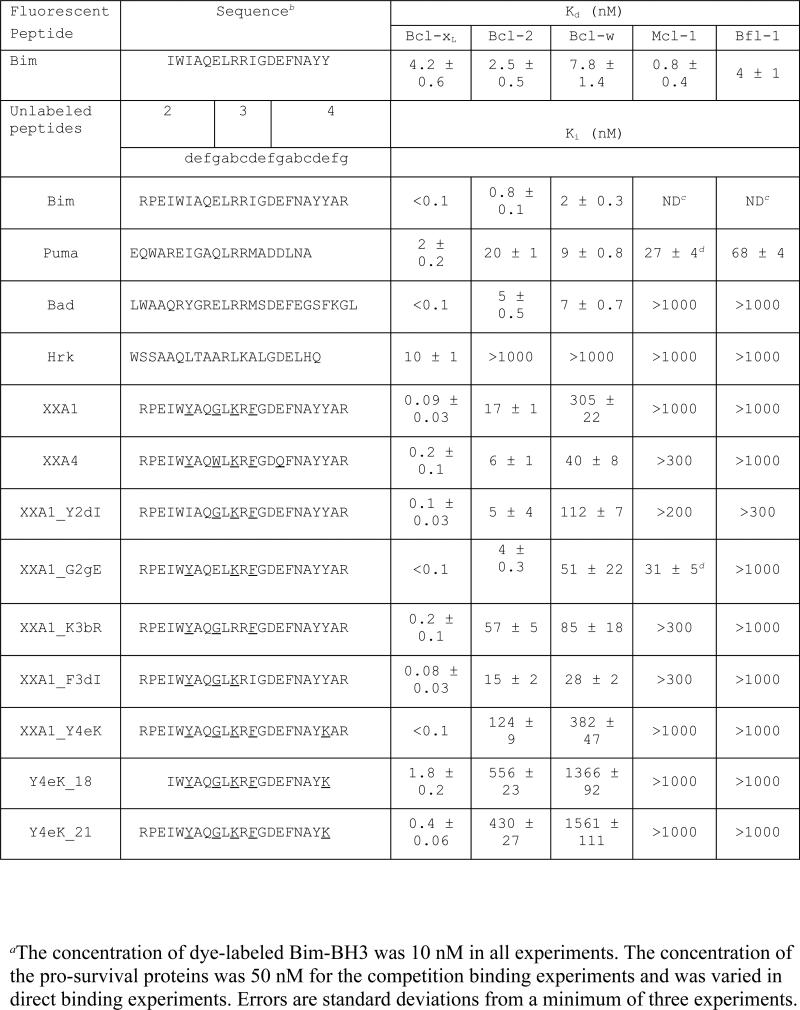
We synthesized XXA1 and XXA4 as 23-residue peptides and measured their binding to Bcl-xL using a fluorescence polarization (FP) assay that involved competition with a fluorescently labeled Bim-BH3 peptide. In agreement with the yeast-display results, both XXA1 and XXA4 bound with sub-nanomolar affinity to Bcl-xL (Table 1, Fig. 2A). In contrast, unlabeled XXA1 or XXA4 did not compete with fluoresceinated Bim-BH3 for binding to Mcl-1 or Bfl-1 up to a concentration of 10 μM (Table 1, Supplemental Fig. 2). XXA1 bound Bcl-xL with ~180-fold specificity over Bcl-2 and ~3,400-fold specificity over Bcl-w (Fig. 2B, C). XXA4 had modest specificity compared to XXA1: ~30 fold for Bcl-2 and ~200 fold for Bcl-w.
Table 1.
Sequences and binding constants for BH3 peptidesa
|

|
Figure 2.

Binding specificity of designed peptides and point mutants. Binding curves show competition between unlabeled BH3 peptides and fluorescently labeled Bim-BH3 for interaction with Bcl-xL (A), Bcl-2 (B) and Bcl-w (C). The concentration of the fluorescent peptide and the pro-survival protein were 10 nM and 50 nM, respectively. (D) Affinities of Bim-BH3 and XXA1, and of peptides with point mutations described in the text, for binding to Bcl-xL, Bcl-2 and Bcl-w. Kd values below 0.1 nM, which could not be reliably quantified, are indicated by asterisks.Error bars represent standard deviations from a minimum of three experiments.
We also measured the affinities of peptides derived from the BH3 regions of Bad, Puma and Hrk for binding to Bcl-2 proteins (Table 1). Puma-BH3 bound with 2-70 nM affinity to all five pro-survival proteins, and most tightly to Bcl-xL (Ki = 2 nM). Hrk-BH3 bound selectively, but not very tightly to Bcl-xL (Ki = 10 nM). Bad-BH3 bound Bcl-xL with affinity similar to or greater than XXA1 and XXA4, and also bound with low-nanomolar affinity to both Bcl-2 and Bcl-w. The Ki values obtained in this study for natural BH3 peptides corresponding to sequences from Bim, Puma, Bad and Hrk differ from some previously published studies 14; 16 It has been demonstrated that peptide length affects the affinity of BH3 peptides for pro-survival proteins 23. We speculate that several factors, such as differences in lengths, sequences (e.g. human vs. mouse sequences), and the presence or absence of fluoresceinated tags are responsible for differences observed. In this work, biochemical affinity measurements were made for the same peptides that were subsequently used in cell-based testing.
Contribution of individual mutations to the specificity of XXA1
XXA1 has four mutations relative to wild-type Bim-BH3. Using position labeling defined in Table 1, and a convention where mutations are described by listing the original residue, the substitution site, and the new residue, the mutants were: I2dY, E2gG, R3bK and I3dF. All of these substitutions were classified as non-disruptive for binding to Bcl-xL based on the SPOT arrays, and three (I2dY, R3bK and I3dF) were classified as potentially providing specificity against Mcl-1 or Bfl-1. None of the residues were classified as potentially providing specificity against Bcl-2 or Bcl-w in the library design procedure (Supplemental Table 1). We made each of these single mutations in Bim-BH3 and also reverted each of the mutated residues to the wild-type Bim residue in the context of XXA1.
Each point mutant of Bim-BH3 bound Bcl-xL with subnanomolar affinity. Due to the limited sensitivity of the assay, we could only assign an upper bound on the Ki (Ki < 0.1 nM) for these interactions (Fig. 2D; Supplemental Table 3 and Supplemental Fig. 3). The two individual mutations I2dY and E2gG modestly weakened binding to Bcl-2, by ~5-fold and 7-fold respectively. Interestingly, all four point mutations had a small, stabilizing effect on binding to Bcl-w, despite the fact that when combined in XXA1 they weakened binding to Bcl-w by ~150 fold, indicating that they act in a non-additive manner.
In the context of XXA1, all single-residue revertants bound strongly to Bcl-xL (Ki ≤ 0.2 nM) (Table 1 and Fig. 2D). Consistent with the influence of I2dY in Bim-BH3, XXA1_Y2dI reduced the specificity for Bcl-xL over Bcl-2 (by ~3-4 fold). The pocket surrounding the 2d-position binding site differs between Bcl-xL and Bcl-2 by a leucine (Leu108 in Bcl-xL) vs. a methionine (Met115 in Bcl-2) (Supplemental Fig. 4). Revertants XXA1_K3bR and XXA1_F3dI showed reduced specificity for Bcl-xL over Bcl-w by ~8-10 fold compared to XXA1. Residues contacting the 3b position in Bcl-xL, Arg 132 and Asp133, are identical between Bcl-xL and Bcl-2 but not Bcl-w, where the equivalent residues are Q88 and E89 (Supplemental Fig. 4). It is noteworthy that substitutions at 3b and 3d weaken binding to Bcl-w in a context dependent manner in XXA1, as R3bK and I3dF did not have a significant effect on Bcl-w binding in the context of Bim (Fig. 2D; Supplemental Table 3).
Revertant XXA1_G2gE bound tighter than XXA1 to Bcl-2 and Bcl-w, and glycine at 2g may contribute to Bcl-xL binding specificity (Table 1). However, we could not quantify differences between very low Ki values for XXA1 vs. XXA1_G2gE binding to Bcl-xL using the 18-mer Bim-BH3 in the competition assay. By using a longer 23-mer fluorescently labeled Bim-BH3 peptide (that binds tighter than the 18-mer Bim-BH3 peptide), we determined that XXA1_G2gE also bound more tightly than XXA1 to Bcl-xL (Supplemental Fig. 5), These data show that glycine at 2g destabilized interactions with each of Bcl-xL, Bcl-2 and Bcl-w. Thus, if this glycine contributes to the specificity of XXA1, it must do so by greater destabilization of binding to Bcl-2 and Bcl-w than to Bcl-xL
Specificity against Mcl-1 was also a criterion in our screen. Tyr substitution at 2d was observed in a high fraction of Bcl-xL specific sequences in a previous screen that included negative selection only against Mcl-124. In addition, a BH3 peptide derived from Bad, which is specific for Bcl-xL/Bcl-2/Bcl-w over Mcl-1/Bfl-1, has a Tyr at the 2d position and a loss of specificity was observed upon reverting Tyr to Ile 28. We could not quantify the effect of the Y2dI mutation in XXA1, given the weak binding to Mcl-1, but this mutation likely contributes to the preference of XXA1 to bind Bcl-xL over Mcl-1. Glycine at position 2g also disfavored binding to Mcl-1. Neither XXA1 nor XXA1_G2gE could compete with fluorescently labeled Bim-BH3 for binding to Mcl-1. However, using a 23-residue fluorescently labeled Hrk-BH3, which has a weaker affinity for Mcl-1 (Kd ~ 33 nM), we measured a Ki value of ~ 31 nM for XXA1_G2gE for Mcl-1, whereas XXA1 did not compete with Hrk up to a concentration of 10 μM (Supplemental Fig. 6). Thus, glycine at position 2g in XXA1 weakened all interactions that we measured.
Increasing the specificity of XXA1 over Bcl-2
Although XXA1 exhibited greater than 100-fold specificity for Bcl-xL over Bcl-2, it still bound Bcl-2 with a Kd of ~17 nM. To reduce affinity for Bcl-2, we introduced point mutation Y4eK (Figure 1A). The 4e position was not included in the SPOT array experiments, and therefore was not included as a position for randomization in our combinatorial library design. However, lysine substitution at 4e in the context of Bim-BH3 has been reported to stabilize binding to Bcl-xL relative to Mcl-123. In addition, substituting the wild-type methionine residue at the 4e position in Bax with lysine imparted ~3-fold specificity for Bcl-xL over Bcl-229. The structure of Bcl-2:Bax illustrates the importance of non-polar interactions at position 4e with Bcl-2, and methionine substitution with alanine at 4e resulted in ~13 fold reduction in affinity for Bcl-230. Introducing lysine into XXA1 at position 4e resulted in ~7 fold reduction in binding to Bcl-2 compared to XXA1, while maintaining the affinity for Bcl-xL unchanged, such that XXA1_Y4eK bound to Bcl-xL ~1200-fold more tightly than to Bcl-2 and ~3800-fold more tightly than to Bcl-w (Table 1).
To explore the possibility that peptide length could differentially influence binding to different proteins23; 31; 32, we investigated the effect of truncating XXA1_Y4eK (Table 1). Y4eK_18, an 18-residue peptide obtained by truncating XXA1_Y4eK from both the N and C terminal ends, bound Bcl-2 ~5-fold weaker than the parent 23-residue XXA1_Y4eK. However, this truncation also reduced the affinity for Bcl-xL by ~18 fold, thereby reducing specificity for Bcl-xL. Shortening XXA1_Y4eK by two residues from only the C terminal end, leading to the peptide Y4eK_21, weakened binding to Bcl-xL and Bcl-2 by similar amounts (Table 1). Y4eK_21 bound to Bcl-xL with Ki = 0.4 nM and to Bcl-2 with Ki = 430 nM, retaining the same ~1000-fold binding preference for Bcl-xL as peptide XXA1_Y4eK.
BH3 profiling with engineered peptides
In BH3 profiling, mitochondrial outer membrane permeabilization (MOMP) in response to a panel of BH3 peptides is monitored and used to identify the pro-survival protein(s) most responsible for apoptotic resistance16; 20. For example, a cell line that depends on Bcl-xL overexpression to inhibit apoptosis should show a response to Bcl-xL binding peptides. Naturally occurring sensitizer BH3 peptides that are currently used for BH3 profiling assays offer a limited range of specificities and potencies. In previous studies, it was demonstrated that the BH3 domains from Bad and Noxa can be used to distinguish the dependence of cancer cells for survival on Bcl-2 vs. Mcl-116. Because our engineered peptides have novel specificity profiles compared to naturally occurring partners, and might therefore have potential to provide additional information about cancer cell states, we tested their efficacy in BH3 profiling assays.
Using a previously reported whole-cell profiling assay21, we monitored loss in membrane potential with increasing concentrations of native or designed BH3 peptides in different cell lines (Figure 3). MDA-MB-231, a human breast cancer cell line, has previously been shown to exhibit a primed Bcl-xL dependent profile, meaning that Bcl-xL antagonizes pro-apoptotic function and treatment with sensitizers targeting Bcl-xL leads to mitochondrial permeabilization33. We tested the response of cells to peptides XXA1, XXA1_Y4eK, XXA1_G2gE, Y4eK_21 and Y4eK_18 along with sensitizer peptides derived from the BH3 regions of Hrk, Bad and Puma that are listed in Table 1 (Figure 3A; Table 2). Puma-BH3 binds to all pro-survival proteins and at concentrations ranging from 10-100 μM depolarizes mitochondria in all cells where pro-survival proteins are holding pro-death factors in check14; 15. Hrk has been used previously as a functional probe for Bcl-xL dependence because it differentiates Bcl-2 and Bcl-xL in BH3 profiling experiments when used at high micromolar concentration16.
Figure 3.

BH3 profiling with native BH3 vs. engineered BH3 peptides. Loss of mitochondrial membrane potential was monitored using the fluorescent probe JC-1 with increasing concentrations of peptides applied to permeabilized MDA-MB-231 cells (A), Bcl-2 3256 cells (B) or OCI-Ly1 cells (C). Depolarization was calculated as a percentage of the value observed with FCCP (see Methods). Error bars are standard deviations from a minimum of three biological replicates for each cell line (see Methods).
Table 2.
Peptide potency and specificity in BH3 profiling assaysa
| Unlabeled peptides (23 residues) | EC50 (nM) | |||
|---|---|---|---|---|
| MDA-MB-231 (Bc1-xL dependent) | Platelets (Bc1-xL dependent) | OCI-Ly1 (Bcl-2 dependent) | Bcl-2 3256 (Bcl-2 dependent) | |
| Bad | 76 ± 25 | 21 ± 3 | 196 ± 48 | 361 ± 125 |
| Puma | 1006 ± 337 | 393 ± 64 | 1967 ± 480 | 663 ± 223 |
| Hrk | >10, 000 | > 1000 | >10,000 | >10,000 |
| XXA1 | 50 ± 16 | 16 ± 3 | 228 ± 56 | 540 ± 183 |
| XXA1_G2gE | 12 ± 4 | 30 ± 5 | 37 ± 9 | 141 ± 50 |
| XXA1_Y4eK | 82 ± 27 | 26 ± 4 | 911 ± 222 | 657 ± 221 |
| Y4eK_18 | 847 ± 284 | not measured | 3325 ± 812 | 8497 ± 3091 |
| Y4eK_21 | 159 ± 53 | not measured | 3435 ± 839 | 5157 ± 1785 |
Error represent standard deviations for a minimum of three biological replicates for each cell line.
The trends in EC50 values for inducing MOMP in MDA-MB-321 cells agreed well with the observed trends in Ki values for Bcl-xL binding, consistent with a model where peptide binding relieves MOMP inhibition that arises from overexpression of pro-survival proteins (Tables 1, 2; Figure 4; Supplemental Table 4). First, all of the engineered Bcl-xL binding peptides were more potent (~50-600 fold) than Hrk for permeabilizing mitochondria (Fig. 3A; Table 2). The potencies of XXA1 and XXA1_Y4eK were comparable to that of the Bad BH3 peptide (EC50 values of 50-80 nM; Ki values ~0.1 nM or lower), whereas XXA1_G2gE was more potent than Bad by ~7-fold, consistent with XXA1_G2gE binding more tightly than XXA1 to Bcl-xL in vitro (Supplemental Fig. 5). The similar responses to Puma (EC50 ~ 1006 nM) and Y4eK_18 (EC50 ~ 847 nM) were consistent with their similar Ki values of ~ 2 nM for solution binding. Y4eK_21 gave an intermediate response, with a Ki value of ~0.4 nM and an EC50 value of ~160 nM.
Figure 4.

Trends in solution Ki values are consistent with EC50 values from BH3 profiling. EC50 values from BH3 profiling are plotted against Ki values measured in solution for natural and engineered BH3 peptides tested in MDA-MB-231 cells (A), Bcl-2 3256 cells (B) or OCI-Ly1 cells (C). Interaction data points for which only an upper or lower bound on the Ki was determined have a red border: upper bounds are plotted for Bad, XXA1_G2gE and XXA1_Y4eK in (A), and lower bounds are shown for Hrk in all three panels. Error bars are standard deviations from a minimum of three biological replicates for each cell line (see Methods).
To assess specificity, we tested the activity of our designed peptides in two murine leukemia lines driven by the overexpression of Myc and dependent on either Mcl-1 (Mcl-1 2640) or Bcl-2 (Bcl-2 3256)34. As expected, the low amount of depolarization observed in the presence of 5 μM XXA1, XXA1_Y4eK or XXA1_G2gE in the Mcl-1 dependent line was similar to that resulting from treatment with Hrk or Bad, whereas natural BH3 peptides from Puma and Noxa gave a strong response at this concentration (Supplemental Fig. 7). In Bcl-2 dependent cells, Bcl-2 3256, all of the designed peptides led to MOMP, although at higher concentrations than those required to induce MOMP in Bcl-xL dependent cells, as expected (Fig. 3B; Table 2). Trends in EC50 values roughly followed trends in Ki values for binding to Bcl-2 (Fig. 4; Supplemental Table 4). However, the response of Bcl-2 3256 cells to XXA1_Y4eK (EC50 ~ 657 nM) was only modestly less than that to Bad BH3 (EC50 ~ 361 nM), even though XXA1_Y4eK bound Bcl-2 ~25 fold weaker than Bad-BH3. Also, XXA1 and XXA1_Y4eK gave similar EC50 values, despite the ~ 7 fold weaker binding to Bcl-2 of XXA1_Y4eK compared to XXA1 in solution. Nevertheless, both Y4eK_18 and Y4eK_21 had significantly weaker responses than XXA1 and XXA1_Y4eK (EC50 values in the 5-8 μM range), consistent with their weaker solution binding (Table 2).
Bcl-2 3256 over-expresses human Bcl-2 but also expresses endogenous murine Bcl-235, and we wondered whether this contributed to differences between the profiling results and in vitro measurements. Therefore, we monitored the response of the peptides in a diffuse large B-cell lymphoma cell line OCI-Ly1, which is solely dependent on human Bcl-2 for survival17 (Figure 3C; Table 2). Quantification of protein levels in this cell line previously showed that Bcl-xL and Bcl-w were present at negligible amounts compared to Bcl-217. We observed that XXA1_Y4eK was less potent in this cell line than Bad (EC50 ~ 911 nM vs. 196 nM) and also less potent than XXA1 (EC50 ~ 228 nM). This trend is in better agreement with the differences in Ki values among Bad and the engineered peptides than what we observed for the Bcl-2 3256 cells (Table 1). Y4eK_18 and Y4eK_21 gave approximately 3-4-fold weaker response than XXA1_Y4eK in OCI-Ly1 cells, which is also in good agreement with the in vitro binding measurements (Figure 4; Supplemental Table 4).
Because Bcl-xL is important for platelet survival36; 37, we subjected primary human platelet samples to the profiling assay with engineered and natural peptides. Not surprisingly, peptides XXA1, XXA1_Y4eK and XXA1_G2gE caused potent depolarization to a similar extent as Bad (Table 2 and Supplemental Figure 8), exceeding the response to the BH3 peptides derived from Hrk and Puma. However, in contrast to depolarization of Bcl-xL dependent MDA-MB-231 cells, the response to XXA1_G2gE was almost identical to that of XXA1 and XXA1_Y4eK in platelets. This could be related to differences in the levels of Bcl-xL and other pro-survival proteins between platelets and MDA-MB-231.
None of XXA1, XXA1_Y4eK or XXA1_G2gE peptides tested showed any non-specific pore forming activities in DHL10 (a Bax- and Bak-deleted cell line), providing evidence that the artificial sensitizer peptides act through the mitochondrial apoptotic pathway (Supplemental Fig. 7)17. In addition, although the engineered peptides are based on Bim BH3, which is an activator peptide that can interact directly with Bax38 and/or Bak39, they did not exhibit any significant response within the relevant concentration range (from 100 nM to 10 μM) in the poorly primed cell line SK-MEL-217, indicating they act primarily as sensitizers in the profiling assay (Supplemental Figure 9).
Conclusions
In this work we used yeast-surface display library screening to identify peptides that bind tightly to Bcl-xL in preference to Bcl-2, Bcl-w, Mcl-1 and Bfl-1. Bcl-xL has different interaction properties compared to Mcl-1 and Bfl-1 with respect to binding native BH3 and BH3-like motifs24; 26 14; 16; 30, and we readily identified tight Bcl-xL binders with much lower affinity for these two pro-survival proteins (specificity > 104). Several prior studies have also reported peptides that can discriminate between Bcl-xL and Mcl-123; 24; 25. It proved more difficult to discriminate among proteins Bcl-xL, Bcl-2 and Bcl-w. The Bim-BH3 mutant SPOT arrays identified only three candidate residues for disfavoring interactions with Bcl-2 or Bcl-w, and these specificity residues predicted by the SPOT experiments were not found in the peptides isolated from our screen. Nevertheless, library screening led to the identification of XXA1 and XXA4, which bound to Bcl-xL 30-190-fold tighter than to Bcl-2, and 200-3400-fold tighter than to Bcl-w (Table 1). Specificity arose from non-additive effects of mutations in the parent Bim-BH3 peptide that could not be easily anticipated based on what is known about Bcl-2 family interaction specificity so far. Subsequent introduction of a tyrosine-to-lysine mutation, and truncation of the resulting peptide, provided even greater Bcl-xL binding specificity.
Our engineered peptides based on XXA1 acted as highly potent apoptotic sensitizers, leading to MOMP in mitochondria from cells that depend on Bcl-xL to block apoptosis. Peptide XXA1, identified by yeast screening, gave an EC50 value of 50 nM in permeabilized MDA-MB-231 cells. Peptide XXA1_G2gE was the most potent peptide we tested, with EC50 values of 12 and 30 nM for inducing MOMP in MDA-MB-231 cells and platelets, respectively. XXA1_G2gE also gave a strong response in Bcl-2 dependent cells, but other peptides from our study could distinguish Bcl-xL from Bcl-2 dependence when used at low nanomolar concentrations. In particular, XXA1_Y4eK and its truncated variant Y4eK_21 showed significantly improved selectivity compared with Bad BH3-for profiling Bcl-2 dependent OCI-Ly1 human cells. These two peptides represent a significant improvement over the weaker but selective Hrk-BH3 peptide that has been used previously for BH3 profiling20. Although not tested in profiling assays, due to the absence of appropriate cell lines, it is noteworthy that our peptides also showed significantly weaker binding to Bcl-w compared to Bad. Thus, we expect that these peptides will have utility for distinguishing patient cancer cells dependent on Bcl-xL vs. Bcl-2 or Bcl-w for survival. High affinity peptides with known binding specificities can also be useful as reagents in basic research and drug discovery, and promising developments in peptide delivery suggest that chemically stabilized peptides that bind to Bcl-2 proteins may have potential as therapeutics11; 40.
Materials and Methods
Computational design of library
The design procedure used in this work is adopted from a previously reported computational library design protocol used in Bfl-1 library design38. Non-disruptive and specific mutations were assigned based on published SPOT array experiments. Residues labeled as non-disruptive were the least destabilizing 50% of mutations tested on SPOT arrays, and residues designated as specific were the top 33% of mutations favoring binding to one protein over another24. As described in Supplementary Table 1, for positions 2d, 2e, 3b, 3e, 3f and 4a, only two types of specificity residues were considered, “Bcl-xL over Mcl-1” and “Bcl-xL over Bfl-1”, whenever such residues exist. For positions 2g, 3a, 3d and 3g, 6 additional types of specificity residues, “Bcl-xL over Bcl-2”, “Bcl-xL over Bcl-w”, “Bcl-2 over Bcl-xL”, “Bcl-2 over Bcl-w”, “Bcl-w over Bcl-xL”, and “Bcl-w over Bcl-2” were predicted. These 4 positions were selected because they contact receptor positions occupied by different amino acids among Bcl-xL, Bcl-2 and Bcl-w based on the crystal structure of the complex between Bcl-xL and Bim (PDB code: 3FDL)41. For Bcl-2 and Bcl-w, the mapping of residues was determined by sequence alignments with Bcl-x L(Supplemental Fig. 4). As for Bfl-1 library design, only codons that encoded both the native Bim-BH3 amino acid and at least one of each type of specificity residues at each design position were considered, whenever such residues existed.
Expression and purification of pro-survival proteins
Pro-survival proteins used in these studies were expressed in Escherichia coli and purified as described previously24; 26. The constructs used in this study expressed the human pro-survival proteins Mcl-1 (residues 172-327), Bcl-xL (residues 1-209), Bcl-2 (residues 1-217), Bcl-w (residues 1-164), and Bfl-1 (residues 1-151).
Yeast library construction
The yeast display library was constructed using previously described procedures24. The randomized BH3 library segment was synthesized using PCR using the mutagenic forward primer Bcl lib (5’ GGC CGT CCG GAA ATT TGG NDK GCG CAG DDK BHC DNA CGT DNS GSC GAT SRA TTT AAT GCG TAT TAT GCG CGT CGC 3’; N represents a mixture of A, T, G and C; D represents a mixture of A, G and T; K represents a mixture of G and T; B represents a mixture of C, G and T; H represents a mixture of A, C and T; R represents a mixture of A and G, S represents a mixture of G and C ) and a reverse primer (5′ CTAAAAGTACAGTGGGAACAAAGTCG 3’) with wild-type Bim-BH3 as a template. The PCR product was further extended at the 5’ end because overlapping ends of greater than 50 base pairs have been shown to increase the efficiency of homologous recombination in yeast42. The product after amplification was purified using the Qiagen PCR purification kit. The purified PCR product was transformed into yeast along with an acceptor vector cut with Nhe1 and Xho1using an established electroporation protocol43.
Yeast-display analysis and flow cytometric sorting
Yeast cells expressing members of the BH3 library were incubated with one or more pro-survival proteins as described previously and stained with primary and fluorescently labeled secondary antibodies24. Expression of BH3 peptides was detected using FITC fluorescence. Binding to Bcl-xL or other pro-survival proteins was monitored using APC fluorescence. Cells were sorted in a BD FacsAria using a 488 nm and/or 561 nm excitation. For positive selection, approximately 108 cells were sorted for binding to 1 μM Bcl-xL for three rounds followed by 100 nM Bcl-xL for one final round. The Bcl-xL-binding yeast population was further sorted for specificity using competition screening, where the relevant pro-survival proteins were pre-mixed before they were added to the yeast cells. The first two competition sorts were carried out in the presence of a mixture of 10 nM Bcl-xL plus 500 nM each of untagged Bcl-w, Bcl-2 and Mcl-1; the top 1% of the expression positive population was collected. This was followed by two additional rounds of sorting at 10 nM Bcl-xL and 1 μM each of Bcl-w, Bcl-2, Mcl-1 and one round with 10 nM Bcl-xL mixed with 2 μM Bcl-2, 5 μM Bcl-w and 2 μM Mcl-1. Because this population still bound to 1 μM Bcl-w, a final competition sorting was carried out with 10 nM Bcl-xL mixed with 10 μM Bcl-w. In the later rounds of the competition screening, the stringency was increased by adjusting the sorting gate to collect ~0.2-0.5% of the expression positive population. Yeast populations isolated during screening were analyzed to assess binding to different populations of receptors.44
Peptide synthesis and purification
All peptides used for fluorescence polarization binding and BH3 profiling assays were synthesized by the Koch Institute Biopolymers and Proteomics Facility. All unlabeled peptides used in this study were synthesized with the N terminus acetylated and the C terminus amidated. Sequences of unlabeled peptides are shown in Table 1; the same peptides were used for fluorescence polarization and BH3 profiling studies. Fluorescently labeled Bim-BH3 was 18 residues long and was synthesized with fluorescein at the N terminus, with the C terminus amidated. Fluorescently labeled Hrk-BH3 was 23 residues long and had the sequence SAAQLTAARLKALGDELHQRTMW with fluorescein at the N terminus and an amidated C terminus. These peptides were purified using reverse phase HPLC with a C18 column and a linear water/acetonitrile gradient and masses were confirmed by MALDI mass spectrometry.
Fluorescence polarization binding assays
The fluorescence polarization binding assays were performed in 20 mM sodium phosphate, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) 0.001% triton X (v/v), and 5% DMSO (v/v), pH 7.8 at 25 °C. 18-residue Bim-BH3 was used as the fluorescently labeled peptide for all direct and competition binding assays, with the exception of competition binding of Puma and XXA1_G2gE to Mcl-1. For the latter two experiments fluorescently labeled Hrk-BH3 was used, as the relatively weaker affinity of fluorescent Hrk-BH3 for Mcl-1allowed the unlabeled peptides to compete for binding within a reasonable range of concentrations. For direct binding experiments, pro-survival proteins were serially diluted in 96-well plates and fluorescently labeled Bim-BH3 was added to a final concentration of 10 nM. The plates were incubated at room temperature and anisotropy was measured over a period of 4-24 hours to ensure that the reactions were equilibrated. Competition binding experiments were performed using previously published protocols44. Briefly, unlabeled peptides were serially diluted, followed by addition of fluorescently labeled Bim-BH3 (or Hrk-BH3) to a final concentration of 10 nM. The plates were then incubated for 15 minutes at room temperature before adding the relevant pro-survival protein. The final concentration of pro-survival proteins was 50 nM for all measurements with fluorescent Bim-BH3. The final concentration of Mcl-1 was 500 nM for the measurements with fluorescent Hrk-BH3, to obtain a significant change of anisotropy signal on competition. Anisotropy was monitored over 2-48 hours to ensure equilibration. As reported previously44, due to the extremely slow off-rates of 23-residue unlabeled Bim-BH3 from Mcl-1 and Bfl-1, we do not report any Ki value for these interactions. Binding curves were fit using Igor Pro 6.02 (Wavemetrics) as described previously24, 38. Binding constants reported are the average and standard deviation for at least three measurements. For strong interactions, ~ 0.1 nM, we inspected the fits to the data and simulated binding curves by fixing Ki values 10-fold higher or lower than the fitted Ki. If the fits did not appear better at 0.1 nM than at a lower Ki value (0.01 nM), then an upper bound of 0.1 nM was reported. In situations where there was a clear difference between the best fit to the data and the curves simulated for different Ki values, we reported the fitted Ki. Error bars represent standard deviations from mean for independent experiments and were taken into consideration when discrimination was made between two Ki values.
Cell Lines and Platelets
Su-DHL10 and MDA-MB-231 were cultured in RPMI 1640 (Life Technologies) supplemented with 10% FBS while MCL1-2640 and BCL2-3256 were cultured in RPMI 1640 with 10% FBS and 7.2 ml/L 2-mercaptoethanol (Sigma). OCI-Ly1 was cultured in DMEM supplemented with 10% FBS. Expired platelets were obtained as bags of platelet rich plasma with ACD-A as the anti-coagulant from the Kraft Family Blood Donor Center.
BH3 Profiling
BH3 profiling was performed in 384-well format as previously described using unlabeled peptides with sequences in Table 121. For cell lines, 2×104 cells were used per well in a final volume of 30 μL in triplicate wells for each peptide concentration. For platelets, 1 ml of platelet rich plasma was pelleted at 700 × g and re-suspended in 3 ml of DTEB, and 15 ml of platelets in DTEB was added to each well of the assay plate in the same manner as the cell lines. Raw fluorescence data were normalized to % depolarization using the equation, 1 – [(DSample-DFCCP)/(DDMSO-DFCCP)] using a minimum of three replicates for all samples, where Dsample, DFCCP and DDMSO are the fluorescence values in the presence of the sample, the mitochondrial uncoupler FCCP and DMSO respectively. Data sets for a specific cell line were fit with Igor Pro's Global Fit Package (Wavemetrics), using the equation D% = Dlower+(Dupper - Dlower)/(1+([BH3]/EC50)), where D% is the percent depolarization, Dlower is the lower baseline, Dupper is the upper baseline, [BH3] is the concentration of the peptide and EC50 is the peptide concentration at half maximal inhibition, with Dlower and Dupper as the linked coefficients between multiple datasets in the fit. Each EC50 measurement of the peptides by BH3 profiling was conducted for one preparation of each cell line per replicate. The average of at least three biological replicates was obtained for each cell line with 2-3 days of culture between measurements.
Supplementary Material
Highlights.
Anti-apoptotic protein Bcl-xL is an important therapeutic target
High-affinity and specific Bcl-xL binding peptides were designed
Engineered peptides discriminate Bcl-xL from related proteins Bcl-2 and Bcl-w
Mechanisms of binding specificity are determined
Cancer cell dependence on Bcl-xL can be detected with the designed peptides
Acknowledgments
We thank the Koch Institute Biopolymers and Proteomics Facilities for peptide synthesis and the Swanson Biotechnology Center Flow Cytometry Facility at MIT for cell sorting. We also thank members of the Keating laboratory for critical reading of the manuscript and helpful suggestions. This work was funded by NIGMS awards P50-GM068762 and GM110048 to AEK and award CA129974 to AL.
Abbreviations used
- FP
Fluorescence Polarization
- BH3
Bcl-2 homology 3
- FITC
Fluorescein isothiocyanate
- APC
allophycocyanin
- MOMP
mitochondrial outer membrane permeabilization
Biography
 Amy Keating
Amy Keating
 Anthony Letai
Anthony Letai
 Jeremy Ryan
Jeremy Ryan
 Sanjib Dutta
Sanjib Dutta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interests.
References
- 1.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 3.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–85. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 4.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–32. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. Embo J. 30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–42. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 8.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 10.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 11.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–70. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJ, Kulasegaram S, Moss RM, Parisot JP, Smith BJ, Street IP, Yang H, Huang DC, Watson KG. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013;9:390–7. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 13.Huang Z. The chemical biology of apoptosis. Exploring protein-protein interactions and the life and death of cells with small molecules. Chem Biol. 2002;9:1059–72. doi: 10.1016/s1074-5521(02)00247-8. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 16.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 17.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, Mitsiades CS, Matulonis UA, Drapkin R, Stone R, Deangelo DJ, McConkey DJ, Sallan SE, Silverman L, Hirsch MS, Carrasco DR, Letai A. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del Gaizo Moore V, Letai A. BH3 profiling - Measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2013;332(2):202–5. doi: 10.1016/j.canlet.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013;61:156–64. doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S, Long A, Link AJ. A comparison of two strategies for affinity maturation of a BH3 peptide toward pro-survival Bcl-2 proteins. ACS Synth Biol. 1:89–98. doi: 10.1021/sb200002m. [DOI] [PubMed] [Google Scholar]
- 23.Boersma MD, Sadowsky JD, Tomita YA, Gellman SH. Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein-protein recognition: application to the Bim BH3 domain. Protein Sci. 2008;17:1232–40. doi: 10.1110/ps.032896.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutta S, Gulla S, Chen TS, Fire E, Grant RA, Keating AE. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J Mol Biol. 2010;398:747–62. doi: 10.1016/j.jmb.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee EF, Czabotar PE, van Delft MF, Michalak EM, Boyle MJ, Willis SN, Puthalakath H, Bouillet P, Colman PM, Huang DC, Fairlie WD. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180:341–55. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBartolo J, Dutta S, Reich L, Keating AE. Predictive Bcl-2 family binding models rooted in experiment or structure. J Mol Biol. 2012;422:124–44. doi: 10.1016/j.jmb.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–7. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 28.Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem. 2005;280:4738–44. doi: 10.1074/jbc.M411434200. [DOI] [PubMed] [Google Scholar]
- 29.Czabotar PE, Lee EF, Thompson GV, Wardak AZ, Fairlie WD, Colman PM. Mutation to Bax beyond the BH3 domain disrupts interactions with pro-survival proteins and promotes apoptosis. J Biol Chem. 286:7123–31. doi: 10.1074/jbc.M110.161281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ku B, Liang C, Jung JU, Oh BH. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–41. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–34. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–6. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 33.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunelle J, Ryan J, Yecies D, Opferman J, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. JOURNAL OF CELL BIOLOGY. 2009;187:429–442. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 38.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–81. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 110:E986–95. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445–54. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee EF, Sadowsky JD, Smith BJ, Czabotar PE, Peterson-Kaufman KJ, Colman PM, Gellman SH, Fairlie WD. High-resolution structural characterization of a helical alpha/beta-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew Chem Int Ed Engl. 2009;48:4318–22. doi: 10.1002/anie.200805761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raymond CK, Pownder TA, Sexson SL. General method for plasmid construction using homologous recombination. Biotechniques. 1999;26:134–8. 140–1. doi: 10.2144/99261rr02. [DOI] [PubMed] [Google Scholar]
- 43.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 44.Dutta S, Chen TS, Keating AE. Peptide ligands for pro-survival protein Bfl-1 from computationally guided library screening. ACS Chem Biol. 2013;8:778–88. doi: 10.1021/cb300679a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.