

Intracellular Ca2+ dysregulation and transport disruption precede cell death in Alzheimer's disease. Mechanisms of AβO-induced Ca2+ elevation are identified that regulate the onset, severity, and spatiotemporal progression of BDNF transport defects. The results challenge dogmatic views on mechanisms of AβO toxicity and subcellular sites of action.
Abstract
Disruption of fast axonal transport (FAT) and intracellular Ca2+ dysregulation are early pathological events in Alzheimer's disease (AD). Amyloid-β oligomers (AβOs), a causative agent of AD, impair transport of BDNF independent of tau by nonexcitotoxic activation of calcineurin (CaN). Ca2+-dependent mechanisms that regulate the onset, severity, and spatiotemporal progression of BDNF transport defects from dendritic and axonal AβO binding sites are unknown. Here we show that BDNF transport defects in dendrites and axons are induced simultaneously but exhibit different rates of decline. The spatiotemporal progression of FAT impairment correlates with Ca2+ elevation and CaN activation first in dendrites and subsequently in axons. Although many axonal pathologies have been described in AD, studies have primarily focused only on the dendritic effects of AβOs despite compelling reports of presynaptic AβOs in AD models and patients. Indeed, we observe that dendritic CaN activation converges on Ca2+ influx through axonal voltage-gated Ca2+ channels to impair FAT. Finally, FAT defects are prevented by dantrolene, a clinical compound that reduces Ca2+ release from the ER. This work establishes a novel role for Ca2+ dysregulation in BDNF transport disruption and tau-independent Aβ toxicity in early AD.
INTRODUCTION
Impaired fast axonal transport (FAT) of organelles correlates with early stages of Alzheimer's disease (AD) progression and is observed before cell death (Goldstein, 2012; Millecamps and Julien, 2013). Neurons cultured from AD mice expressing disease-associated mutations exhibit FAT defects (Pigino et al., 2003; Lazarov et al., 2007; Stokin et al., 2008). These findings are corroborated in vivo by manganese-enhanced magnetic resonance imaging of FAT, which reveal defects that precede amyloid plaque deposition and extensive neurofibrillary tangle formation (Minoshima and Cross, 2008; Smith et al., 2011). Collectively these data support a causal role for FAT disruption in AD. Although the mechanisms of dendritic transport are less well characterized, they are also critical for neuronal function and survival. Transport of glutamate receptors, endosomes, and brain-derived neurotrophic factor (BDNF) within dense-core vesicles (DCVs) is essential for spine growth, learning, and memory (Kennedy et al., 2010; Lu et al., 2013; Yoshii et al., 2013). Thus transport dysregulation in both axons and dendrites has significant physiological consequences in diseased neurons.
According to the “calcium hypothesis of AD,” amyloid-β (Aβ) overproduction and toxicity are induced by aberrant Ca2+ signaling before accumulation of hyperphosphorylated tau (p-tau) and cognitive decline (Berridge, 2010; Chakroborty and Stutzmann, 2011). Aβ oligomers (AβOs) increase Ca2+ influx through several membrane receptors, including N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors and axonal voltage-gated Ca2+ channels (VGCCs; Ferreira and Klein, 2011). This triggers a persistent elevation in resting cytosolic Ca2+, which is primarily maintained by Ca2+-induced Ca2+ release (CICR) from the endoplasmic reticulum (ER; Paula-Lima et al., 2011). Elevated cytosolic Ca2+ activates the Ca2+/calmodulin-dependent phosphatase calcineurin (CaN). Activated CaN relieves inhibition of protein phosphatase-1 (PP1), which in turn overactivates glycogen synthase kinase 3β (GSK3β), ultimately leading to synapse failure (Reese and Taglialatela, 2011).
In axons, GSK3β activation can induce hyperphosphorylation of tau and disrupt FAT by inhibiting motor protein activity and cargo binding (Morfini et al., 2002; Weaver et al., 2013). AβOs reduce vesicular transport of BDNF in primary neurons through an NMDA receptor (NMDAR)–dependent mechanism, which is mediated by GSK3β (Decker et al., 2010b). Of note, we found that AβO-induced transport defects occur independent of tau, microtubule destabilization, and acute cell death (Ramser et al., 2013). We rescued FAT defects by inhibiting CaN and prevented them by inhibiting PP1 and GSK3β. Our findings implicated dysregulated Ca2+ signaling in BDNF transport disruption when the contribution of pathogenic tau is likely minimal.
The Ca2+-dependent mechanisms that regulate the onset, severity, and spatiotemporal progression of BDNF transport defects from dendritic and axonal AβO binding sites are unknown. In particular, it is unclear how the binding of AβOs to postsynaptic sites leads to tau-independent FAT impairment. We investigated these mechanisms in primary hippocampal neurons cultured from wild-type (tau+/+) and tau-knockout (tau−/−) mice. This model system is ideal for high-resolution imaging of organelle transport and detection of spatiotemporal changes in protein localization. Furthermore, cultured neurons treated with AβOs recapitulate AD pathologies such as calcium dyshomeostasis, kinase signaling cascade dysregulation, tau hyperphosphorylation, and FAT defects (De Felice et al., 2007, 2008; Decker et al., 2010b; Vossel et al., 2010; Zempel et al., 2010), which are central to this study. Here we show that defects in dendritic and axonal transport of BDNF are induced simultaneously but decline at different rates: maximal impairment of dendritic transport precedes maximal impairment of FAT. This correlates with Ca2+ elevation and CaN activation in dendrites and subsequently in axons. Postsynaptic CaN activation converges on axonal calcium dysregulation to impair FAT. Specifically, AβOs colocalize with axonal VGCCs, and blocking VGCCs prevents FAT defects. Finally, BDNF transport defects are prevented by dantrolene, a compound that reduces CICR through ryanodine receptors in the ER membrane. This work establishes a novel role for Ca2+ dysregulation in BDNF transport disruption and tau-independent Aβ toxicity.
RESULTS
AβOs induce dendritic, calcineurin-dependent transport defects that precede maximal impairment of FAT
We previously showed that AβOs impair axonal BDNF transport independent of tau by nonexcitotoxic activation of CaN, a Ca2+/calmodulin-dependent phosphatase implicated in AD (Ramser et al., 2013). It is unknown how the binding of AβOs to dendritic synaptic sites leads to FAT impairment. Because CaN, its effectors (PP1 and GSK3β), and KIF1A, the primary kinesin motor required for BDNF transport (Lo et al., 2011), are present in both dendrites and axons (Lee et al., 2003; Mansuy, 2003), we asked whether AβOs induce dendritic, CaN-dependent transport defects that precede FAT disruption. In addition, to determine whether AβO-induced mislocalization of axonal tau to dendrites (Zempel et al., 2010; Ittner and Gotz, 2011) impairs dendritic transport, we imaged tau+/+ and tau−/− hippocampal neurons expressing BDNF-monomeric red fluorescent protein (mRFP) 4–26 h after exposure to 500 nM AβOs (Figure 1, Supplemental Table S1, and Supplemental Video S1). Irreversible AβO binding was confirmed retrospectively by immunocytochemistry (Figure 1A) using an oligomer-specific antibody (NU-4; Lambert et al., 2007). Representative kymographs illustrate differences between BDNF transport in control (vehicle-treated) and AβO-treated neurons (Figure 1, B and C) and are used to calculate vesicle flux, an index of transport (see Materials and Methods for a detailed explanation). Total dendritic flux was similarly and markedly reduced in AβO-treated cells, both in the presence and absence of tau (Figure 1, B and C, and Supplemental Table S1). Treatment with 1 μM FK506, a highly specific, potent inhibitor of CaN, rescued these defects (Schreiber and Crabtree, 1992). A complete list of transport statistics is provided in Supplemental Table S1. To assess the spatiotemporal progression of transport defects, we measured and compared dendritic and axonal transport after 4–26 h of AβO treatment (Figure 1C). BDNF transport defects were induced simultaneously in both compartments but exhibited different rates of decline: maximal impairment of dendritic transport defects were observed within 5–12 h of AβO treatment, before maximal impairment of FAT after 18 h of AβO exposure. As we previously reported, we observed no concomitant reduction in cell viability or structural alterations of the microtubule network (Decker et al., 2010b; Ramser et al., 2013). Further control experiments include treatment of cultured neurons with a nonaggregated, scrambled form of the Aβ1-42 peptide, which failed to induce transport defects (Decker et al., 2010b). Thus, under our experimental conditions, BDNF transport defects arise before overt neurotoxicity and are likely an early and specific consequence of AβO treatment. Collectively our results show that AβO-induced dendritic transport defects precede FAT disruption and occur by a common tau-independent mechanism that is reversible upon CaN inhibition.
FIGURE 1:

AβOs induce dendritic, calcineurin-dependent transport defects that precede maximal impairment of FAT. (A) Expression of soluble blue fluorescent protein (BFP) and BDNF-mRFP in an AβO-treated tau−/− neuron (from left to right). Overlay of BFP and AβO images shows binding of AβOs to dendrites (MAP2 positive) and axons. Immunocytochemistry shows that AβOs remain oligomeric after 18 h in culture. Arrows indicate axons; arrowheads indicate dendrites. (B) Total dendritic flux was markedly reduced in AβO-treated tau−/− cells. Treatment with 1 μM FK506 rescued these defects. Representative kymographs illustrate differences between BDNF transport in control and treated neurons. (C) BDNF transport defects were induced simultaneously in both compartments but exhibited different rates of decline: significant dendritic transport defects were observed within 5–12 h of AβO treatment, before maximal impairment of FAT after 18 h of AβO exposure. Green trace indicates anterograde transport; red trace indicates retrograde transport. Graphs show means ± SEM. A minimum of 20 cells from three different cultures were analyzed per condition; ***p < 0.001 relative to controls. Tau+/+ transport data are presented in Supplemental Table S1. Complete statistical evaluation is presented in Supplemental Table S1. Scale bar, 25 μm.
AβO-induced elevation of intracellular calcium correlates with the spatiotemporal progression of BDNF transport defects
AβOs increase Ca2+ influx through several membrane receptors, including NMDARs, AMPA receptors (AMPARs), and voltage-gated Ca2+ channels (Ferreira et al., 2007). This triggers a persistent elevation in resting cytosolic Ca2+, which activates downstream Ca2+/calmodulin-dependent proteins, such as CaN (Reese and Taglialatela, 2011). Within minutes of AβO treatment, CaN activation is observed, first in dendritic spines and minutes to hours later in the cell body (Wu et al., 2012). Because AβOs impair dendritic and axonal transport by a similar CaN-dependent mechanism, we asked whether Ca2+ and active CaN are elevated first in dendrites and later in axons, reflecting the spatiotemporal progression of BDNF transport defects. To detect AβO-induced changes in neuronal cytosolic Ca2+, we expressed a genetically encoded Ca2+ sensor termed cameleon (D3cpV) in neurons before treatment with either vehicle or 500 nM AβOs (Figure 2). In comparison to control neurons, a significant increase in fluorescence resonance energy transfer (FRET) between the calmodulin–cyan fluorescent protein (CFP) donor and the M13-cpVenus acceptor was observed exclusively in the cell body and primary dendrites after 5 h of AβO treatment (Figure 2, A and B). After 13–18 h of AβO exposure, FRET ratios were also markedly increased in proximal axon segments within 300 μm of the cell body (Figure 2, A and B), indicated by the increased red shift of the thermoscale. Therefore, AβOs trigger an increase in cytosolic Ca2+ that is initially restricted to the somatodendritic domain and subsequently spreads through the axon. This coincides with the spatiotemporal progression of BDNF transport defects from dendrites to axons.
FIGURE 2:

AβO-induced elevation of intracellular calcium correlates with the spatiotemporal progression of BDNF transport defects. (A, B) Representative images and quantification of cameleon FRET ratios in control and AβO-treated tau−/− neurons. In comparison to control neurons, a significant increase in FRET between the calmodulin-CFP donor and the M13-cpVenus acceptor was observed exclusively in the cell body and primary dendrites after 5 h of AβO treatment. After 13–18 h of AβO exposure, FRET ratios were also markedly increased in proximal axon segments within 300 μm of the cell body. Graphs show means ± SEM. A minimum of 15 cells from three independent cultures were analyzed per condition; *p < 0.05, **0.001 < p < 0.05, and ***p < 0.001 relative to controls. Scale bar, 50 μm.
AβO-induced calcineurin activation coincides with the spatiotemporal progression of BDNF transport defects
Calmodulin (CaM) binds free Ca2+ ions and directly activates CaN (Reese and Taglialatela, 2011). Previously we used in vitro phosphatase assays to detect elevation of CaN activity in cultured neurons treated with 500 nM AβOs for 18 h (Ramser et al., 2013). To determine the spatiotemporal progression of AβO-induced CaN activation, we performed in situ proximity ligation assays (PLAs) on control and AβO-treated neurons (Figure 3). First, CaM and CaN-specific primary antibodies for PLA analyses were validated by immunocytochemistry. As expected, CaM appeared soluble and ubiquitous, whereas CaN appeared punctate and localized predominantly to cell bodies and dendrites in control cells (Mansuy, 2003; Wu et al., 2012; Figure 3A). We visualized and quantified PLA puncta, indicative of CaN activation, in 100-μm segments of dendrites and axons after 4, 8, and 18 h of AβO exposure (Figure 3, B and C). In comparison to control cells, a significant increase in CaN activation was observed exclusively in the cell body and dendrites after 4 h of AβO treatment. After 18 h of AβO exposure, marked CaN activation was also detected in proximal axon segments (Figure 3, B and C). Ionomycin, a Ca2+ ionophore, induced global CaN activation within 30 min of treatment. Results show that AβO-induced CaN activation is concomitant with cytosolic Ca2+ elevation in dendrites and axons and strongly suggests that this response mediates the spatiotemporal progression of BDNF transport defects.
FIGURE 3:

AβO-induced calcineurin activation coincides with the spatiotemporal progression of BDNF transport defects. (A) CaN- and CaM-specific primary antibodies for PLA analyses were validated by immunocytochemistry. As expected, CaM appeared soluble and ubiquitous, whereas CaN appeared punctate and localized predominantly to dendrites in control cells. (B) Representative images and (C) quantification of CaN-CaM puncta in control and AβO-treated tau−/− neurons. In comparison to control cells, a significant increase in CaN activation was observed by PLA exclusively in the cell body and dendrites after 5 h of AβO treatment. After 13–18 h of AβO exposure, marked CaN activation was also detected in proximal axon segments. Graphs show means ± SEM. A minimum of 15 cells from three independent cultures were analyzed per condition; *p < 0.05, **0.001< p < 0.05, and ***p < 0.001 relative to controls. Scale bar, 25 μm.
AβOs bind to axons and colocalize with presynaptic voltage-gated calcium channels
Of interest, although they decline at different rates, dendritic and axonal transport defects are induced simultaneously (Figure 1). This may be attributed to a novel, presynaptic mechanism of AβO-induced Ca2+ dysregulation that converges on postsynaptic mechanisms of CaN activation to impair FAT. Although AβOs are believed to bind exclusively to dendritic membrane proteins (Cochran et al., 2013), in vitro and in vivo evidence suggests that AβOs also modulate presynaptic VGCC activity (Cataldi, 2013). If Ca2+ elevation is restricted to the cell body and dendrites by extensive buffering mechanisms, axonal AβO binding may induce aberrant Ca2+ influx through VGCCs and contribute to FAT impairment. By immunocytochemistry with an AβO-specific antibody (NU-4), we discovered that AβOs bind along the entire length of the axon in cultured neurons (Figure 4A). To determine whether axonal AβOs are also present in vivo, we performed immunohistochemistry on brain sections from double-transgenic AD mice (LaFerla 3xTg and APP23/PS45; unpublished data for the latter genotype). NU-4 staining revealed a punctate distribution of AβOs along dendrites and, strikingly, axons in the cortex (Figure 4B). AβOs were not detected in age-matched wild-type control mice (Figure 4B). On the basis of reports that AβOs shift the steady-state activation of VGCCs toward more hyperpolarized values in HEK cells and increase Ca2+ influx in Xenopus oocytes (Hermann et al., 2013), we asked whether the P/Q and N-types, expressed abundantly in hippocampal neurons, constitute binding targets. Immunocytochemistry revealed a punctate distribution of both channel types in hippocampal neurons and a high degree of colocalization with AβOs (P/Q type, 83.5%; N type, 73.3%; Figure 4C and unpublished data). These results suggest that axonal AβOs associate directly or indirectly with presynaptic VGCCs and modulate their activity.
FIGURE 4:

AβOs bind to axons and colocalize with presynaptic voltage-gated calcium channels. (A) Representative images of immunocytochemistry with an AβO-specific antibody (NU-4) show that AβOs bind along the entire length of the axon in cultured neurons. (B) Immunohistochemistry on coronal sections from 12-mo-old transgenic AD mouse brain (LaFerla 3xTg) reveal a punctate distribution of AβOs along axons in the cortex. Axons are distinguished by the absence of MAP2 staining (inset). AβOs were not detected in age-matched wild-type control mice. (C) Representative images of AβO and CaV 2.1 (P/Q-type VGCC) immunocytochemistry. Of axonal AβOs, 83.5% colocalize with P/Q-type VGCCs. A minimum of 15 cells from three independent cultures were analyzed. Arrows indicate axons, arrowheads indicate dendrites, and asterisks indicate regions of overlapping puncta. Scale bar, 100 μm.
Inhibition of presynaptic voltage-gated Ca2+ channels prevents axonal, but not dendritic, BDNF transport defects
To determine whether AβO-induced activation of presynaptic VGCCs impairs FAT, we incubated tau+/+ and tau−/− neurons with 50 nM ω-agatoxin IVA (P/Q-type channel blocker) or 100 nM ω-conotoxin GVIA (N-type channel blocker) for 30 min. As a negative control, we exposed neurons similarly to 10 μM nimodipine, which inhibits postsynaptic L-type Ca2+ channels (spatial distribution of VGCCs reviewed in Dolphin, 2012). Subsequently we treated tau+/+ and tau−/− neurons with 500 nM AβOs for 18 h. Exposure to VGCC inhibitors did not prevent AβO binding (Figure 5A). Remarkably, inhibition of P/Q- and N-type VGCCs prevented axonal BDNF transport defects, independent of tau (Figure 5B and Supplemental Table S2). ω-Agatoxin and ω-conotoxin maintained normal anterograde and retrograde flux in the presence of AβOs (Figure 5B). Average vesicle velocity and run length were also similar in control and pretreated cells. Consistent with the low abundance of L-type Ca2+ channels in axons (Hell et al., 1993; Leitch et al., 2009), nimodipine pretreatment did not prevent AβO-induced transport defects (Figure 5B). To confirm that Ca2+ influx mediates transport disruption, we chelated extracellular Ca2+ with 1.5 mM ethylene glycol tetraacetic acid (EGTA) before AβO addition. Indeed, extracellular Ca2+ chelation precluded FAT disruption (Figure 5B). By contrast, in dendrites, inhibition of P/Q- and N-type VGCCs failed to prevent AβO-induced transport defects (Figure 5C). A complete list of transport statistics is provided in Supplemental Table S2. Similar trends were observed upon inhibition of L-type channels, despite their predominantly somatodendritic distribution (Figure 5C). This result suggests that Ca2+ influx through L-type channels may be negligible compared with NMDARs and other abundant postsynaptic channels and receptors and thus does not regulate dendritic BDNF transport. Collectively our findings indicate that AβO-induced Ca2+ influx through presynaptic P/Q- and N-type VGCCs specifically blocks BDNF transport in axons.
FIGURE 5:

Inhibition of presynaptic voltage-gated Ca2+ channels prevents axonal but not dendritic BDNF transport defects. (A) Representative images of MAP2 and AβO immunocytochemistry. Pretreatment of tau−/− neurons with 50 μM ω-agatoxin IVA (P/Q-type channel blocker), 100 μM ω-conotoxin GVIA (N-type channel blocker), 10 μM nimodipine, or 1.5 mM EGTA did not prevent AβO binding. (B) Inhibition of P/Q- and N-type VGCCs prevented axonal BDNF transport defects independent of tau. Consistent with the absence of L-type Ca2+ channels in axons, nimodipine pretreatment did not prevent AβO-induced transport defects. Extracellular Ca2+ chelation with EGTA precluded FAT disruption. (C) By contrast, in dendrites, inhibition of P/Q- and N-type VGCCs failed to prevent AβO-induced transport defects. Pretreatment with nimodipine or EGTA also did not prevent AβO-induced transport defects. Graphs show means ± SEM. A minimum of 15 cells from three different cultures were analyzed per condition; ***p < 0.001 relative to controls, and +++p < 0.001 relative to AβO-treated cells. Tau+/+ transport data are presented in Supplemental Table S2. Complete statistical evaluation is presented in Supplemental Table S2. Scale bar, 25 μm.
Ryanodine receptor inhibition prevents axonal BDNF transport defects
Although there are many possible extracellular routes for AβO-induced Ca2+ influx, they may converge on CICR from the ER. CICR is required for sustained Ca2+ elevation and signaling dysregulation in AD pathology (Demuro et al., 2010). Compounds that restore Ca2+ homeostasis can improve learning and memory in transgenic AD animal models (Oules et al., 2012). Traditionally, neuronal ER was believed to exist only in the cell body and dendrites; however, we and others have localized ER to axons of mammalian CNS neurons (Shimizu et al., 2008; Deng et al., 2013). We detected ER in the dendrites and axons of primary hippocampal neurons by staining for endogenous ryanodine receptors (RyRs) and reticulon-3 (Ret3), well-defined markers for the ER membrane (Figure 6, A and B). Axons were distinguished by standard morphological criteria and by the absence of MAP2. To determine whether CICR contributes to FAT impairment, we exposed tau+/+ and tau−/− neurons to 0.5 μM dantrolene, a clinical compound that selectively blocks RyRs (Chakroborty et al., 2012). Subsequently we treated neurons with 500 nM AβOs for 18 h. Remarkably, inhibition of RyRs prevented AβO-induced transport defects independent of tau (Figure 6C and Supplemental Video S2). Dantrolene treatment maintained normal anterograde and retrograde flux in the presence of AβOs (Figure 6C and Supplemental Video S2). A complete list of transport statistics is provided in Supplemental Table S3. Taken together, our data demonstrate a central role for CICR in the disruption of FAT by AβOs and indicate that restoring Ca2+ homeostasis prevents these FAT defects.
FIGURE 6:

Ryanodine receptor inhibition prevents axonal BDNF transport defects. (A, B) Representative images of Ret3 and RyR immunocytochemistry. ER was detected in the dendrites and axons of tau−/− neurons. Axons were distinguished by standard morphological criteria and by the absence of MAP2. (C) Inhibition of RyRs prevented AβO-induced transport defects independent of tau. Dantrolene treatment maintained normal anterograde and retrograde flux in the presence of AβOs. Graphs show means ± SEM. A minimum of 15 cells from three different cultures were analyzed per condition; ***p < 0.001 relative to controls. Tau+/+ transport data are presented in Supplemental Table S3. Complete statistical evaluation is presented in Supplemental Table S3. Scale bar, 25 μm.
DISCUSSION
Intracellular Ca2+ dysregulation and FAT disruption are early pathological manifestations that lead to loss of synapse function and axonal degeneration in AD (Berridge, 2010; Millecamps and Julien, 2013). Here we correlate the spatiotemporal progression of transport defects with Ca2+ elevation and CaN activation in dendrites and subsequently in axons. Postsynaptic CaN activation converges on axonal calcium dysregulation to impair FAT. Specifically, AβOs colocalize with axonal VGCCs, and blocking VGCCs prevents FAT defects. Finally, BDNF transport defects are prevented by dantrolene, a compound that reduces CICR through RyRs. Collectively this work establishes a novel role for Ca2+ dysregulation in BDNF transport disruption and in tau-independent Aβ toxicity during early AD pathogenesis.
Dendritic BDNF transport defects may contribute to AβO-induced cellular toxicity
Although substantial evidence implicates axonal transport deficits in neurodegeneration, less is known about the roles and regulation of dendritic transport in normal and disease states. KIF1A coordinates dendrite branch morphogenesis and regulates the apposition of active zones and postsynaptic densities by controlling site-specific deposition of its cargo (Kern et al., 2013). In rat primary neurons, BDNF synthesized in the cell body is trafficked to proximal dendrites, where it promotes spine formation (Orefice et al., 2013), increases dendritic branching, and modulates synaptic function (Kuczewski et al., 2009). Alternatively, dendritic BDNF might constitute a reserve pool for presynaptic BDNF, which could be rapidly recruited to or from axon terminals upon changes in synapse activity (Maeder et al., 2014). Collectively these studies demonstrate critical roles for dendritic BDNF transport in postsynaptic development, function, and plasticity. Here we show that AβOs impair bidirectional transport of BDNF in dendrites. Ultimately, this may compromise postsynaptic BDNF secretion, reduce synaptic efficacy, and lead to neurodegeneration in AD. Restoring BDNF transport increases its release and promotes neuronal survival (Pineda et al., 2009). To our knowledge, we are the first to implicate reduced dendritic transport of BDNF in AD pathogenesis.
Dendritic and axonal sources of calcium elevation converge to disrupt BDNF transport
We demonstrate that dendritic and axonal BDNF transport defects are induced concomitantly but exhibit different rates of decline: significant dendritic transport defects precede maximal impairment of FAT. These findings suggest that dendritic and axonal sources of Ca2+ elevation converge to disrupt BDNF transport. AβOs are believed to interact preferentially with postsynaptic membrane receptors and modulate their activity (Cochran et al., 2013). Glutamate receptors, which mediate dendritic Ca2+ elevation, appear to be centrally involved (Ferreira and Klein, 2011); NMDARs coimmunoprecipitate with AβOs from rat synaptosomal membranes (De Felice et al., 2007), and AβO binding is reduced in dendrites of NMDAR-knockdown neurons (Decker et al., 2010a). We previously showed that NMDARs mediate AβO-induced disruption of BDNF transport by activation of CaN-GSK3β signaling. Dendritic transport may decline more rapidly than FAT due to the abundance and density of postsynaptic glutamate receptors at spines and therefore greater proximity of the transport apparatus and its regulators to sites of Ca2+influx and CICR from somatodendritic ER.
Of interest, although they decline at different rates, dendritic and axonal transport defects are induced simultaneously. This may be attributed to a novel, presynaptic mechanism of AβO-induced Ca2+ dysregulation. Presynaptic AβO binding has not been investigated extensively, and specific axonal binding sites and protein interactions remain uncharacterized. Here we report that AβOs bind to axons in culture and transgenic AD mouse brain. Consistently, immuno–electron microscopy studies indicate that AβOs localize to axons and presynaptic terminals at higher density in AD mice and patients than in wild-type mice or nondemented individuals (Kokubo et al., 2005a, b). Furthermore, we show that AβOs colocalize with axonal VGCCs. Although other work failed to demonstrate direct or indirect binding, AβOs markedly increase VGCC currents in cultured cortical and hippocampal neurons (Ramsden et al., 2002; Hermann et al., 2013). Treatment with antagonists rectifies Ca2+influx (Bobich et al., 2004) and protects against Aβ-induced cellular toxicity (Anekonda and Quinn, 2011; Copenhaver et al., 2011). In the present study, inhibition of P/Q- and N-type channels precludes axonal BDNF transport defects, further implicating VGCCs in AD progression.
Although there are many possible extracellular routes for AβO-induced Ca2+influx, they may converge on CICR from the ER to disrupt transport. Indeed, we prevented axonal BDNF transport defects by inhibiting RyRs with dantrolene, attesting to ER involvement. In accordance with this finding, activity-dependent capture of DCVs at synaptic boutons requires CICR by presynaptic RyRs and activation of Ca2+/calmodulin-dependent kinase II (CaMKII; Wong et al., 2009). Moreover, in young 3xTg-AD mice, a compensatory increase in RyR expression reduces the threshold for CICR, such that basal NMDAR activation elevates Ca2+ efflux from the ER in both dendrites and axons (Chakroborty et al., 2012). It is possible that AβO-induced Ca2+ release from axonal RyRs contributes to FAT disruption, notably in the presence or absence of tau. Collectively these results strongly support a role for Ca2+ dysregulation during early neurodegeneration.
Calcium-dependent mechanisms of motor protein regulation
There are several Ca2+-dependent mechanisms by which AβOs might disrupt BDNF transport. One mechanism could involve CaN-dependent inhibition of motor protein activity, mediated by GSK3β. GSK3β is implicated in many aspects of AD pathogenesis (Hooper et al., 2008) and negatively regulates axonal transport in squid axoplasm (Pigino et al., 2003), Drosophila neurons (Shaw and Chang, 2013), and mammals (Cantuti Castelvetri et al., 2013; Ramser et al., 2013). Negative regulation of kinesin-1 (KIF5) and cytoplasmic dynein is accomplished by reducing the number of motors that are bound to microtubules (Dolma et al., 2014). A second mechanism might comprise disruption of motor protein–cargo binding. Dendritic trafficking of NMDAR-containing vesicles is perturbed upon phosphorylation of KIF17 by CaMKII, which attenuates the interaction between KIF17 and its cargo adaptor, Mint1 (Yin et al., 2011). Similarly, it is possible that activation of CaN-GSK3β signaling phosphorylates KIF1A and blocks BDNF transport. A third mechanism could require activation of a Ca2+-sensing protein that directly inhibits motor motility. For example, the Ca2+-sensitive mitochondrial protein, Miro, interacts with the motor domain of KIF5 to dissociate it from microtubules (Wang and Schwarz, 2009). KIF1A motility and BDNF transport might be impaired by an analogous mechanism.
On the basis of our present findings and other current models of AD pathogenesis, we propose the following mechanism for Ca2+-dependent disruption of dendritic and axonal BDNF transport (Figure 7). AβOs bind to dendrites and axons, enhancing Ca2+ influx through dendritic glutamate receptors and axonal VGCCs. In turn, this induces CICR from postsynaptic and presynaptic ER to elevate resting cytosolic Ca2+. CaN-GSK3β signaling may disrupt BDNF transport by phosphorylating and inhibiting motor proteins and/or disrupting motor–DCV interactions. Alternatively, a Ca2+-sensing adaptor protein may directly impair motor protein motility. Additional research is required to determine whether transport impairment contributes to synapse loss and axonal degeneration in AD. In the adult brain, BDNF enhances synaptic transmission, facilitates synaptic plasticity, and increases the size and number of dendritic spines (Lu et al., 2013; Rothman and Mattson, 2013). Impaired transport may deprive processes of BDNF, inducing cytoskeletal-based retraction, increasing endocytosis, and promoting microtubule destabilization (Lu et al., 2013). Finally, from a clinical perspective, our findings are significant because intracellular Ca2+ dysregulation and transport impairment precede severe tau pathology and microtubule destabilization (Stutzmann et al., 2007; Goldstein, 2012). Compounds targeted to these early disease mechanisms may be more effective at preventing or reversing cell death.
FIGURE 7:

Proposed mechanism for Ca2+-dependent disruption of dendritic and axonal BDNF transport. AβOs bind to dendrites and axons, enhancing Ca2+ influx through dendritic glutamate receptors and axonal VGCCs. In turn, this induces CICR from postsynaptic and presynaptic ER to elevate resting cytosolic Ca2+. Calmodulin binds free Ca2+ ions and subsequently activates CaN-GSK3β signaling in dendrites and axons. On activation, GSK3β may disrupt BDNF transport by directly phosphorylating and inhibiting motor proteins and/or disrupting motor–DCV interactions. Alternatively, a Ca2+-sensing adaptor protein may directly impair motor protein motility.
MATERIALS AND METHODS
Hippocampal cell culture and expression of transgenes
Primary hippocampal neuronal cultures from E16 wild-type (tau+/+) and tau-knockout (tau−/−) embryonic mice (Jackson Laboratory, Bar Harbor, ME) of either sex were prepared as described by Kaech and Banker (2006) and kept in PNGM primary neuron growth medium (Lonza, Basel, Switzerland). The glial feeder layer was derived from murine neural stem cells as described by Miranda et al. (2012). At 10 d in vitro (DIV), cells were cotransfected with pβ-actin-BDNF-mRFP and pmUBa–enhanced blue fluorescent protein (BFP; from Gary Banker, Oregon Health and Sciences University, Portland, OR) using Lipofectamine (Invitrogen, Carlsbad, CA). Cells expressed the plasmids for 24–36 h before live imaging. The absence of tau in tau−/− mice was previously confirmed by immunoblotting with the antibodies PHF-1 and tau-46 (Supplemental Figure S1 in Ramser et al., 2013). All experiments with animals were approved by and followed the guidelines set out by the Simon Fraser University Animal Care Committee, Protocol 943-B05.
AβO, FK506, VGCC inhibitor, and RyR inhibitor treatments
Full-length, synthetic Aβ 1–42 peptides (AβOs; American Peptide, Sunnyvale, CA) were prepared exactly according to the method of Lambert et al. (1998) and applied to 11–13 DIV cells at a final concentration of 500 nM for 18 h. We use synthetic AβOs in our studies for the following reasons. First, they mimic the toxic properties of natural oligomers (brain or cell derived) as described previously (Jin et al., 2011; Welzel et al., 2014). Second, unlike natural oligomers, synthetic AβOs can be detected by immunocytochemistry. Confirmation of AβO binding is crucial in our experiments because it varies considerably between neurons. Although they may not be identical to natural oligomers, synthetic AβOs are a tractable tool for investigating mechanisms of AD pathogenesis (Ferreira and Klein, 2011). After AβO or vehicle exposure, cells were incubated with 1 μM FK506 (Sigma-Aldrich, St. Louis, MO) or equivalent volumes of vehicle (ethanol) for 1–3 h before imaging of transport. For all VGCC inhibition experiments, cells were incubated with 100 μM conotoxin GVIA (Alomone Labs, Jerusalem, Israel), 50 μM agatoxin IVA (Alomone Labs), or 10 μM nimodipine (Tocris Bioscience, Bristol, United Kingdom) for 30 min before AβO treatments. For all RyR inhibition experiments, cells were incubated with 0.5 μM dantrolene (Sigma-Aldrich) for 1 h before AβO treatment.
Live imaging and analysis of BDNF-mRFP transport
BDNF-mRFP transport was analyzed using a standard wide-field fluorescence microscope equipped with a cooled charge-coupled device camera and controlled by MetaMorph (Molecular Devices, Sunnyvale, CA) as described previously (Kwinter and Silverman, 2009). All imaging—typically 100 frames—was recorded by the “stream acquisition module” in MetaMorph. Briefly, cells were sealed in a heated imaging chamber, and recordings were acquired from double transfectants at an exposure time of 250 ms for 90 s. This captured dozens of transport events per cell in 50-μm segments of the dendrite or 100-μm segments of the axon. Dendrites and axons were initially identified based on morphology and confirmed retrospectively by immunostaining against MAP2, a dendritic cytoskeletal protein. Soluble BFP detection was necessary to determine the orientation of the cell body relative to the axon and thus distinguish between anterograde and retrograde transport events. Vesicle flux, velocity, and run lengths were obtained through tracing kymographs in MetaMorph. Flux was defined as the total distance traveled by vesicles standardized by the length and duration of each movie (in micrometer-minutes), 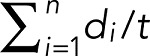 , where di are the individual DCV run lengths, l is the length of axon imaged, and t is the duration of the imaging session. A vesicle was defined as undergoing a directed run if it traveled a distance of ≥2 μm. This distance was determined as a safe estimate of the limit of diffusion based on the assumption that root-mean-squared displacement equals 2Dt, where D is the diffusion coefficient (D = 0.01 μm2/s for DCVs) and t is the duration of the imaging period (t = 50 s; Abney et al., 1999). A run was defined as terminating if the vesicle remained in the same position for at least four consecutive frames. In healthy control neurons, a significant fraction of vesicles typically pause while navigating microtubule track intersections and maneuvering around obstacles or when captured at synaptic sites of release (Trybus, 2013; Bulgari et al., 2014). Percentage flux represents the flux in treated neurons normalized to controls (100%).
, where di are the individual DCV run lengths, l is the length of axon imaged, and t is the duration of the imaging session. A vesicle was defined as undergoing a directed run if it traveled a distance of ≥2 μm. This distance was determined as a safe estimate of the limit of diffusion based on the assumption that root-mean-squared displacement equals 2Dt, where D is the diffusion coefficient (D = 0.01 μm2/s for DCVs) and t is the duration of the imaging period (t = 50 s; Abney et al., 1999). A run was defined as terminating if the vesicle remained in the same position for at least four consecutive frames. In healthy control neurons, a significant fraction of vesicles typically pause while navigating microtubule track intersections and maneuvering around obstacles or when captured at synaptic sites of release (Trybus, 2013; Bulgari et al., 2014). Percentage flux represents the flux in treated neurons normalized to controls (100%).
Immunocytochemistry
Neurons were fixed in 4% paraformaldehyde and blocked with 0.5% fish-skin gelatin (Kwinter et al., 2009). To confirm AβO binding to dendrites and verify qualitatively that AβOs remained oligomeric after 18 h in culture, cells were stained with an AβO-specific antibody (NU-4, 1:1000; from W. L. Klein, Northwestern University, Evanston, IL) or 6E10 (1:1000; Covance, Berkeley, CA) and anti-MAP2 (1:2000; Millipore, Billerica, MA). To assess axonal AβO binding and the presence of axonal ER, neurons were stained with anti–α-tubulin (1:1000, Sigma-Aldrich) or anti–reticulon 3 (1:1000; R458, from Riqiang Yan, Cleveland Clinic, Cleveland, OH) and anti–ryanodine receptor 2 (1:300; Alomone Labs), respectively. To determine AβO colocalization with VGCCs, AβO-treated cells were stained with anti-CaV 2.1, anti-CaV 2.2, and anti-CaV 2.3 (1:100; Alomone Labs). Neurons were subsequently incubated with compatible secondary antibodies conjugated to Cy3 (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA), Alexa 488 (1:500, Invitrogen), or Cy5 (1:500; Jackson ImmunoResearch Laboratories). To semiquantify AβO colocalization with VGCCs, line scans of overlapping signal intensity peaks were generated using MetaMorph. Appropriate thresholds were applied to eliminate background signal before analysis.
FRET analysis of intracellular calcium
AβO-induced changes in cytosolic Ca2+ were detected using a genetically encoded Ca2+ sensor termed cameleon (D3cpV; gift from T. Pozzan, University of Padua, Padua, Italy). The cameleon comprises two Ca2+-responsive elements, calmodulin and M13, which alter the efficiency of FRET between their respective CFP donor and cpVenus acceptor fluorophores (Palmer and Tsien, 2006). Neurons were transfected with D3cpV and mounted in heated chambers for imaging as described in the previous section. Using a scanning confocal microscope equipped with argon 457/514-nm lasers (A1R, Nikon, Melville, NY; Simon Fraser University Imaging Centre), we acquired CFP, cpVenus, and FRET (CFP excitation, cpVenus emission) images of the soma, dendrites, and axons. To assess cross-talk between the CFP and cpVenus channels, we expressed calmodulin-CFP and M13-cpVenus separately and measure donor and acceptor fluorescence through corresponding and opposing channels. Using Nikon Elements AR 3.2 software, we generated maximum-intensity projections from each Z-stack, defined regions of interest (ROIs), and calculated background-corrected FRET ratios (FRET − FRETbackground/CFP − CFPbackground) within each ROI. Each experiment was performed on 12–15 cells from at least three independent cultures. Significance was determined using Student's t test.
In situ proximal ligation assay
Calcineurin activation in dendrites and axons was detected in situ using the Duolink PLA (Sigma-Aldrich). Control and AβO-treated neurons were fixed and stained with monoclonal anti-calmodulin (1:200; EMD Millipore, Billerica, MA) and polyclonal anti–calcineurin A (1:100; Enzo Life Sciences, Farmingdale, NY) as described previously. Primary antibodies were detected with proximity probes composed of secondary antibodies conjugated to oligonucleotides, which hybridized to form circular DNA strands when CaN and CaM were in close proximity. These strands served as templates for localized rolling-circle amplification and detection with fluorescently labeled oligonucleotides. PLA probe hybridization, ligation, and amplification were performed in 40-μl open droplet reactions exactly according to the manufacturer's protocol. PLA puncta were quantified in 100-μm segments of primary dendrites and proximal axons using the Count Nuclei application in MetaMorph. Each experiment was performed on 15–20 cells from at least two independent cultures. Significance was determined using Student's t test.
Immunohistochemistry
Coronal brain sections from 3-mo-old transgenic AD mice (APP23/PS45) and age-matched, wild-type control animals were obtained from Weihong Song (University of British Columbia, Vancouver, Canada). Coronal brain sections from 12-mo-old transgenic AD mice (LaFerla 3xTg) and age-matched, wild-type control animals were obtained from Charles Krieger (Simon Fraser University, Burnaby, Canada). To detect AβOs, sections were rinsed in phosphate-buffered saline/Tween 20 (PBST), blocked in 10% donkey serum and 0.1% bovine serum albumin for 1 h, and incubated with NU-4 primary antibody (1:1000) overnight at 4°C. After further washes in PBST, sections were incubated with a compatible secondary antibody conjugated to Cy3 (1:500; Jackson ImmunoResearch Laboratories) for 1.5 h at room temperature and counterstained with 4′,6-diamidino-2-phenylindole. Images were acquired on a Nikon A1R scanning confocal system equipped with multiple laser lines (Simon Fraser University Imaging Facility).
Supplementary Material
Acknowledgments
This research was supported by grants from the National Science and Engineering Research Council of Canada (327100-06) and the Canadian Institutes of Health Research (90396) to M.A.S. We are grateful to L. Chen for expert technical assistance and D. Poburko for essential discussions. We thank P. Copenhaver and G. Morfini for critical reading of the manuscript, C. Krieger for the Laferla 3xTg mice brains, and W. Song for the APP23/PS45 mice brains. K.J.G. is funded by a C.D. Nelson Memorial Graduate Scholarship from Simon Fraser University and a National Science and Engineering Research Council of Canada Postgraduate Scholarship. We also thank the Simon Fraser University Animal Care Services staff for their essential assistance in our experiments.
Abbreviations used:
- AβO
amyloid-β oligomer
- AD
Alzheimer's disease
- BDNF
brain-derived neurotrophic factor
- CaN
calcineurin
- DCV
dense-core vesicle
- FAT
fast axonal transport
- VGCC
voltage-gated calcium channel.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-12-1612) on January 21, 2015.
K.J.G. designed the study, performed all experiments, analyzed and interpreted data, and cowrote the article. M.A.S. designed and supervised the study, interpreted data, and cowrote the article.
REFERENCES
- Abney JR, Meliza CD, Cutler B, Kingma M, Lochner JE, Scalettar BA. Real-time imaging of the dynamics of secretory granules in growth cones. Biophys J. 1999;77:2887–2895. doi: 10.1016/S0006-3495(99)77120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anekonda TS, Quinn JF. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: the case for isradipine. Biochim Biophys Acta. 2011;1812:1584–1590. doi: 10.1016/j.bbadis.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium hypothesis of Alzheimer's disease. Pflugers Arch. 2010;459:441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- Bobich JA, Zheng Q, Campbell A. Incubation of nerve endings with a physiological concentration of Abeta1-42 activates CaV2.2(N-Type)-voltage operated calcium channels and acutely increases glutamate and noradrenaline release. J Alzheimers Dis. 2004;6:243–255. doi: 10.3233/jad-2004-6305. [DOI] [PubMed] [Google Scholar]
- Bulgari D, Zhou C, Hewes RS, Deitcher DL, Levitan ES. Vesicle capture, not delivery, scales up neuropeptide storage in neuroendocrine terminals. Proc Natl Acad Sci USA. 2014;111:3597–3601. doi: 10.1073/pnas.1322170111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A, Lopez-Rosas A, Morfini G, Pigino G, Sands M, et al. The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3beta and deregulation of molecular motors. J Neurosci. 2013;33:10048–10056. doi: 10.1523/JNEUROSCI.0217-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldi M. The changing landscape of voltage-gated calcium channels in neurovascular disorders and in neurodegenerative diseases. Curr Neuropharmacol. 2013;11:276–297. doi: 10.2174/1570159X11311030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, et al. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PLoS One. 2012;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Stutzmann GE. Early calcium dysregulation in Alzheimer's disease: setting the stage for synaptic dysfunction. Sci China Life Sci. 2011;54:752–762. doi: 10.1007/s11427-011-4205-7. [DOI] [PubMed] [Google Scholar]
- Cochran JN, Hall AM, Roberson ED. The dendritic hypothesis for Alzheimer's disease pathophysiology. Brain Res Bull. 2013;103:18–28. doi: 10.1016/j.brainresbull.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver PF, Anekonda TS, Musashe D, Robinson KM, Ramaker JM, Swanson TL, Wadsworth TL, Kretzschmar D, Woltjer RL, Quinn JF. A translational continuum of model systems for evaluating treatment strategies in Alzheimer's disease: isradipine as a candidate drug. Dis Model Mech. 2011;4:634–648. doi: 10.1242/dmm.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker H, Jurgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-beta peptide oligomers. J Neurochem. 2010a;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010b;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, He W, Tan Y, Han H, Hu X, Xia K, Zhang Z, Yan R. Increased expression of reticulon 3 in neurons leads to reduced axonal transport of beta site amyloid precursor protein-cleaving enzyme 1. J Biol Chem. 2013;288:30236–30245. doi: 10.1074/jbc.M113.480079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma K, Iacobucci GJ, Hong Zheng K, Shandilya J, Toska E, White JA, 2nd, Spina E, Gunawardena S. Presenilin influences glycogen synthase kinase-3 beta (GSK-3beta) for kinesin-1 and dynein function during axonal transport. Hum Mol Genet. 2014;23:1121–1133. doi: 10.1093/hmg/ddt505. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13:542–555. doi: 10.1038/nrn3311. [DOI] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- Goldstein LS. Axonal transport and neurodegenerative disease: can we see the elephant. Prog Neurobiol. 2012;99:186–190. doi: 10.1016/j.pneurobio.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann D, Mezler M, Muller MK, Wicke K, Gross G, Draguhn A, Bruehl C, Nimmrich V. Synthetic Abeta oligomers (Abeta(1-42) globulomer) modulate presynaptic calcium currents: prevention of Abeta-induced synaptic deficits by calcium channel blockers. Eur J Pharmacol. 2013;702:44–55. doi: 10.1016/j.ejphar.2013.01.030. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Gotz J. Amyloid-beta and tau—a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JV, Zhang YV, Kramer S, Brenman JE, Rasse TM. The kinesin-3, unc-104 regulates dendrite morphogenesis and synaptic development in Drosophila. Genetics. 2013;195:59–72. doi: 10.1534/genetics.113.151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo H, Kayed R, Glabe CG, Yamaguchi H. Soluble Abeta oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer's disease brain. Brain Res. 2005a;1031:222–228. doi: 10.1016/j.brainres.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Kokubo H, Saido TC, Iwata N, Helms JB, Shinohara R, Yamaguchi H. Part of membrane-bound Abeta exists in rafts within senile plaques in Tg2576 mouse brain. Neurobiol Aging. 2005b;26:409–418. doi: 10.1016/j.neurobiolaging.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwinter D, Silverman MA. Live imaging of dense-core vesicles in primary cultured hippocampal neurons. J Vis Exp. 2009;27:1144. doi: 10.3791/1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, Ho H, Brady ST, Sisodia SS. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci. 2007;27:7011–7020. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Shin H, Ko J, Choi J, Lee H, Kim E. Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. J Biol Chem. 2003;278:2624–2629. doi: 10.1074/jbc.M211152200. [DOI] [PubMed] [Google Scholar]
- Leitch B, Szostek A, Lin R, Shevtsova O. Subcellular distribution of L-type calcium channel subtypes in rat hippocampal neurons. Neuroscience. 2009;164:641–657. doi: 10.1016/j.neuroscience.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Lo KY, Kuzmin A, Unger SM, Petersen JD, Silverman MA. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci Lett. 2011;491:168–173. doi: 10.1016/j.neulet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Maeder CI, San-Miguel A, Wu EY, Lu H, Shen K. In vivo neuron-wide analysis of synaptic vesicle precursor trafficking. Traffic. 2014;15:273–291. doi: 10.1111/tra.12142. [DOI] [PubMed] [Google Scholar]
- Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Cross D. In vivo imaging of axonal transport using MRI: aging and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35((Suppl 1)):S89–S92. doi: 10.1007/s00259-007-0707-8. [DOI] [PubMed] [Google Scholar]
- Miranda CJ, Braun L, Jiang Y, Hester ME, Zhang L, Riolo M, Wang H, Rao M, Altura RA, Kaspar BK. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell. 2012;11:542–552. doi: 10.1111/j.1474-9726.2012.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orefice LL, Waterhouse EG, Partridge JG, Lalchandani RR, Vicini S, Xu B. Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J Neurosci. 2013;33:11618–11632. doi: 10.1523/JNEUROSCI.0012-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oules B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Brechot P, Trebak M, Checler F, et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- Paula-Lima AC, Adasme T, SanMartin C, Sebollela A, Hetz C, Carrasco MA, Ferreira ST, Hidalgo C. Amyloid beta-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal. 2011;14:1209–1223. doi: 10.1089/ars.2010.3287. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol Brain. 2009;2:33. doi: 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Henderson Z, Pearson HA. Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1-40) is dependent on solubility status. Brain Res. 2002;956:254–261. doi: 10.1016/s0006-8993(02)03547-3. [DOI] [PubMed] [Google Scholar]
- Ramser EM, Gan KJ, Decker H, Fan EY, Suzuki MM, Ferreira ST, Silverman MA. Amyloid-beta oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell. 2013;24:2494–2505. doi: 10.1091/mbc.E12-12-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Taglialatela G. A role for calcineurin in Alzheimer's disease. Curr Neuropharmacol. 2011;9:685–692. doi: 10.2174/157015911798376316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Mattson MP. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience. 2013;239:228–240. doi: 10.1016/j.neuroscience.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Shaw JL, Chang KT. Nebula/DSCR1 upregulation delays neurodegeneration and protects against APP-induced axonal transport defects by restoring calcineurin and GSK-3beta signaling. PLoS Genet. 2013;9:e1003792. doi: 10.1371/journal.pgen.1003792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Fukaya M, Yamasaki M, Watanabe M, Manabe T, Kamiya H. Use-dependent amplification of presynaptic Ca2+ signaling by axonal ryanodine receptors at the hippocampal mossy fiber synapse. Proc Natl Acad Sci USA. 2008;105:11998–12003. doi: 10.1073/pnas.0802175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Paylor R, Pautler RG. R-flurbiprofen improves axonal transport in the Tg2576 mouse model of Alzheimer's disease as determined by MEMRI. Magn Reson Med. 2011;65:1423–1429. doi: 10.1002/mrm.22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Almenar-Queralt A, Gunawardena S, Rodrigues EM, Falzone T, Kim J, Lillo C, Mount SL, Roberts EA, McGowan E, et al. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum Mol Genet. 2008;17:3474–3486. doi: 10.1093/hmg/ddn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann NY Acad Sci. 2007;1097:265–277. doi: 10.1196/annals.1379.025. [DOI] [PubMed] [Google Scholar]
- Trybus KM. Intracellular transport: the causes for pauses. Curr Biol. 2013;23:R623–R625. doi: 10.1016/j.cub.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver C, Leidel C, Szpankowski L, Farley NM, Shubeita GT, Goldstein LS. Endogenous GSK-3/shaggy regulates bidirectional axonal transport of the amyloid precursor protein. Traffic. 2013;14:295–308. doi: 10.1111/tra.12037. [DOI] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, et al. Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry. 2014;53:3908–3921. doi: 10.1021/bi5003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Shakiryanova D, Levitan ES. Presynaptic ryanodine receptor-CamKII signaling is required for activity-dependent capture of transiting vesicles. J Mol Neurosci. 2009;37:146–150. doi: 10.1007/s12031-008-9080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-beta revealed by a novel fluorescence resonance energy transfer assay. J Neurosci. 2012;32:5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Takei Y, Kido MA, Hirokawa N. Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron. 2011;70:310–325. doi: 10.1016/j.neuron.2011.02.049. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Zhao JP, Pandian S, van Zundert B, Constantine-Paton M. A Myosin Va mutant mouse with disruptions in glutamate synaptic development and mature plasticity in visual cortex. J Neurosci. 2013;33:8472–8482. doi: 10.1523/JNEUROSCI.4585-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.