

GCN2 exerts its proapoptotic function in cancer cell death by posttranslational mechanisms. Modulation of GCN2 expression can be used for molecular targeted cancer therapy and drug development. Na+,K+-ATPase ligands are the first identified small-molecule drugs that can trigger cancer cell death by modulating GCN2 signaling.
Abstract
General control nonderepressible kinase 2 (GCN2) is a promising target for cancer therapy. However, the role of GCN2 in cancer cell survival or death is elusive; further, small molecules targeting GCN2 signaling are not available. By using a GCN2 level-based drug screening assay, we found that GCN2 protein level critically determined the sensitivity of the cancer cells toward Na+,K+-ATPase ligand–induced apoptosis both in vitro and in vivo, and this effect was largely dependent on C/EBP homologous protein (CHOP) induction. Further analysis revealed that GCN2 is a short-lived protein. In A549 lung carcinoma cells, cellular β-arrestin1/2 associated with GCN2 and maintained the GCN2 protein level at a low level by recruiting the E3 ligase NEDD4L and facilitating consequent proteasomal degradation. However, Na+,K+-ATPase ligand treatment triggered the phosphorylation of GCN2 at threonine 899, which increased the GCN2 protein level by disrupting the formation of GCN2–β-arrestin–NEDD4L ternary complex. The enhanced GCN2 level, in turn, aggravated Na+,K+-ATPase ligand–induced cancer cell apoptosis. Our findings reveal that GCN2 can exert its proapoptotic function in cancer cell death by posttranslational mechanisms. Moreover, Na+,K+-ATPase ligands emerge as the first identified small-molecule drugs that can trigger cancer cell death by modulating GCN2 signaling.
INTRODUCTION
Pathological stress is a hallmark of cancer. Owing to poor vascularization, cancer cells normally stay in a stressful tumor microenvironment, including hypoxia, low nutrient availability, and immune infiltrates. These conditions, however, activate cellular stress response pathways to promote tumor survival and aggressiveness (Fulda et al., 2010). Targeting these stress pathways—for example, hypoxia inducible factor (HIF)–mediated signaling—in cancer cells has proven to be very helpful in the development of novel anticancer drugs.
To handle the external stress, a reduction in global protein synthesis is initiated by the phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) at serine 51 (Wek et al., 2006). This adaptive response provides cancer cells the opportunity to save energy and regulate signaling pathways to facilitate survival. Four eIF2α kinases have been identified: heme-regulated inhibitor (HRI), endoplasmic reticulum–resident kinase (PERK), RNA-dependent protein kinase (PKR), and general control nonderepressible kinase 2 (GCN2). All of these kinases share a conserved kinase domain but have different regulatory domains that enable them to be reactive under specific stress conditions (Wek et al., 2006).
GCN2 is an eIF2α protein kinase with high molecular weight (>200 kDa) and remains inactive in normal conditions. When confronted with amino acid deficiency, GCN2 is activated by uncharged tRNA, leading to the phosphorylation of eIF2α (Wek et al., 1995), which then increases the affinity with eIF2B, the guanine exchange factor (Pavitt et al., 1998, 2005). Activation of GCN2 is proposed to involve a transition from an inhibited to a catalytically active conformation, followed by autophosphorylation at threonine residues 882 and 887 (yeast GCN2) in the activation loop (Romano et al., 1998). This process of autophosphorylation needs GCN2 dimerization (Narasimhan et al., 2004). The extreme carboxy terminus of GCN2 can facilitate GCN2 association with ribosomes and binding of the HisRS-related domain to uncharged tRNA (Ramirez et al., 1991; Zhu and Wek, 1998; Dong et al., 2000). Phosphorylation of eIF2α by GCN2 translationally up-regulates GCN4 in yeast or active transcription factor 4 (ATF4) in mammals, which in turn stimulates the expression of many amino acid biosynthetic genes (Harding et al., 2000, 2003; Baird and Wek, 2012). The structure and function of yeast GCN2 have been intensively investigated (Padyana et al., 2005; He et al., 2014), but little information is available for mammalian GCN2.
The importance of GCN2 in cancer development has been increasingly appreciated; for example, a study clearly demonstrated that GCN2-ATF4 pathway is activated in cancer cells to promote tumor survival and aggressiveness by maintaining metabolic homeostasis (Ye et al., 2010). GCN2 activation in cancer cells also led to vascular endothelial growth factor expression (Wang et al., 2013). Further, GCN2 was highly expressed in human primary tumors, including breast, liver, and lung cancer (Ye et al., 2010). All of these facts suggest that GCN2 could be a novel target for cancer therapy. However, GCN2 is also involved in cell death (Gentz et al., 2013). These results indicate that the regulatory mechanisms for GCN2 in cancer cell are elusive and need to be addressed. Further, in contrast to the several small molecules found to have anticancer effects by targeting HIFs, small molecules that can target GCN2 signaling in cancer cells are lacking. To address these deficiencies, we initiated this study.
RESULTS
GCN2 expression level determines the sensitivity of cells to Na+,K+-ATPase ligand–induced apoptosis
By examining GCN2 protein level in a variety of cell lines, we found that GCN2 was differentially expressed in three colon cancerous cell lines, HCT116, HT29, and LoVo (Figure 1A). Using these cell lines, we evaluated the apoptotic effects of hundreds of chemicals that have cytotoxic effects. Greater than 95% of the test chemicals had no influence or produced comparable apoptosis-inducing effects on these three cell lines. However, when these cells were challenged with Na+,K+-ATPase ligands, including ouabain, oleandrin, and foliandrin, cellular apoptosis was well correlated with the GCN2 protein level. Ouabain, oleandrin, and foliandrin triggered significant apoptotic response in HCT116 cells but failed to induce apoptosis in LoVo cells (Figure 1B). Apoptosis of HT29 cell induced by ouabain, oleandrin, and foliandrin was exactly in between that observed in HCT116 and LoVo cells. We also observed that a distinct poly(ADP-ribose) polymerase (PARP) cleavage occurred in ouabain-treated HCT116 cells but not in LoVo cells (Figure 1C). PARP cleavage in HT29 cells was evident but was obviously less potent than that in HCT116 cells. The specificity of the effect of GCN2 was indicated by the significant attenuation of ouabain-induced apoptosis in HCT116 cells after the silencing of GCN2 expression (Figure 1, D-1 and D-2). In contrast, when ectopic hemagglutinin (HA)-GCN2 was expressed in LoVo cells (Figure 1E-1), the lack of sensitivity of LoVo cells toward ouabain treatment was significantly reversed (Figure 1E-2). Further, Na+,K+-ATPase ligand–induced apoptosis was largely attenuated in GCN2 but not in PERK- or PKR-silenced cells (Figure 1F and Supplemental Figure S1).
FIGURE 1:

Involvement of GCN2 in Na+,K+-ATPase ligand–induced cell apoptosis. (A) GCN2 protein levels in HCT116, HT29, and LoVo under normal growth conditions. (B) Cell apoptosis and (C) PARP cleavage in HCT116, HT29, and LoVo cells after treatment with ouabain, oleandrin, or foliandrin at 200 nM for 24 h. Data are expressed as mean ± SD, n = 3. HCT116 cells were transfected with GCN2 siRNA or control siRNA (D), and LoVo cells were transfected with HA-GCN2 or empty vector (E), and cells were further treated in the absence or presence of ouabain at 200 nM for additional 24 h. GCN2 level was detected by immunoblot analysis. Cell apoptosis was analyzed by flow cytometry. (F) The control, GCN2, PERK, and PKR siRNA–transfected A549 cells were treated in the absence or presence of ouabain at 200 nM for 24 h. Cell apoptosis was examined by the flow cytometry. Data represent mean ± SD, n = 3; ns, not significant, ***p < 0.001. (G) Nude mice were injected with 3 × 106 HCT116 cells or 5 × 106 LoVo cells per mouse to produce the tumor model. At 25 d after drug treatment, tumor growth curves were plotted. (H) Tumor tissues were removed from animals and homogenized for the assay of caspase 3 cleavage activity by flow cytometry. FITC-DEVD-FMK was used as a specific fluorescent substrate for caspase 3.
To confirm these results in vivo, we inoculated HCT116 and LoVo cells into nude mice to produce tumor-bearing models. Of interest, ouabain treatment significantly slowed the tumor growth of HCT116 but not LoVo xenografts (Figure 1G). After experiments, tumor tissues were removed and homogenized; the caspase 3 cleavage activities of tumor tissues were measured by flow cytometry by using fluorescein isothiocyanate (FITC)–conjugated caspase 3 substrate. The results demonstrated that ouabain administration led to significant caspase 3 activation in HCT116 cells but not in LoVo cells isolated from the tumor tissue (Figure 1H).
C/EBP homologous protein is required for the proapoptotic function of GCN2
To understand why GCN2 is able to enhance apoptosis, we profiled gene expression in cells after ouabain treatment by microarray analysis. The data demonstrated that ouabain significantly increased mRNA expression of C/EBP homologous protein (CHOP) in A549 cells (Supplemental Table S1). This result was also verified by reverse transcription (RT) PCR analysis (Figure 2A). Of note, ouabain-induced CHOP expression was critically dependent on GCN2 (Figure 2, B and C), and apoptosis induction by ouabain was also largely attenuated in A549 cells after the silencing of CHOP expression (Figure 2D) or in CHOP−/− MEF cells (Figure 2E). A previous study suggested that death receptor 5 (DR5) is able to induce Fas-associated death domain–dependent cell apoptosis (Chaudhary et al., 1997). However, surface DR5 expression in A549 cells is relatively low, and therefore A549 cells are resistant to apoptosis induced by tumor necrosis factor–related apoptosis-inducing ligand (TRAIL), a promising antineoplastic agent that can selectively kill cancerous cells after ligation with surface DR5. Up-regulation of DR5 can increase apoptotic sensitivity of cancer cells toward TRAIL (Liu et al., 2004), and several monoclonal antibodies against DR5 are in clinical trials (Wiezorek et al., 2010). Of interest, Na+,K+-ATPase ligands were able to significantly increase the DR5 protein expression in non–small-cell lung carcinoma cells, including A549, but the underlying mechanism is largely unresolved (Frese et al., 2006). By using flow cytometry analysis to measure the abundance of membrane-bound DR5, we found here that the ouabain-induced up-regulation of DR5 is indeed largely dependent on the presence of GCN2 and CHOP (Figure 2F).
FIGURE 2:

Involvement of CHOP in Na+,K+-ATPase ligand–induced cell apoptosis. (A) A549 cells were treated with ouabain or dimethyl sulfoxide at 400 nM for 12 h. CHOP mRNA was examined by RT-PCR analysis. (B) Control or GCN2-silenced A549 cells were treated with ouabain at 400 nM for 12 h, and (C) GCN2 WT and GCN2−/− MEF cells were treated with ouabain at 10 μM for 12 h, and CHOP expression was examined by immunoblot analysis. (D) A549 cells after silencing of CHOP expression were treated with ouabain at 400 nM for 12 h. Cell apoptosis in control siRNA transfectant was arbitrarily set at 1.0. (E) WT or CHOP−/− MEF cells were treated with ouabain at 10 μM for 12 h; cell apoptosis in CHOP WT MEF cells was arbitrarily set at 1.0. (F) Control, GCN2, or CHOP siRNA–transfected cells were treated with ouabain at 400 nM for 8 h; after treatment, membrane-bound DR5 expression was examined by flow cytometry. Similar results from three independent experiments are shown.
β-Arrestin–mediated GCN2 ubiquitination and degradation
To further characterize the importance of GCN2 in apoptosis, we unexpectedly observed that ouabain dose-dependently increased GCN2 protein levels (Figure 3A). Because eIF2α is the substrate of GCN2, ouabain treatment also led to concentration-dependent phosphorylation of eIF2α, but had no effect on the total eIF2α expression. Further analysis revealed that GCN2 is a protein with a short half-life, because both exogenous and endogenous GCN2 levels were significantly up-regulated in proteasome inhibitor MG132-treated cells but not in cysteine protease inhibitor E64-treated cells, suggesting that ubiquitin pathways probably mediate rapid GCN2 degradation (Figure 3, B-1 and B-2). Cycloheximide (CHX) is commonly used to halt protein synthesis. In this study, progressive degradation of GCN2 in cells after addition of CHX was observed (Figure 3C). Both ouabain and MG132 potently suppressed GCN2 degradation, suggesting that ouabain may influence ubiquitin-mediated GCN2 degradation.
FIGURE 3:
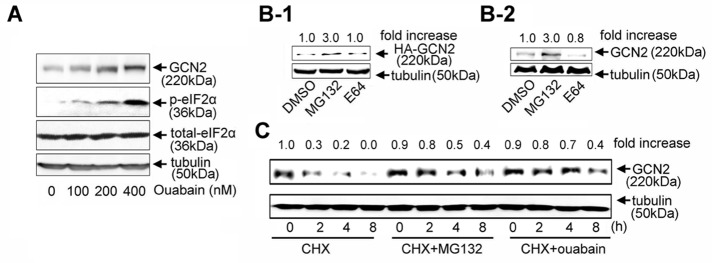
GCN2 ubiquitination and degradation. (A) A549 cells were treated with ouabain for 8 h at 0, 100, 200, and 400 nM. (B) A549 cells transfected (B-1) or nontransfected with HA-GCN2 (B-2) were treated with MG132 (20 μM), E64 (5 μM), or dimethyl sulfoxide for 12 h and the lysates subjected to immunoblot analysis by using antibodies against GCN2 and tubulin. (C) A549 cells were treated with CHX (2 μM) plus vehicle, CHX (2 μM) plus MG132 (20 μM), and CHX (2 μM) plus ouabain (100 nM) for the indicated times and GCN2 protein levels examined by immunoblot analysis. Similar results from three independent experiments are shown.
To further confirm that GCN2 is a ubiquitinated protein, we used anti-ubiquitin immunoglobulin Gs (IgGs) and protein A/G–conjugated agarose beads in immunoprecipitation experiments to pull down cellular ubiquitinated proteins. The results, as shown in Figure 4A, demonstrated that cellular GCN2 ubiquitination was also detected in cells after MG132 treatment. This GCN2 ubiquitination was greatly attenuated when cellular GCN2 expression was reduced by small interfering RNA (siRNA)–mediated gene silencing (Supplemental Figure S2). Ubiquitin (Ub)-mediated protein degradation needs assembly of E3 ubiquitin ligase complex (Lu and Hunter, 2009). To identify the components of E3 ligase complex for GCN2, we established A549 cell lines stably overexpressing green fluorescent protein (GFP)–tagged GCN2 and used this system to screen adaptor proteins that can potentially suppress GCN2 level by monitoring GFP fluorescence in flow cytometry. As a result, we found that β-arrestin1/2 was critically involved in GCN2 degradation, because overexpression of β-arrestin1/2 dose-dependently suppressed GCN2 protein level (Figure 4B). Moreover, this effect was blocked by MG132, suggesting that β-arrestin1/2 may influence GCN2 ubiquitination. Of note, as a commonly used proteasome inhibitor, MG132, can also trigger eIF2α phosphorylation and inhibit general protein synthesis (Mazroui et al., 2007), which may explain why MG132 failed to completely reverse β-arrestin1/2–mediated GCN2 down-regulation as occurred in Figure 4B. In addition, in the GCN2 overexpression experiment, we also found that β-arrestin1/2 potently suppressed cellular GCN2 level (Figure 4C). In contrast, siRNA-mediated silencing of β-arrestin1/2 in A549 cells led to a significant increase of cellular GCN2 protein level (Figure 4D) and a corresponding decrease in cellular GCN2 ubiquitination (Figure 4E).
FIGURE 4:

Effect of β-arrestin1/2 on GCN2 degradation. (A) Cells were transfected with HA-Ub or empty vector and further treated or untreated with MG132 (50 μM) for an additional 5 h before being harvested. Lysates were immunoprecipitated using anti-HA IgG, followed by immunoblot analysis using the purified anti-GCN2 IgG. (B) A549 cells were cotransfected with HA-GCN2 plus FLAG-β-arrestin1, FLAG-β-arrestin2, or empty vector. After 36 h, cells were treated with MG132 (20 μM) or vehicle for 8 h. Lysates were detected by immunoblot analysis using the indicated antibodies. (C) A549 cells were transfected with FLAG-β-arrestin1, FLAG-β-arrestin2, or empty vector. After 36 h, cells were harvested, and lysates were subjected to Western blot analysis using indicated antibodies. (D) A549 cells were transfected with β-arrestin1 siRNA, β-arrestin2 siRNA, or control siRNA. After 36 h, the lysates were detected by using the indicated antibodies. (E) Cells were cotransfected with HA-Ub plus siRNAs or control siRNA. After 30 h, cells were treated with MG132 (50 μM) for 5 h before being harvested. Lysates were immunoprecipitated using anti-HA IgG, followed by immunoblot analysis using anti-GCN2 and other indicated IgGs. (F) A549 cells were cotransfected with vectors as indicated. After 36 h, the cells were treated with ouabain at 200 nM for 12 h. The cell lysates were examined by immunoblot analysis with the indicated antibodies. (G) A549 cells were treated with MG132 (20 μM) plus vehicle or MG132 (20 μM) plus ouabain (100 nM) for 5 h, and the lysates were immunoprecipitated with anti-GCN2 IgG or normal rabbit IgG as a negative control. The immunpellets were detected by immunoblot analysis with anti-Ub and anti-GCN2 IgGs. Each experiment was performed at least in triplicate.
In examining whether ouabain influences GCN2 ubiquitination, we found that β-arrestin1/2–mediated suppression of GCN2 level was attenuated by ouabain treatment (Figure 4F). Moreover, ouabain treatment significantly suppressed cellular GCN2 ubiquitination (Figure 4G, lane 2).
β-Arrestins interact with GCN2
As scaffold proteins, β-arrestins regulate a variety of signaling events (Wisler et al., 2007). We found that β-arrestin1/2 colocalized with GCN2 in the cytoplasm (Figure 5A). Analysis by immunoprecipitation indicated strong association between β-arrestin1/2 and GCN2 (Figure 5, B-1 and B-2). However, this association was detected in MG132- but not ouabain-treated cells, indicating that the underlying regulatory mechanisms for the effects of MG132 and ouabain on GCN2 level are different, although both of them were able to delay GCN2 degradation (Figure 3C) and suppress β-arrestin1/2–mediated GCN2 down-regulation (Figure 4, B and F). The physical association between β-arrestin1/2 and GCN2 was not an artifact of protein overexpression, because cellular β-arrestin was also found to readily associate with endogenous GCN2 (Figure 5C). Glutathione S-transferase (GST) pull-down analysis was performed to examine the association between β-arrestin1/2 and GCN2. The outcome of this analysis indicated that although GST alone was unable to pull down GCN2, GST-fused β-arrestin1/2 can immunoprecipitate GCN2, and the association between β-arrestin2 with GCN2 was significantly stronger than that of β-arrestin1, as judged by the band intensity of yellow fluorescent protein (YFP)–tagged GCN2 (Figure 5D). To map the interacting domain between GCN2 and β-arrestin1/2, we constructed truncated GCN2 mutants (Figure 5E-1). Immunoprecipitation analysis demonstrated that the kinase domain of GCN2 is required for interaction with β-arrestin1/2 (Figure 5, F-1 and F-2). Truncated β-arrestin1/2 mutants were also constructed (Figure 5E-2). The results demonstrated that the C- or N-terminus is sufficient for binding, but both are required for strong binding with GCN2 (Figure 5, G-1 and G-2). Consistently, the protein level of GCN2 mutants with a kinase domain—for example, HA-PK2 and HA-GCN2-N—was greatly reduced by β-arrestin1/2. However, GCN2 mutants without a kinase domain—for example, HA-RWD and HA-PK1—were not suppressed by β-arrestin1/2 (Figure 5H).
FIGURE 5:

The protein interaction between β-arrestin1/2 and GCN2. (A) A549 cells were cotransfected with YFP-tagged GCN2 and FLAG-tagged β-arrestin1/2. Immunofluorescence analysis was performed as described in Materials and Methods. Scale bar, 20 μm. (B) Cells were cotransfected with expression vectors harboring HA-GCN2 plus FLAG-β-arrestin1 (B-1) or FLAG-β-arrestin2 for 36 h (B-2) and untreated or treated with MG132 (20 μM) or ouabain (200 nM) for 8 h before being harvested. The cell lysates were immunoprecipitated with anti-HA IgG, and the immune pellets were detected by immunoblot analysis with anti-FLAG IgGs. (C) After treatment with 20 μM MG132, A549 cell lysates were used for immunoprecipitation with an anti-GCN2 IgG or normal rabbit IgG and analyzed by immunoblot analysis with anti–β-arrestin1/2 IgGs. (D) YFP-GCN2–transfected HEK293 cells lysates were precipitated with glutathione beads and incubated with purified GST, GST-β-arrestin1, or GST-β-arrestin2. Precipitates were analyzed by immunoblot analysis with an anti-YFP IgG. (E) Deletion mutants for GCN2 (E-1) and β-arrestin1/2 (E-2). (F) HEK293 cells were cotransfected with GCN2 deletion mutants plus FLAG-β-arrestin1 (F-1) or FLAG-β-arrestin2 (F-2). The cells were treated with MG132 (20 μM) for 5 h before being harvested, and lysates were immunoprecipitated with an anti-HA IgG, and the immune pellets were detected by immunoblot analysis using anti-FLAG IgG. (G) A549 cells were cotransfected with HA-GCN2 plus β-arrestin1 deletion mutants (G-1) or β-arrestin2 deletion mutants (G-2). The cells were treated with MG132 (20 μM) for 5 h before being harvested, and lysates were immunoprecipitated with anti-FLAG IgG; the immune pellets were detected by immunoblot analysis with anti-HA IgG. (H) HEK293 cells were cotransfected with HA-RWD, HA-PK1, HA-PK2, or HA-GCN2-N plus FLAG tagged β-arrestin1/2. The lysates were subjected to immunoblot analysis by using the indicated antibodies. A representative Western blot for each treatment from three independent experiments is shown.
Neural precursor cell–expressed, developmentally down-regulated 4–like is the specific E3 ligase for GCN2 ubiquitination and degradation
β-Arrestins are unable to directly transfer ubiquitin to the target protein. Identification of E3 ligase specifically for GCN2 degradation is therefore critical. By using the aforementioned GFP screening method, we found that neural precursor cell–expressed, developmentally down-regulated 4–like (NEDD4L) was critically involved in GCN2 ubiquitination, because NEDD4L dose-dependently suppressed GCN2 protein level (Figure 6A). This result delivered other important messages, that is, NEDD4L is an easily degradable E3 ligase and thus has a relatively low basal level, because NEDD4L expression level was greatly up-regulated in the presence of MG132 treatment. NEDD4L-mediated GCN2 degradation was not an artifact of protein overexpression, because the silencing of NEDD4L expression in cells led to enhanced cellular GCN2 level (Figure 6B) and corresponding decreased GCN2 ubiquitination (Figure 4E). In immunoprecipitation experiments, strong interaction between β-arrestin1/2 and NEDD4L was detected (Figure 6, C-1 and C-2). Furthermore, GST pull-down experiments revealed that purified GST–β-arrestin1/2 can immunoprecipitate both GCN2 and NEDD4L from the cell lysate, suggesting the existence of a complex consisting of GCN2, β-arrestin1/2, and NEDD4L (Figure 6D). The association between NEDD4L and GCN2 was rather weak when β-arrestin1/2 was absent; however, in the presence of β-arrestin1/2, the interaction between NEDD4L and GCN2 was significantly strengthened, as judged by the band intensity of YFP-tagged GCN2 (Figure 6E). Similarly, although NEDD4L alone can increase GCN2 ubiquitination, overexpression of β-arrestin1/2 significantly strengthened this effect, as judged by the intensity of the ubiquitinated form of GCN2 (Figure 6F, lanes 4 and 5). Therefore β-arrestins promote the assembly of GCN2–β-arrestin–NEDD4L complex by recruiting NEDD4L E3 ligase to GCN2, thereby facilitating ubiquitin-mediated GCN2 degradation.
FIGURE 6:

Involvement of NEDD4L in GCN2 ubiquitination and degradation. (A) A549 cells were transfected with HA-NEDD4L or empty vector for 36 h and further treated with MG132 (20 μM) or vehicle for 8 h. The lysates were detected by immunoblot analysis using the indicated antibodies. (B) A549 cells were transfected with NEDD4L or control siRNAs for 36 h, and the lysates were detected by immunoblot analysis using anti-GCN2, anti-NEDD4L, and anti-tubulin IgGs. (C) A549 cells were cotransfected with HA-NEDD4L plus FLAG-β-arrestin1 (C-1) or FLAG-β-arrestin2 (C-2) for 36 h and further treated with MG132 (20 μM) for 5 h before being harvested. The cell lysates were immunoprecipitated with anti-HA IgG or normal mouse IgG as a negative control, and the immune pellets were detected by immunoblot analysis using anti-FLAG IgG. (D) A549 cells lysate was precipitated with glutathione beads incubated with purified GST, GST-β-arrestin1, or GST-β-arrestin2, and precipitates were analyzed by immunoblot analysis using anti-GCN2, anti-NEDD4L, and anti-GST IgGs. (E) A549 cells were cotransfected with YFP-GCN2, HA-NEDD4L plus FLAG-β-arrestin1, or FLAG-β-arrestin2 for 36 h and further treated with MG132 (20 μM) for 5 h before being harvested. The cell lysates were immunoprecipitated with anti-HA IgG, and the immunpellets were detected by immunoblot analysis using anti-YFP, anti-FLAG, and anti-HA IgGs. (F) A549 cells were treated as described in E, and the lysates were immunoprecipitated using anti-YFP IgG, followed by immunoblot analysis using anti-Ub IgG and other indicated IgGs. A representative immunoblot analysis for each treatment from three independent experiments is shown.
Disruption of GCN2–β-arrestin–NEDD4L complex by Na+,K+-ATPase ligand
Consistent with the foregoing finding that ouabain treatment can prevent β-arrestin1/2–mediated suppression of GCN2 level, we found that the interaction between β-arrestin1/2 and GCN2 was greatly reduced in ouabain-treated cells, either by using GCN2 as a bait to immunoprecipitate β-arrestin1/2 (Figure 7A), or vice versa, by using β-arrestin1/2 as a bait to immunoprecipitate GCN2 (Figure 7B). Of note, MG132 treatment was used here because it was required for the occurrence of interaction between β-arrestin1/2 and GCN2 (Figure 5, B-1 and B-2); however, note that both MG132 and ouabain were toxic to cells, especially when they were used at high concentrations or for a relatively long period of time. Treatment of cells with ouabain plus MG132 likely produce toxic or other nonspecific effects that can mask the real effect of ouabain on the interaction between β-arrestin1/2 and GCN2. Therefore, to assess objectively the effect of ouabain, we treated cells with MG132 and ouabain at low concentrations and for a short period of time.
FIGURE 7:

Effect of ouabain on GCN2–β-arrestin1/2–NEDD4L degradation complex. (A) A549 cells were cotransfected with HA-GCN2 plus FLAG-β-arrestin1 or FLAG-β-arrestin2 and further treated with ouabain (100 nM, 6 h) and MG132 (20 μM, 5 h) as indicated. The cell lysates were immunoprecipitated with an anti-HA IgG, and the immunpellets were detected by immunoblot analysis with anti-FLAG and other indicated IgGs. (B) The reverse immunoprecipitation proceeded with an anti-FLAG IgG, and the immunpellets were detected by immunoblot analysis with anti-HA and other indicated IgGs. (C) A549 cells were treated with MG132 (20 μM, 5 h) plus vehicle or MG132 (20 μM, 5h) plus ouabain (100 nM, 6 h), and the lysates were immunoprecipitated with anti-GCN2 or normal rabbit IgG. The immune pellets were detected by immunoblot analysis with the indicated antibodies.
The suppressive effect of ouabain on the interaction between β-arrestin1/2 and GCN2 was not unique to cells in which GCN2 and β-arrestin1/2 were overexpressed. In A549 cells without plasmid transfection, ouabain was also able to disrupt the assembly of endogenous GCN2–β-arrestin–NEDD4L complex, because when ouabain was present in cells, the immunoprecipitated NEDD4L and β-arrestin1/2 by anti-GCN2 IgG were significantly less than in the control cells (Figure 7C).
Phosphorylation of GCN2 suppresses ubiquitin-mediated GCN2 degradation
To understand why ouabain is able to disrupt the GCN2–β-arrestin–NEDD4L degradation complex, we noticed that immunoprecipitated GCN2 in ouabain-treated cells migrated more slowly than in the control cells, which raises the possibility that ouabain might regulate the posttranslational modification of GCN2 (Figure 7C). Previous study demonstrated that GCN2 activation needs the phosphorylation of threonine 899 (Romano et al., 1998). Phosphorylation and ubiquitination are two closely linked events in the process of protein degradation. We therefore set out to determine whether phosphorylation of GCN2 at threonine 899 is also relevant to GCN2 degradation. By using an antibody specifically recognizing GCN2 phosphorylation at Thr-899 (Harding et al., 2000), we unexpectedly observed a significant increase in GCN2 phosphorylation in the immunoprecipitate from ouabain-treated cells as compared with that of the control cells (Figure 7C).
To examine the biological significance of this, we substituted Thr-899 of GCN2 by alanine (T to A) or asparagine (T to D) to mimic the nonphosphorylated or phosphorylated form of GCN2, respectively. GST pull-down analysis demonstrated that GCN2 T899A associated with more β-arrestin1/2 than did GCN2 wild type (WT); however, the association between GCN2 T899D and β-arrestin1/2 was much weaker than that of GCN2 WT and GCN2 T899A (Figure 8, A-1 and A-2). Moreover, β-arrestin1/2–mediated GCN2 (T899D) degradation was significantly less potent than in GCN2 (WT) or GCN2 (T899A; Figure 8B), and, correspondingly, GCN2 (T899A) degradation by β-arrestin1/2 was stronger than that of GCN2 WT. In addition, the ubiquitinated form of GCN2 (T899D) was weaker than that of GCN2 (T899A; Figure 8C). In cells overexpressing HA-GCN2, GCN2 ubiquitination was significantly suppressed by ouabain; however, ouabain failed to influence GCN2 (T899A) ubiquitination, as judged by the intensity of the ubiquitinated form of GCN2 (Supplemental Figure S3). Collectively, these data suggest that phosphorylation of GCN2 at Thr-899 triggered by ouabain impairs its ability to associate with β-arrestins, thereby rendering this protein less susceptible to ubiquitin-mediated degradation.
FIGURE 8:

Effect of GCN2 phosphorylation on ubiquitin-mediated GCN2 degradation. (A) HEK293 cells were transfected with YFP-tagged GCN2 (WT), GCN2 (T899A), GCN2 (T899D), or empty vector. Cell lysates were precipitated with glutathione beads incubated with purified GST–-β-arrestin1 (A-1) or GST–β-arrestin2 (A-2). Precipitates were analyzed by immunoblot analysis with the indicated antibodies. (B) A549 cells were cotransfected with YFP-tagged GCN2 (WT), GCN2 (T899A), or GCN2 (T899D) plus FLAG-β-arrestin1/2. After 36 h, lysates were examined by immunoblot analysis with the indicated IgGs. (C) A549 cells were cotransfected with HA-Ub and YFP-tagged GCN2 (WT), GCN2 (T899A), or GCN2 (T899D) and further treated with MG132 (50 μM) for 5 h before being harvested. Cells lysates were immunoprecipitated with anti-HA IgG, and the immune pellets were detected by immunoblot analysis with the indicated antibodies. (D) A549 and HeLa cells were treated in the presence or absence of ouabain at 100 nM for 12 h. Phosphorylation of GCN2 T899, phosphorylation of eIF2α, and the total eIF2α were assessed by immunoblot analysis. (E) A549 cells were treated with 400 nM ouabain for 0, 4, 8, and 12 h, respectively. Cell apoptosis was detected by flow cytometry. Data are expressed as the fold of cell apoptosis in ouabain-treated cells relative to control cells (E-1).The lysates were detected by immunoblot analysis with the indicated antibodies. For the quantitation of the band densities of immunoblot, values were calculated as the ratio of p-GCN2 (T899) or GCN2 to tubulin and normalized to the values obtained in control cells (1.0; E-2). A representative immunoblot analysis for each treatment from three independent experiments is shown. (F) Regulatory roles of GCN2 in cells sensing the mild or severe stress of Na+,K+-ATPase impairment by its ligands.
Phosphorylation of GCN2 at T899 triggered by ouabain is not unique to A549 cells. Western blot analysis revealed that ouabain treatment can also lead to a dramatic increase in GCN2 phosphorylation in HeLa cells, as shown in Figure 8D. Further, GCN2 phosphorylation in A549 and HeLa cells correlated with enhanced GCN2 protein level and eIF2α phosphorylation, suggesting that GCN2 phosphorylation likely couples GCN2 activation and degradation. To confirm this, we compared cell apoptosis, GCN2 phosphorylation, and GCN2 levels in cells after ouabain treatment (Figure 8, E-1 and E-2). The results demonstrated that GCN2 phosphorylation was also well correlated with apoptosis due to ouabain. Of interest, cellular GCN2 level rapidly increased in <4 h when apoptosis was not apparent but gradually decreased from 4 to 12 h when the apoptosis program was initiated.
DISCUSSION
The Na+,K+-ATPase ligands are a family of compounds derived from the foxglove plant. They are potent Na+,K+-ATPase inhibitors that can bind with the extracellular portion of Na+,K+-ATPase α subunits and compete for K+-binding site. Na+,K+-ATPase ligands are not exclusively plant derived; endogenous Na+,K+-ATPase ligands such as endogenous ouabain have are also been isolated and identified from human plasma (Hamlyn et al., 1991; Mathews et al., 1991). Amazingly, the chemical structure of plant-derived ouabain is identical to the endogenous ouabain secreted by human adrenal gland (Hamlyn et al., 1991). Given these facts, identification of the biological activities of ouabain, as well as of other Na+,K+-ATPase ligands, is meaningful.
Recently, increasing evidence has critically implicated Na+,K+-ATPase in cancer progression (Rajasekaran et al., 2001; Yin et al., 2007; Lefranc and Kiss, 2008). Many Na+,K+-ATPase ligands have potent antitumor effects in clinical or experimental settings (Lefranc and Kiss, 2008; Newman et al., 2008; Prassas and Diamandis, 2008). Ouabain is a case in point. Previous studies suggested that ouabain has a death-inducing effect on a variety of cancer cell lines (Kulikov et al., 2007; Winnicka et al., 2008; Ozdemir et al., 2012; Tailler et al., 2012; Pezzani et al., 2014). In addition to directly triggering apoptosis, Na+,K+-ATPase ligands also sensitize cancerous cells to apoptotic insults, including TRAIL (Frese et al., 2006), CD95 (McCarthy and Cotter, 1997; Bortner and Cidlowski, 1999; Nobel et al., 2000; Bortner et al., 2001), and C2-ceramide (Xiao et al., 2002). All of these findings indicate that Na+,K+-ATPase ligands are promising anticancer drugs. However, although most Na+,K+-ATPase ligands have strong apoptosis-inducing effects on cancer cells in vitro, many of them lose their effects in vivo (Stenkvist, 1999; Biggar et al., 2011; Mijatovic and Kiss, 2013), indicating that the sensitivity of tumor cells toward Na+,K+-ATPase ligands largely depends on the cellular environment. We found here that GCN2 expression level specifically regulates cancer cell sensitivity toward Na+,K+-ATPase ligands. In addition, Na+,K+-ATPase ligands increase GCN2 level by suppressing its degradation. These results indicate that GCN2 level, as well as GCN2 signaling, is critical for Na+,K+-ATPase ligands to have an effect. Of interest, Na+,K+-ATPase ligands not only modulate GCN2 signaling, but also target HIF pathways, which sense the stress of oxygen deficiency in cancerous cells (Zhang et al., 2008). Taking the results together, it is reasonable to conclude that Na+,K+-ATPase ligands can preferentially modulate survival-promoting signaling in cancer cells caused by the stress of nutrient and oxygen deficiency.
The regulatory mechanism for GCN2 expression at the posttranslational level is largely unknown. We found that GCN2 has a short half-life. The precise regulation of GCN2 at a low basal level by the GCN2–β-arrestin1/2–NEDD4L degradation complex has at least three purposes: 1) GCN2 protein has a tendency to promote apoptosis, 2) this regulatory process meets the requirement for rapid GCN2 induction after Na+,K+-ATPase ligand treatment because suppression of GCN2 degradation is obviously less time and energy consuming than that by de novo protein synthesis, and 3) β-arrestins are critically involved in cancer cell growth and chemotaxis (Hu et al., 2013), and GCN2 degradation is likely a strategy adopted by β-arrestins to promote cancer cell survival and growth.
As a mammalian E3 ubiquitin ligase, NEDD4L shares common domain architecture with its yeast homologue, Rsp5. The WW domains of NEDD4L bind to a PPxY or PY motif, where x represents any amino acid (Staub et al., 1996). In general, the WW domain is composed of 38–40 amino acids characterized by two highly conserved tryptophans and folded as a three-strand β-sheet, which is known to mediate specific interactions by binding to proline-rich sequences and may be repeated up to four times in some proteins (Sudol et al., 1995). The WW domain has been found in various important signaling and structural proteins (Chen and Sudol, 1995), and epithelial amiloride-sensitive sodium channel (ENaC) and epidermal growth factor receptor ErbB4 are well characterized (Staub et al., 1996; Zeng et al., 2009). Consistently, GCN2 contains the PY motif in its N-terminus. However, unlike ENaC and ErbB4, which have multiple PY motifs, only a PPTY motif is present in GCN2, which offers a possible explanation for why the association between NEDD4L and GCN2 needs the assistance of β-arrestins.
Under the stress of nutrient deficiency, the uncharged tRNAs bind the C-terminal of GCN2, leading to GCN2 activation, subsequent eIF2α phosphorylation, and global reduction in protein translation. Basically, GCN2 phosphorylation exerts a protective role for cells to recover from the stressful conditions. However, we found here that phosphorylation of GCN2 at Thr-899 has another important function—regulating GCN2 level by disrupting the GCN2–β-arrestin1/2–NEDD4L degradation complex. In addition, GCN2 phosphorylation correlated with ouabain-induced apoptosis. Overexpression of GCN2(T899D) in cells led to a more pronounced apoptosis-inducing effect than that of wild-type GCN2 (Supplemental Figure S4). All of these facts suggest that GCN2 phosphorylation at T899 integrates its activation and degradation and has dual roles in regulating cell fate. We found here that ouabain treatment at 400 nM induced GCN2-dependent CHOP up-regulation and CHOP-dependent apoptosis. However, we also noticed that ouabain used at 100 nM failed to trigger CHOP up-regulation and apoptosis but still could induce GCN2 phosphorylation (unpublished data). Therefore it seems that the concrete role of GCN2 phosphorylation in cell survival and death largely depends on the intensity of stress. If the cells are challenged with ouabain at a relatively low concentration, GCN2 phosphorylation mainly protects cells; however, if the stress of ouabain treatment is severe, GCN2 phosphorylation switches its function from prosurvival to death by CHOP up-regulation. More experiments need to be done to confirm this and examine whether this mechanism is also applicable to stressful conditions of nutrient deficiency. Of note, although GCN2 phosphorylation at T899 significantly impaired the formation of GCN2 degradation complex, a small fraction of GCN2 (T899D) was still degraded in the presence of β-arrestin overexpression (Figure 8B), suggesting that GCN2 phosphorylation at T899 is probably necessary but not sufficient for suppressing GCN2 degradation. GCN2 is a protein kinase with a relatively large molecular weight. Bioinformatic analysis reveals that many potential phosphorylation sites exist in the amino acid sequence of GCN2. Whether the phosphorylation of GCN2 at T899 is primed for another phosphorylation event deserves further investigation.
We noticed in this study that in cells after treatment with ouabain at 400 nM for 12 h, GCN2 protein level was not increased as expected but instead was decreased (Figure 8E-2). Of note, many cells underwent apoptosis at this stage (Figure 8E-1). Meanwhile, the decrease in GCN2 level in 400 nM ouabain–treated cells could be blocked by the pan-caspase inhibitor z-VAD.fmk (Y.W., unpublished data). Therefore ubiquitination is probably not the sole mechanism for GCN2 degradation. When cell apoptosis occurs, the cellular GCN2 is likely subject to caspase-mediated cleavage and degradation. If this is the case, the regulatory mechanism for GCN2 in sensing stress could be rather flexible and complex. When the intensity of stressful stimuli is mild, a small fraction of GCN2 is phosphorylated as a defense mechanism to protect cells. As the stress intensity increases, more GCN2 proteins are phosphorylated, which leads to an enhanced GCN2 level. This dramatic increase in GCN2 level can help cells to maximally adapt to stress. However, if the stress becomes intolerable, GCN2 phosphorylation promotes cell apoptosis by increasing CHOP expression. In the course of apoptosis, the protective role of GCN2 is no longer needed, and so it will be degraded by caspases to facilitate rapid apoptotic clearance (Figure 8F).
How GCN2 is involved in Na+,K+-ATPase ligand–induced cancer cell death remains an important question. We speculate that Na+,K+-ATPase ligands may produce a stressful environment similar to that of abnormal amino acid metabolism, because cellular amino acid uptake is indeed tightly controlled by plasma membrane Na+,K+-ATPase activity (Hyde et al., 2003). Na+,K+-ATPase impairment by Na+,K+-ATPase ligands will influence amino acid transport by destroying the Na+ gradient. Our cDNA microarray analysis supported this, because treatment of A549 cells with 100 nM ouabain for 12 h up-regulated the expression of many amino acid transporters and aminoacyl-tRNA synthetase genes (Supplemental Table S1).
In summary, Na+,K+-ATPase ligands are the first identified small-molecule drugs that can modulate GCN2 degradation machinery in cancer cells. By using these small molecules, we uncovered novel regulatory mechanisms for GCN2 in cancer cell death. The posttranslational mechanisms modulating GCN2 expression can be used for molecular-targeted cancer therapy and drug development.
MATERIALS AND METHODS
Cell culture and transfection
Human embryonic kidney 293 (HEK293), GCN2 wild-type mouse embryonic fibroblast (MEF), GCN2−/− MEF, CHOP wild-type MEF, and CHOP−/− MEF cells and cancer cell lines, including A549, HeLa, HCT116, HT29, and LoVo, were all obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured under standard procedures. Transient transfection was performed by the polyJet reagent (Signagen, Gaithersburg, MD).
Plasmids
To make PCI-HA-GCN2, the coding region of human GCN2 (mRNA RefSeq: NM_001013703) was amplified from 293T cDNA by using primers 5′-tatctcgagtaatggctgggggccgtggg-3′ and 5′-cgcgcggccgcttaaaataagattctgtagt-3′ and was ligated as an XhoI–NotI fragment into the XhoI–NotI sites of PCI-HA mammalian expressing vector. To produce HA-tag fusions with the RWD domain (amino acids [aa] 1–201), PK1 domain (aa 202–569), PK2 domain (aa 570–1010), and GCN2-N terminal region (aa 1–1010) of GCN2 (aa 1–1649), fragments containing XhoI (5′-end) and NotI (3′-end) restriction sites were generated by PCR amplification from the PCI-HA-GCN2 constructs described and inserted into the PCI-HA plasmid downstream of the HA-tag reading frame. For YFP-GCN2, the human GCN2 was excised as an XhoI–NotI/blunt fragment from PCI-HA-GCN2 and inserted into the XhoI–SmaI sites of peYFP-c1 downstream of the YFP reading frame. GCN2(T899A) and GCN2(T899D) mutant expression plasmids were constructed using the point mutation PCR method. Briefly, mutagenesis primers encoding threonine-to- alanine and threonine-to–aspartic acid mutations for T899 were designed as follows: hGCN2-T899A-R(1677): agtgccaaccatcccagctaagtgacctgaagg; hGCN2-T899A-F(238): acccttcaggtcacttagctgggatggttggc; hGCN2-T899D-R(1677): agtgccaaccatcccatttaagtgacctgaagg; and hGCN2-T899D-F(238): acccttcaggtcacttaaatgggatggttggc.
To make PRK5-Flag-β-arrestin1– and PRK5-Flag-β-arrestin2–expressing plasmids, we amplified human β-arrestin1 (NM_004041.3) using primers 5′-tatctcgagccatgggcgacaaaggga-3′ and 5′-cccaagcttctatctgttgttgagctgtgg-3′ and ligated it as an XhoI–HindIII fragment into the XhoI–HindIII sites of PRK5-FLAG–expressing plasmids; human β-arrestin2 (NM_004313.3) was amplified using primers 5′-tatggatccatgggggagaaacccggga-3′ and 5′-cccaagcttcgcttcctagcagagttgatcatc-3′ and ligated as a BamHI–HindIII fragment into the BamHI–HindIII sites of a PRK5-FLAG–expressing plasmid. PRK5-FLAG-hβarr1N(1–200 aa) and PRK5-FLAG-hβarr1C(201–418 aa) constructs were generated by subcloning the XhoI–HindIII fragments of PRK5-FLAG-β-arrestin1. PRK5-FLAG-hβarr2N(1–185 aa) and PRK5-FLAG-hβarr2C(186–409 aa) constructs were generated by subcloning the BamHI–HindIII fragments of PRK5-FLAG-β-arrestin2; then these regions were inserted into PRK5-FLAG vector. For pGEX-4T-1-β-arrestin1– and pGEX-4T-1-β-arrestin2–expressing vectors, human β-arrestin1 and β-arrestin2 cDNAs were amplified from plasmid PRK5-FLAG-β-arrestin1 and PRK5-FLAG-β-arrestin2 by using primers; the PCR products were then digested with SalI and NotI and inserted into the SalI–NotI sites of pGEX-4T-1. All plasmids tags were N-terminal. HA-tagged human NEDD4L–expressing plasmid was purchased from Addgene (Cambridge, MA).
Antibodies
Specific anti–phospho-eIF2α at Ser-52, anti–total eIF2α, and anti-GCN2 IgGs were purchased from Cell Signaling Technology (Danvers, MA). Rabbit polyclonal antibody against β-arrestin1/2 was purchased from Calbiochem (La Jolla, CA). Antibodies against NEDD4L, GFP, Ub, tubulin, and secondary IgGs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-PARP IgG was purchased from Millipore (Billerica, MA). Anti–GST monoclonal IgG was purchased from Oncogene Science (Cambridge, MA). Anti-FLAG and HA IgG was purchased from Sigma-Aldrich (St. Louis, MO).
siRNA experiments
Cells were transfected with the siRNAs of target genes and negative control siRNA using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Chemically synthesized double-stranded siRNAs were purchased from Genepharma (Shanghai, China). GCN2 siRNA (h) (sc-45644), PERK siRNA (h) (sc-36213), PKR siRNA (h) (sc-36263), and CHOP siRNA (h) (sc-35437) were purchased from Santa Cruz Biotechnology. These siRNA products consist of pools of three to five target-specific 19- to 25-nucleotide siRNAs designed to knock down gene expression.
Protein extraction and Western blotting
The detailed procedures were as previously described (Feng et al., 2011). In brief, cells were lysed in cell lysis buffer (50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 50 mM NaF, 5 mM EDTA, 5 mM β-glycerophosphate, 1 mM sodium vanadate, 1% Nonidet P-40, and protease inhibitors). The proteins were quantified with the Bradford assay. The extract was then removed and mixed with loading buffer. Proteins were boiled, resolved in SDS–polyacrylamide gel, transferred to polyvinylidene difluoride (PVDF) membranes, blocked with blocking buffer (5% skimmed milk in phosphate-buffered saline [PBS] with 0.02% Tween-20), and incubated with the indicated primary antibodies at 4°C overnight or at room temperature for 2 h. Blots were washed three times for 5 min each in PBS with 0.02% Tween-20 (PBST) and then incubated with horseradish peroxide–conjugated secondary antibodies in blocking buffer. After three washes with PBST, the blots were visualized by the ChemiQ 4800 mini Imaging System (Bioshine, Shanghai, China).
RT-PCR
In brief, the total RNAs were extracted with TRIZOL reagent (Invitrogen) following the manufacturer's instructions. Reverse transcription reactions were performed using the PrimeScript RT Reagent Kit (Takara, Otsu, Japan). PCR was performed for 22–30 cycles in 20 μl of reaction mixture. PCR products were resolved on 1% agarose gels and stained with ethidium bromide. Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control.
Coimmunoprecipitation experiment
After transfection, cells were lysed in the lysis buffer supplemented with complete protease inhibitor cocktail (Sigma-Aldrich) and N-ethylmaleimide (Sigma-Aldrich) at 4°C. Lysates were centrifuged at 12,000 rpm at 4°C for 10 min. Proteins (0.5 mg/500 μl) were immunoprecipitated with the indicated antibodies. The precleared protein A/G -agarose beads (Millipore) or TrueBlot IP beads (eBioscience, San Diego, CA) were incubated with immunocomplexes for another 2 h and washed four times with the lysis buffer. The immunoprecipitates were subjected to SDS–PAGE, followed by transferring onto PVDF membranes.
GST pull-down assay
GST-β-arrestin1, GST-β-arrestin2, and GST were heterologously expressed and purified from Rosetta-gami B strains. Each 5 μg of GST-β-arrestin1, GST-β-arrestin2, or GST was coincubated for 1 h with 20 μl of 50% GST Sepharose 4B slurry at 4°C in 1 ml of binding buffer (PBS and 10 mM dithiothreitol) and the beads were washed four times with 1 ml of wash buffer (PBS and 0.5% Triton X-100). Whole 293T cell extracts were then added into the mixture and further incubated at 4°C overnight with rotation. The beads were washed, separated by SDS–PAGE, and analyzed by immunoblot analysis.
Measurement of surface DR5 expressions by flow cytometry
The detailed procedures were as previously described (Frese et al., 2006). In brief, cells were harvested, washed, and resuspended with ice-cold PBS containing 1% bovine serum albumen. Then 5 μg of primary anti-DR5 antibody or control IgG was added per sample. After 30 min of incubation on ice, the cells were washed twice and incubated with FITC-labeled secondary antibody. At least 10,000 cells were analyzed by flow cytometry.
Xenograft tumor models
LoVo and HCT116 cells were implanted subcutaneously in the right flank of each mouse. Once tumor volume reached >50 mm3, animals were randomized so that all groups had similar starting mean tumor volumes. Tumor measurements were taken three times per week, and tumor volume (in cubic millimeters ) was calculated using the ellipsoid formula D(d2), where D is the largest diameter of the tumor and d the smallest. At the end of the experiments, the animals were killed, and the tumor growth curves were plotted. Tumors were removed, homogenized, and stained with 1–2 μl of FITC-DEVD-FMK for detection of caspase 3 activity based on the manufacturer's instruction (CaspGLOW Fluorescein Active Caspase-3 Staining Kit; BioVision, Milpitas, CA).
Cell apoptosis assay
Cell apoptosis was measured by the annexin V-FITC/propidium iodide double-staining method following the manufacturer's instructions (Abcam, Cambridge, UK). A total of 10,000 cells per sample were analyzed by flow cytometry.
Ubiquitination assay
The cells were lysed in a stringent cell lysis buffer containing 2% SDS, 150 mM NaCl, 10 mM Tris-HCl (pH 8.0), 2 mM sodium orthovanadate, 50 mM sodium fluoride, protease inhibitors, and 2 mM N-ethylmaleimide (Choo and Zhang, 2009). The cell lysates were boiled for 10 min, sonicated for 10–20 s, added to 900 μl of dilution buffer containing 150 mM NaCl, 10 mM Tris-HCl (pH 8.0), 2 mM EDTA, 1% Triton X, protease inhibitors, and 2 mM N-ethylmaleimide, and further rotated at 4°C for 1 h. For immunoprecipitation experiments, the supernatants were collected after centrifugation, 1 μg of anti-ubiquitin IgGs or anti-GCN2 IgGs was added to 1.5 mg of the supernatant, and the mixture was rotated at 4°C overnight; then 30 μl of prepared Protein A or G agarose beads was added to the samples. The samples were rotated for 2 h. After rotation, beads were washed three times with washing buffer containing 10 mM Tris-HCl (pH 8.0), 1 M NaCl, 1 mM EDTA, and 1% NP-40, boiled, and subjected to Western blotting analysis.
Statistical analysis
The results are expressed as mean ± SD. Statistical analysis involving two groups was performed by means of Student's t test. Analysis of variance followed by Dunnett's multiple comparison tests was used to compare more than two groups. All data were processed with SPSS 10.0 software (IBM, Armonk, NY).
Supplementary Material
Acknowledgments
We express our gratitude to the reviewers for valuable comments and suggestions. This project was sponsored by the Natural Science Fund of China (31071250, 81171843, 81473293,81421091, and J1103521), the National Key Basic Research Program of the Ministry of Science and Technology of China (2012CB967004, 2014CB744501), an open project of the National Key Lab of Natural Medicines (SKLNMKF201303, G140014), an open project of the National Key Lab of Drug Discovery (SIMM1106KF-01), the Jiangsu Province Administration of Traditional Chinese Medicine (LZ13230), and Fundamental Research Funds for the Central Universities, Nanjing 321 Talents Project, to Y.W.
Abbreviations used:
- ATF4
active transcription factor 4
- CHOP
C/EBP homologous protein
- CHX
cycloheximide
- eIF2α
eukaryotic initiation factor 2α
- FITC
fluorescein isothiocyanate
- GCN2
general control nonderepressible kinase 2
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HA
hemagglutinin
- HEK293
human embryonic kidney 293
- NEDD4L
neural precursor cell–expressed, developmentally down-regulated 4–like
- PARP
poly(ADP-ribose) polymerase
- PERK
endoplasmic reticulum–resident kinase
- PKR
RNA-dependent protein kinase
- Ub
ubiquitin
- WT
wild type
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-10-1438) on January 14, 2015.
*These authors contributed equally to this work
The authors declare no potential conflicts of interest.
Y.W. conceived the project and provided the financial support; Y.W. and C.W. designed the projects, performed experiments and data analysis, and wrote the paper; M.L. performed WB analysis; F.S. contributed to GST protein expressions and the initiation of the project; C.D. performed animal experiments; H.Z.C. and B.J.J. provided partial financial support and some reagents; H.Z.C., Y.J., and Z.J. provided valuable discussions.
REFERENCES
- Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr. 2012;3:307–321. doi: 10.3945/an.112.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar RJ, Wohlfahrt J, Oudin A, Hjuler T, Melbye M. Digoxin use and the risk of breast cancer in women. J Clin Oncol. 2011;29:2165–2170. doi: 10.1200/JCO.2010.32.8146. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. Caspase independent/dependent regulation of K(+), cell shrinkage, and mitochondrial membrane potential during lymphocyte apoptosis. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Gomez-Angelats M, Cidlowski JA. Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis. J Biol Chem. 2001;276:4304–4314. doi: 10.1074/jbc.M005171200. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- Chen HI, Sudol M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci USA. 1995;92:7819–7823. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo YS, Zhang Z. Detection of protein ubiquitination. 2009:J Vis Exp 30, 1293. doi: 10.3791/1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–279. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- Feng S, Chen W, Cao D, Bian J, Gong FY, Cheng W, Cheng S, Xu Q, Hua ZC, Yin W. Involvement of Na(+), K (+)-ATPase and its inhibitors in HuR-mediated cytokine mRNA stabilization in lung epithelial cells. Cell Mol Life Sci. 2011;68:109–124. doi: 10.1007/s00018-010-0444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese S, Frese-Schaper M, Andres AC, Miescher D, Zumkehr B, Schmid RA. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 2006;66:5867–5874. doi: 10.1158/0008-5472.CAN-05-3544. [DOI] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentz SH, Bertollo CM, Souza-Fagundes EM, da Silva AM. Implication of eIF2alpha kinase GCN2 in induction of apoptosis and endoplasmic reticulum stress-responsive genes by sodium salicylate. J Pharm Pharmacol. 2013;65:430–440. doi: 10.1111/jphp.12002. [DOI] [PubMed] [Google Scholar]
- Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA. 1991;88:6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- He H, Singh I, Wek SA, Dey S, Baird TD, Wek RC, Georgiadis MM. Crystal structures of GCN2 protein kinase C-terminal domains suggest regulatory differences in yeast and mammals. J Biol Chem. 2014;289:15023–15034. doi: 10.1074/jbc.M114.560789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Wang D, Wu J, Jin J, Wei W, Sun W. Involvement of beta-arrestins in cancer progression. Mol Biol Rep. 2013;40:1065–1071. doi: 10.1007/s11033-012-2148-0. [DOI] [PubMed] [Google Scholar]
- Hyde R, Taylor PM, Hundal HS. Amino acid transporters: roles in amino acid sensing and signalling in animal cells. Biochem J. 2003;373:1–18. doi: 10.1042/bj20030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov A, Eva A, Kirch U, Boldyrev A, Scheiner-Bobis G. Ouabain activates signaling pathways associated with cell death in human neuroblastoma. Biochim Biophys Acta. 2007;1768:1691–1702. doi: 10.1016/j.bbamem.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Lefranc F, Kiss R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia. 2008;10:198–206. doi: 10.1593/neo.07928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–1780. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews WR, DuCharme DW, Hamlyn JM, Harris DW, Mandel F, Clark MA, Ludens JH. Mass spectral characterization of an endogenous digitalislike factor from human plasma. Hypertension. 1991;17:930–935. doi: 10.1161/01.hyp.17.6.930. [DOI] [PubMed] [Google Scholar]
- Mazroui R, Di Marco S, Kaufman RJ, Gallouzi IE. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol Biol Cell. 2007;18:2603–2618. doi: 10.1091/mbc.E06-12-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JV, Cotter TG. Cell shrinkage and apoptosis: a role for potassium and sodium ion efflux. Cell Death Differ. 1997;4:756–770. doi: 10.1038/sj.cdd.4400296. [DOI] [PubMed] [Google Scholar]
- Mijatovic T, Kiss R. Cardiotonic steroids-mediated Na+/K+-ATPase targeting could circumvent various chemoresistance pathways. Planta Medica. 2013;79:189–198. doi: 10.1055/s-0032-1328243. [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Staschke KA, Wek RC. Dimerization is required for activation of eIF2 kinase Gcn2 in response to diverse environmental stress conditions. J Biol Chem. 2004;279:22820–22832. doi: 10.1074/jbc.M402228200. [DOI] [PubMed] [Google Scholar]
- Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- Nobel CS, Aronson JK, van den Dobbelsteen DJ, Slater AF. Inhibition of Na+/K(+)-ATPase may be one mechanism contributing to potassium efflux and cell shrinkage in CD95-induced apoptosis. Apoptosis. 2000;5:153–163. doi: 10.1023/a:1009684713784. [DOI] [PubMed] [Google Scholar]
- Ozdemir T, Nar R, Kilinc V, Alacam H, Salis O, Duzgun A, Gulten S, Bedir A. Ouabain targets the unfolded protein response for selective killing of HepG2 cells during glucose deprivation. Cancer Biother Radiopharm. 2012;27:457–463. doi: 10.1089/cbr.2011.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padyana AK, Qiu H, Roll-Mecak A, Hinnebusch AG, Burley SK. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2alpha protein kinase GCN2. J Biol Chem. 2005;280:29289–29299. doi: 10.1074/jbc.M504096200. [DOI] [PubMed] [Google Scholar]
- Pavitt GD. eIF2B, a mediator of general and gene-specific translational control. Biochem Soc Trans. 2005;33:1487–1492. doi: 10.1042/BST0331487. [DOI] [PubMed] [Google Scholar]
- Pavitt GD, Ramaiah KV, Kimball SR, Hinnebusch AG. eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev. 1998;12:514–526. doi: 10.1101/gad.12.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzani R, Rubin B, Redaelli M, Radu C, Barollo S, Cicala MV, Salva M, Mian C, Mucignat-Caretta C, Simioni P, et al. The antiproliferative effects of ouabain and everolimus on adrenocortical tumor cells. Endocr J. 2014;61:41–53. doi: 10.1507/endocrj.ej13-0225. [DOI] [PubMed] [Google Scholar]
- Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov. 2008;7:926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- Rajasekaran SA, Palmer LG, Quan K, Harper JF, Ball WJ, Jr, Bander NH, Peralta Soler A, Rajasekaran AK. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell. 2001;12:279–295. doi: 10.1091/mbc.12.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, Hinnebusch AG. Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:3027–3036. doi: 10.1128/mcb.11.6.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol. 1998;18:2282–2297. doi: 10.1128/mcb.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- Stenkvist B. Is digitalis a therapy for breast carcinoma. Oncol Rep. 1999;6:493–496. [PubMed] [Google Scholar]
- Sudol M, Chen HI, Bougeret C, Einbond A, Bork P. Characterization of a novel protein-binding module—the WW domain. FEBS Lett. 1995;369:67–71. doi: 10.1016/0014-5793(95)00550-s. [DOI] [PubMed] [Google Scholar]
- Tailler M, Senovilla L, Lainey E, Thepot S, Metivier D, Sebert M, Baud V, Billot K, Fenaux P, Galluzzi L, et al. Antineoplastic activity of ouabain and pyrithione zinc in acute myeloid leukemia. Oncogene. 2012;31:3536–3546. doi: 10.1038/onc.2011.521. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ning Y, Alam GN, Jankowski BM, Dong Z, Nor JE, Polverini PJ. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia. 2013;15:989–997. doi: 10.1593/neo.13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497–4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res. 2010;16:1701–1708. doi: 10.1158/1078-0432.CCR-09-1692. [DOI] [PubMed] [Google Scholar]
- Winnicka K, Bielawski K, Bielawska A, Surazynski A. Antiproliferative activity of derivatives of ouabain, digoxin and proscillaridin A in human MCF-7 and MDA-MB-231 breast cancer cells. Biol Pharm Bull. 2008;31:1131–1140. doi: 10.1248/bpb.31.1131. [DOI] [PubMed] [Google Scholar]
- Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao AY, Wang XQ, Yang A, Yu SP. Slight impairment of Na+,K+-ATPase synergistically aggravates ceramide- and beta-amyloid-induced apoptosis in cortical neurons. Brain Res. 2002;955:253–259. doi: 10.1016/s0006-8993(02)03472-8. [DOI] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29:2082–2096. doi: 10.1038/emboj.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Cheng W, Shen W, Shu L, Zhao J, Zhang J, Hua ZC. Impairment of Na(+),K(+)-ATPase in CD95(APO-1)-induced human T-cell leukemia cell apoptosis mediated by glutathione depletion and generation of hydrogen peroxide. Leukemia. 2007;21:1669–1678. doi: 10.1038/sj.leu.2404791. [DOI] [PubMed] [Google Scholar]
- Zeng F, Xu J, Harris RC. Nedd4 mediates ErbB4 JM-a/CYT-1 ICD ubiquitination and degradation in MDCK II cells. FASEB J. 2009;23:1935–1945. doi: 10.1096/fj.08-121947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Wek RC. Ribosome-binding domain of eukaryotic initiation factor-2 kinase GCN2 facilitates translation control. J Biol Chem. 1998;273:1808–1814. doi: 10.1074/jbc.273.3.1808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.